
MCPH1: A Novel Case Report and a Review of the Literature

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
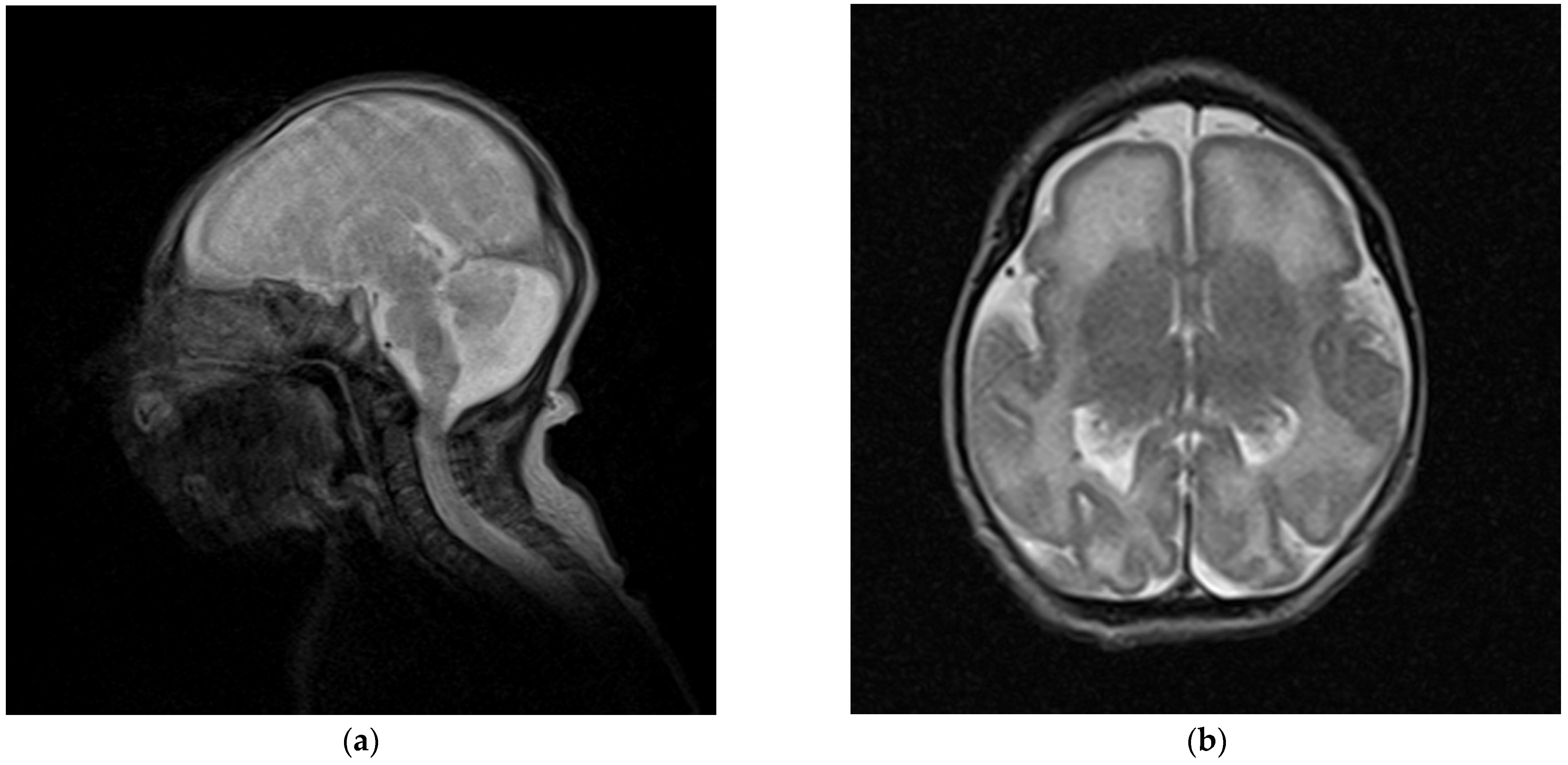
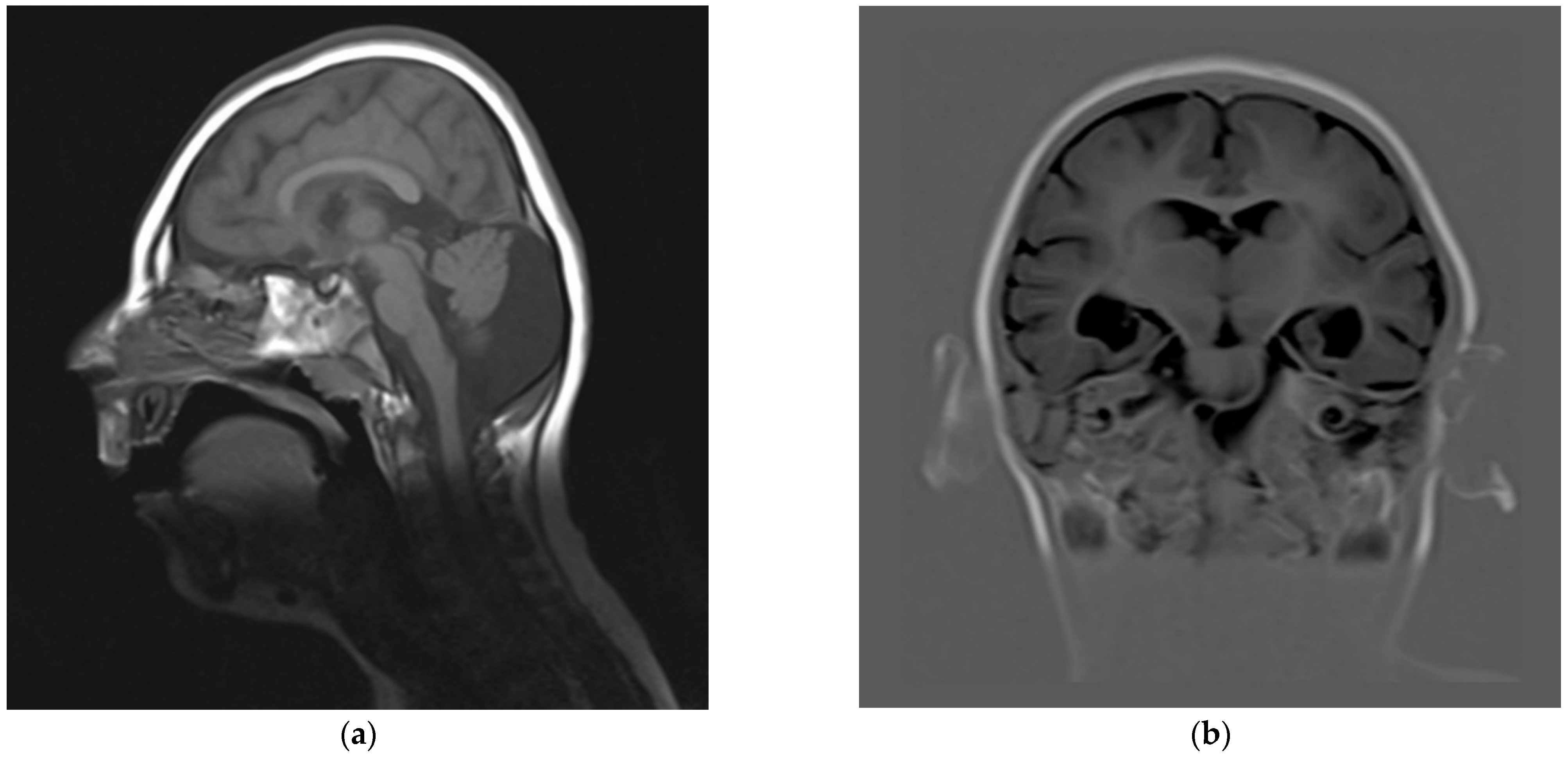
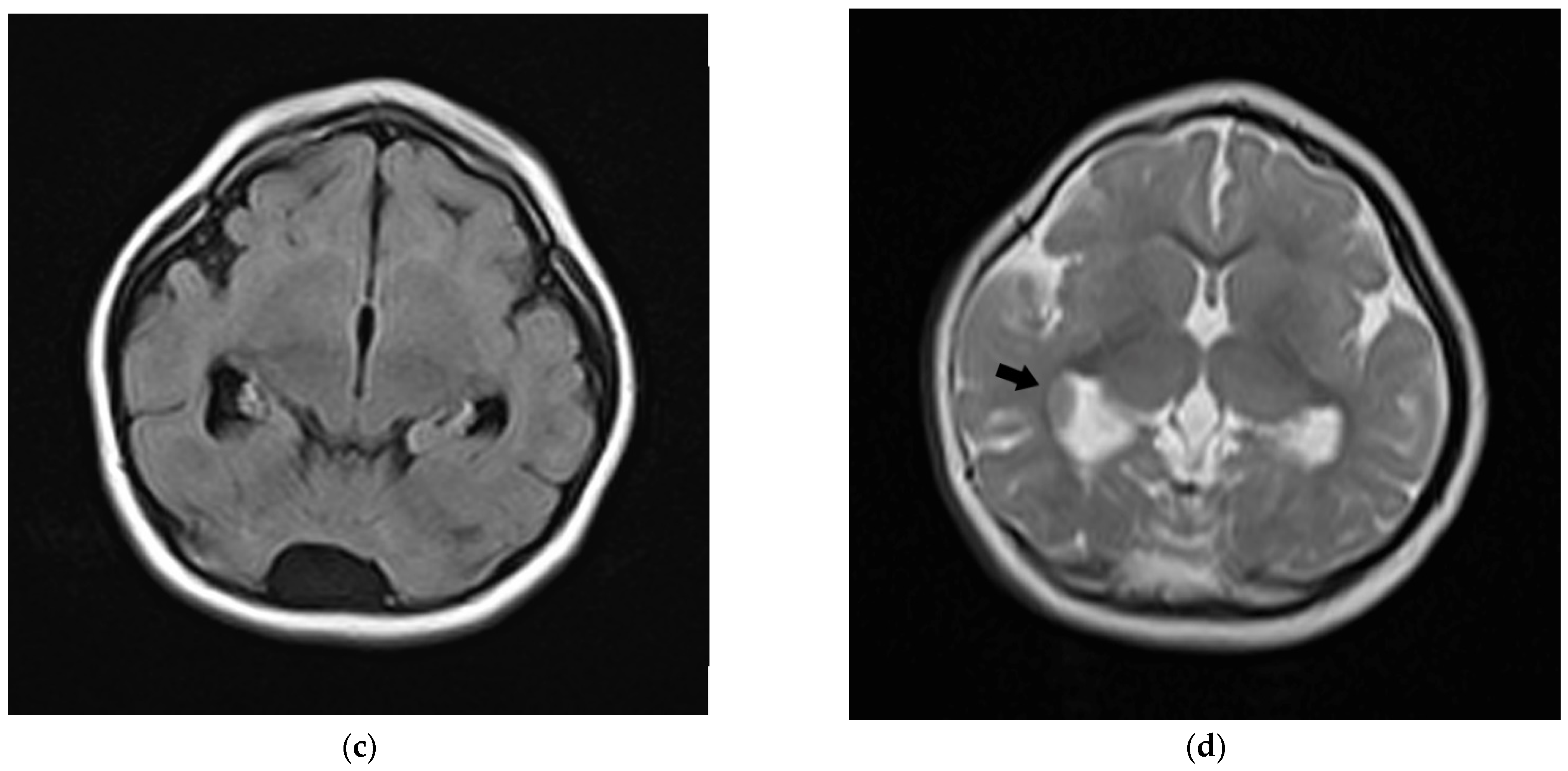
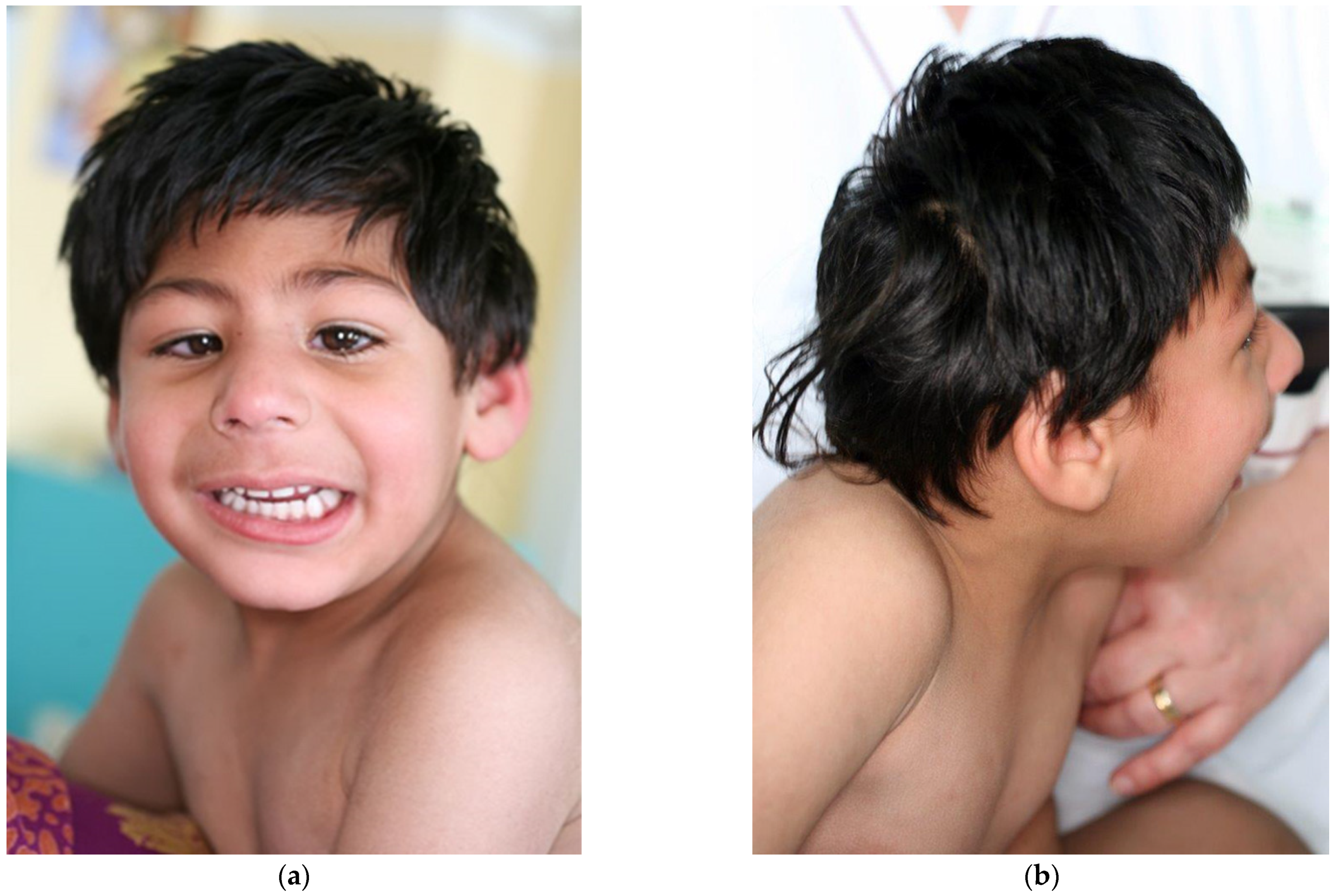
2.1. Clinical Report
2.2. Molecular Diagnosis
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jayaraman, D.; Bae, B.-I.; Walsh, C.A. The Genetics of Primary Microcephaly. Annu. Rev. Genom. Hum. Genet. 2018, 19, 177–200. [Google Scholar] [CrossRef] [Green Version]
- Farooq, M.; Lindbæk, L.; Krogh, N.; Doganli, C.; Keller, C.; Mönnich, M.; Gonçalves, A.B.; Sakthivel, S.; Mang, Y.; Fatima, A.; et al. RRP7A Links Primary Microcephaly to Dysfunction of Ribosome Biogenesis, Resorption of Primary Cilia, and Neurogenesis. Nat. Commun. 2020, 11, 5816. [Google Scholar] [CrossRef]
- Duerinckx, S.; Meuwissen, M.; Perazzolo, C.; Desmyter, L.; Pirson, I.; Abramowicz, M. Phenotypes in Siblings with Homozygous Mutations of TRAPPC9 and/or MCPH1 Support a Bifunctional Model of MCPH1. Mol. Genet. Genom. Med. 2018, 6, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Duerinckx, S.; Abramowicz, M. The Genetics of Congenitally Small Brains. Semin. Cell Dev. Biol. 2018, 76, 76–85. [Google Scholar] [CrossRef]
- Thornton, G.K.; Woods, C.G. Primary Microcephaly: Do All Roads Lead to Rome? Trends Genet. 2009, 25, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Pulvers, J.N. MCPH1: A Window into Brain Development and Evolution. Front. Cell. Neurosci. 2015, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Darvish, H.; Esmaeeli-Nieh, S.; Monajemi, G.B.; Mohseni, M.; Ghasemi-Firouzabadi, S.; Abedini, S.S.; Bahman, I.; Jamali, P.; Azimi, S.; Mojahedi, F.; et al. A Clinical and Molecular Genetic Study of 112 Iranian Families with Primary Microcephaly. J. Med. Genet. 2010, 47, 823–828. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.P.; McHale, D.P.; Campbell, D.A.; Jafri, H.; Rashid, Y.; Mannan, J.; Karbani, G.; Corry, P.; Levene, M.I.; Mueller, R.F.; et al. Primary Autosomal Recessive Microcephaly (MCPH1) Maps to Chromosome 8p22-Pter. Am. J. Hum. Genet. 1998, 63, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.P.; Eastwood, H.; Bell, S.M.; Adu, J.; Toomes, C.; Carr, I.M.; Roberts, E.; Hampshire, D.J.; Crow, Y.J.; Mighell, A.J.; et al. Identification of Microcephalin, a Protein Implicated in Determining the Size of the Human Brain. Am. J. Hum. Genet. 2002, 71, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Gavvovidis, I.; Rost, I.; Trimborn, M.; Kaiser, F.J.; Purps, J.; Wiek, C.; Hanenberg, H.; Neitzel, H.; Schindler, D. A Novel MCPH1 Isoform Complements the Defective Chromosome Condensation of Human MCPH1-Deficient Cells. PLoS ONE 2012, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Jeffers, L.J.; Coull, B.J.; Stack, S.J.; Morrison, C.G. Distinct BRCT Domains in Mcph1/Brit1 Mediate Ionizing Radiation-Induced Focus Formation and Centrosomal Localization. Oncogene 2008, 27, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.-Y.; Rai, R.; Li, K.; Xu, Z.-X.; Elledge, S.J. BRIT1/MCPH1 Is a DNA Damage Responsive Protein That Regulates the Brca1-Chk1 Pathway, Implicating Checkpoint Dysfunction in Microcephaly. Proc. Natl. Acad. Sci. USA 2005, 102, 15105–15109. [Google Scholar] [CrossRef] [Green Version]
- Rai, R.; Dai, H.; Multani, A.S.; Li, K.; Chin, K.; Gray, J.; Lahad, J.P.; Liang, J.; Mills, G.B.; Meric-Bernstam, F.; et al. BRIT1 Regulates Early DNA Damage Response, Chromosomal Integrity, and Cancer. Cancer Cell 2006, 10, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Cicconi, A.; Rai, R.; Xiong, X.; Broton, C.; Al-Hiyasat, A.; Hu, C.; Dong, S.; Sun, W.; Garbarino, J.; Bindra, R.S.; et al. Microcephalin 1/BRIT1-TRF2 Interaction Promotes Telomere Replication and Repair, Linking Telomere Dysfunction to Primary Microcephaly. Nat. Commun. 2020, 11, 5861. [Google Scholar] [CrossRef]
- Gruber, R.; Zhou, Z.; Sukchev, M.; Joerss, T.; Frappart, P.-O.; Wang, Z.-Q. MCPH1 Regulates the Neuroprogenitor Division Mode by Coupling the Centrosomal Cycle with Mitotic Entry through the Chk1–Cdc25 Pathway. Nat. Cell Biol. 2011, 13, 1325–1334. [Google Scholar] [CrossRef]
- Barbelanne, M.; Tsang, W.Y. Molecular and Cellular Basis of Autosomal Recessive Primary Microcephaly. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef]
- Yamashita, D.; Shintomi, K.; Ono, T.; Gavvovidis, I.; Schindler, D.; Neitzel, H.; Trimborn, M.; Hirano, T. MCPH1 Regulates Chromosome Condensation and Shaping as a Composite Modulator of Condensin II. J. Cell Biol. 2011, 194, 841–854. [Google Scholar] [CrossRef] [Green Version]
- Houlard, M.; Cutts, E.E.; Shamim, M.S.; Godwin, J.; Weisz, D.; Presser Aiden, A.; Lieberman Aiden, E.; Schermelleh, L.; Vannini, A.; Nasmyth, K. MCPH1 Inhibits Condensin II during Interphase by Regulating Its SMC2-Kleisin Interface. eLife 2021, 10, e73348. [Google Scholar] [CrossRef]
- Arroyo, M.; Kuriyama, R.; Trimborn, M.; Keifenheim, D.; Cañuelo, A.; Sánchez, A.; Clarke, D.J.; Marchal, J.A. MCPH1, Mutated in Primary Microcephaly, Is Required for Efficient Chromosome Alignment during Mitosis. Sci. Rep. 2017, 7, 13019. [Google Scholar] [CrossRef] [Green Version]
- Neitzel, H.; Neumann, L.M.; Schindler, D.; Wirges, A.; Tönnies, H.; Trimborn, M.; Krebsova, A.; Richter, R.; Sperling, K. Premature Chromosome Condensation in Humans Associated with Microcephaly and Mental Retardation: A Novel Autosomal Recessive Condition. Am. J. Hum. Genet. 2002, 70, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Zaqout, S.; Morris-Rosendahl, D.; Kaindl, A. Autosomal Recessive Primary Microcephaly (MCPH): An Update. Neuropediatrics 2017, 48, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, D.; O’Driscoll, M. Congenital Microcephaly: AMERICAN JOURNAL OF MEDICAL GENETICS PART C (SEMINARS IN MEDICAL GENETICS). Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 124–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavone, P.; Pappalardo, X.G.; Praticò, A.D.; Polizzi, A.; Ruggieri, M.; Piccione, M.; Corsello, G.; Falsaperla, R. Primary Microcephaly with Novel Variant of MCPH1 Gene in Twins: Both Manifesting in Childhood at the Same Time with Hashimoto’s Thyroiditis. J. Pediatr. Genet. 2020, 09, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Trimborn, M.; Bell, S.M.; Felix, C.; Rashid, Y.; Jafri, H.; Griffiths, P.D.; Neumann, L.M.; Krebs, A.; Reis, A.; Sperling, K.; et al. Mutations in Microcephalin Cause Aberrant Regulation of Chromosome Condensation. Am. J. Hum. Genet. 2004, 75, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E. Autosomal Recessive Primary Microcephaly: An Analysis of Locus Heterogeneity and Phenotypic Variation. J. Med. Genet. 2002, 39, 718–721. [Google Scholar] [CrossRef] [Green Version]
- Trimborn, M.; Richter, R.; Sternberg, N.; Gavvovidis, I.; Schindler, D.; Jackson, A.P.; Prott, E.-C.; Sperling, K.; Gillessen-Kaesbach, G.; Neitzel, H. The First Missense Alteration in the MCPH1 Gene Causes Autosomal Recessive Microcephaly with an Extremely Mild Cellular and Clinical Phenotype. Hum. Mutat. 2005, 26, 496. [Google Scholar] [CrossRef]
- Garshasbi, M.; Motazacker, M.M.; Kahrizi, K.; Behjati, F.; Abedini, S.S.; Nieh, S.E.; Firouzabadi, S.G.; Becker, C.; Rüschendorf, F.; Nürnberg, P.; et al. SNP Array-Based Homozygosity Mapping Reveals MCPH1 Deletion in Family with Autosomal Recessive Mental Retardation and Mild Microcephaly. Hum. Genet. 2006, 118, 708–715. [Google Scholar] [CrossRef]
- Farooq, M.; Baig, S.; Tommerup, N.; Kjaer, K.W. Craniosynostosis-Microcephaly with Chromosomal Breakage and Other Abnormalities Is Caused by a Truncating MCPH1 Mutation and Is Allelic to Premature Chromosomal Condensation Syndrome and Primary Autosomal Recessive Microcephaly Type 1. Am. J. Med. Genet. A 2010, 152, 495–497. [Google Scholar] [CrossRef]
- Tommerup, N.; Mortensen, E.; Nielsen, M.H.; Wegner, R.-D.; Schindler, D.; Mikkelsen, M. Chromosomal Breakage, Endomitosis, Endoreduplication, and Hypersensitivity toward Radiomimetric and Alkylating Agents: A Possible New Autosomal Recessive Mutation in a Girl with Craniosynostosis and Microcephaly. Hum. Genet. 1993, 92, 339–346. [Google Scholar] [CrossRef]
- Leung, J.W.; Leitch, A.; Wood, J.L.; Shaw-Smith, C.; Metcalfe, K.; Bicknell, L.S.; Jackson, A.P.; Chen, J. SET Nuclear Oncogene Associates with Microcephalin/MCPH1 and Regulates Chromosome Condensation. J. Biol. Chem. 2011, 286, 21393–21400. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, M.M.; Tonekaboni, S.H.; Papari, E.; Bahman, I.; Behjati, F.; Kahrizi, K.; Najmabadi, H. A Novel Mutation in MCPH1 Gene in an Iranian Family with Primary Microcephaly. J. Pak. Med. Assoc. 2012, 62, 4. [Google Scholar]
- Ghani-Kakhki, M.; Robinson, P.N.; Morlot, S.; Mitter, D.; Trimborn, M.; Albrecht, B.; Varon, R.; Sperling, K.; Neitzel, H. Two Missense Mutations in the Primary Autosomal Recessive Microcephaly Gene MCPH1 Disrupt the Function of the Highly Conserved N-terminal BRCT Domain of Microcephalin. Mol. Syndromol. 2012, 3, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfau, R.B.; Thrush, D.L.; Hamelberg, E.; Bartholomew, D.; Botes, S.; Pastore, M.; Tan, C.; del Gaudio, D.; Gastier-Foster, J.M.; Astbury, C. MCPH1 Deletion in a Newborn with Severe Microcephaly and Premature Chromosome Condensation. Eur. J. Med. Genet. 2013, 56, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Perche, O.; Menuet, A.; Marcos, M.; Liu, L.; Pâris, A.; Utami, K.H.; Kervran, D.; Cacheux, V.; Laudier, B.; Briault, S. Combined Deletion of Two Condensin II System Genes (NCAPG2 and MCPH1) in a Case of Severe Microcephaly and Mental Deficiency. Eur. J. Med. Genet. 2013, 56, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Sajid Hussain, M.; Marriam Bakhtiar, S.; Farooq, M.; Anjum, I.; Janzen, E.; Reza Toliat, M.; Eiberg, H.; Kjaer, K.; Tommerup, N.; Noegel, A.; et al. Genetic Heterogeneity in Pakistani Microcephaly Families: Pakistani Microcephaly Mutations. Clin. Genet. 2013, 83, 446–451. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Fardaei, M.; Gholami, M.; Miryounesi, M. A Case Report: Autosomal Recessive Microcephaly Caused by a Novel Mutation in MCPH1 Gene. Gene 2015, 571, 149–150. [Google Scholar] [CrossRef]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, I.; Baig, S.M.; Abdulkareem, A.R.; Hussain, M.S.; Sur, I.; Toliat, M.R.; Nürnberg, G.; Dalibor, N.; Moawia, A.; Waseem, S.S.; et al. Genetic Heterogeneity in Pakistani Microcephaly Families Revisited: Genetic Heterogeneity in Pakistani Microcephaly Families. Clin. Genet. 2017, 92, 62–68. [Google Scholar] [CrossRef]
- Hemmat, M.; Rumple, M.J.; Mahon, L.W.; Morrow, M.; Zach, T.; Anguiano, A.; Elnaggar, M.M.; Wang, B.T.; Boyar, F.Z. CMA Analysis Identifies Homozygous Deletion of MCPH1 in 2 Brothers with Primary Microcephaly-1. Mol. Cytogenet. 2017, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Naseer, M.; Rasool, M.; Abdulkareem, A.; Bassiouni, R.; Algahtani, H.; Chaudhary, A.; Al-Qahtani, M. Novel Compound Heterozygous Mutations in MCPH1 Gene Causes Primary Microcephaly in Saudi Family. Neurosciences 2018, 23, 347–350. [Google Scholar] [CrossRef]
- McSherry, M.; Masih, K.E.; Elcioglu, N.H.; Celik, P.; Balci, O.; Cengiz, F.B.; Nunez, D.; Sineni, C.J.; Seyhan, S.; Kocaoglu, D.; et al. Identification of Candidate Gene FAM183A and Novel Pathogenic Variants in Known Genes: High Genetic Heterogeneity for Autosomal Recessive Intellectual Disability. PLoS ONE 2018, 13, e0208324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budday, S.; Steinmann, P.; Kuhl, E. Physical Biology of Human Brain Development. Front. Cell. Neurosci. 2015, 9, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germanaud, D.; Lefèvre, J.; Fischer, C.; Bintner, M.; Curie, A.; des Portes, V.; Eliez, S.; Elmaleh-Bergès, M.; Lamblin, D.; Passemard, S.; et al. Simplified Gyral Pattern in Severe Developmental Microcephalies? New Insights from Allometric Modeling for Spatial and Spectral Analysis of Gyrification. NeuroImage 2014, 102, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-W.; Tapias, A.; Bruhn, C.; Gruber, R.; Sukchev, M.; Wang, Z.-Q. DNA Damage Response in Microcephaly Development of MCPH1 Mouse Model. DNA Repair 2013, 12, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Yim, E.-K.; Dai, H.; Jackson, A.P.; van der Burgt, I.; Pan, M.-R.; Hu, R.; Li, K.; Lin, S.-Y. BRIT1/MCPH1 Links Chromatin Remodelling to DNA Damage Response. Nat. Cell Biol. 2009, 11, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Trimborn, M.; Ghani, M.; Walther, D.J.; Dopatka, M.; Dutrannoy, V.; Busche, A.; Meyer, F.; Nowak, S.; Nowak, J.; Zabel, C.; et al. Establishment of a Mouse Model with Misregulated Chromosome Condensation Due to Defective Mcph1 Function. PLoS ONE 2010, 5, e9242. [Google Scholar] [CrossRef]
- Venkatesh, T.; Suresh, P.S. Emerging Roles of MCPH1: Expedition from Primary Microcephaly to Cancer. Eur. J. Cell Biol. 2014, 93, 98–105. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Jackson, A.; Jeggo, P.A. Microcephalin: A Causal Link Between Impaired Damage Response Signalling and Microcephaly. Cell Cycle 2006, 5, 2339–2344. [Google Scholar] [CrossRef]
- Sharma, R.; Lewis, S.; Wlodarski, M.W. DNA Repair Syndromes and Cancer: Insights Into Genetics and Phenotype Patterns. Front. Pediatr. 2020, 8, 570084. [Google Scholar] [CrossRef]
- Liang, Y.; Gao, H.; Lin, S.-Y.; Peng, G.; Huang, X.; Zhang, P.; Goss, J.A.; Brunicardi, F.C.; Multani, A.S.; Chang, S.; et al. BRIT1/MCPH1 Is Essential for Mitotic and Meiotic Recombination DNA Repair and Maintaining Genomic Stability in Mice. PLoS Genet. 2010, 6, e1000826. [Google Scholar] [CrossRef] [Green Version]
- Gavvovidis, I.; Pöhlmann, C.; Marchal, J.A.; Stumm, M.; Yamashita, D.; Hirano, T.; Schindler, D.; Neitzel, H.; Trimborn, M. MCPH1 Patient Cells Exhibit Delayed Release from DNA Damage-Induced G 2 /M Checkpoint Arrest. Cell Cycle 2010, 9, 4893–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Ref. | Patients #a | Variant Types | Birth OFC, in SD | OFC (at Age), in SD | Stature, in SD | DD/ID | MRI Findings | Other Findings | Ethnic Origin |
|---|---|---|---|---|---|---|---|---|---|
| [8,9,24] | 7 (2 peds) | ns | MC | −5 to −10 (13–28 y) | variable | mild to moderate ID | reduced cortex size, mild cerebellar hypoplasia | no | Pakistani (Mirpur) |
| [20,24] | 2 sibs | fs | −3.5 to −4 | −8 to −10 (5−7 y) | −2 to −5 | DD, profound ID (poor or no language) | reduced cortex size, pachygyria, mild cerebellar hypoplasia, PNH, ventriculomegaly | slightly upslanted palpebral fissures, thin upper lip (hyperreflexia in one sib) | nk |
| [25] | 7 (2 peds) | linkage only | nk | −7 to −10 (0−50 y entire MCPH cohort) | nk | mild to moderate ID (language delay?) | nk | sloping forehead in some cases | Pakistani (north) |
| [26] | 1 | mis in BRCT1 | −2 | −3 (6 y) | −0.5 (normal) | mild ID (normal language, mild fine motor delay) | nk | mild PCC effect; 2–3 toe syndactyly, upslanted palpebral fissures | German and nk |
| [7,27] | 7 (1 ped) | del ex 1−6 | nk | −3 (18−32 y) | −2.5 to −3 | moderate ID | nk | no | Iranian (north) |
| [28,29] | 1 | ns | nk | −9 (16 y) | −3 | moderate ID | nk | craniosynostosis, ptosis, mild micrognathia, mild exotropia | Danish |
| [7] | 2 related | del ex 4 | nk | −10 to −11 | nk | moderate ID | nk | no | Iranian |
| [7] | 3 related | fs | nk | −6 | nk | moderate ID | nk | no | Iranian |
| [7] | 3 related | del ex 2−3 | nk | −6 to −8 | nk | mild to moderate ID | nk | no | Iranian |
| [7] | 2 related | splice site | nk | −9 | nk | severe ID | nk | no | Iranian |
| [7] | 4 related | mis in BRCT1 | nk | −7 to −9 | nk | moderate ID | nk | no | Iranian |
| [7] | 2 related | del ex 3 | nk | −6 to −10 | nk | mild to moderate ID | nk | no | Iranian |
| [7] | 3 related | mis in BRCT1 | nk | −6 to −7 | nk | mild to moderate ID | nk | (ataxia and autism, not linked with MCPH1 locus) | Iranian |
| [30] | 1 | mis in BRCT1 | nk | <−3.5 (6 m) | nk | nk | nk | prenatal cystic hygroma | nk |
| [31] | 1 | ns | −5 | −4 (2 y 4 m) | normal | DD/ID (no language) | pachygyria, mild corpus callosum hypoplasia | no | Iranian (northwest) |
| [32] | 1 (pt.1) | mis in BRCT1 | −3.5 | −6 (5 y 2 m) | −3 | mild DD/ID | mild ventriculomegaly, small frontal lobes | mild hypotonia | Iraqi |
| [32] | 1 (pt.2) | mis in BRCT1 | −6 | −5 (6 y 9 m) | −0.5 (normal) | mild DD/ID | nk | no | Turkish |
| [33] | 1 | del ex 1−11 | −3 | −5 (10 m) | −2 | nk (infant) | coarsened gyral pattern with reduced number of sulci (normal cortex thickness and cerebellum) | small anterior fontanelle, closed posterior fontanelle | Indian |
| [34] | 1 | del ex 6−9 + 7qter del | −1.5 | −5 (8 y) | −2 | severe DD/ID (no language) | normal | failure to thrive, GERD, downslanted palpebral fissures, large ears, scoliosis | nk |
| [35] | 4 related | fs | nk | −8 to −10 (10−18 y) | normal | ID (no language) | nk | aggressive behavior | Pakistani |
| [36] | 2 sibs | splicing/del in-frame | −2.5 to −3 | −5 to −12 (15−18 y) | −1 to −3 | none to severe ID | normal | no | Iranian |
| [37] | 2 related | del ex 1−2 | nk | MC | nk | DD/ID | nk | nk | nk |
| [38] | 7 related | del ex 1−2 | nk | −6 to −7 (18−44 y) | normal | mild ID | nk | no | Pakistani (Punjabi) |
| [38] | 3 related | del ex 1−11 | nk | −6 to −7 (10−27 y) | normal | mild ID | nk | irritability | Pakistani (Baloch) |
| [39] | 2 sibs | del ex 1−8 | nk | −3.5 (3−14 y) | short | mild DD, ID (poor language) | normal (small lipoma) | epicanthus, esotropia, low hairline, large ears, thin upper lip (right lung hypoplasia in 1 sib) | Hispanic |
| [40] | 2 sibs | mis central region | MC | −5 to −6 (5−10 y) | nk | severe ID (poor language) | nk | sloping forehead | Saudi |
| [41] | 1 | fs | nk | MC (1.5 m) | normal | nk (infant) | cortical atrophy, deep sulcation | long philtrum, large ears, dermatitis | Turkish |
| [3] | 2 sibs | ns b + biallelic TRAPPC9 mis | MC | −3 to −4 (13−16 y) | −1.5 (low normal) | DD, severe ID (poor or no language) | corpus callosum and cerebellum hypoplasia/atrophy, mild colpocephaly | hyperkinetic movements, epilepsy | Moroccan |
| This report | 1 | del ex 1−8 | −3.5 | −5 (10 y) | −2 | mild DD, profound ID (no language) | fronto-polar simplified gyral pattern, PNH, mild ventriculomegaly, enlarged posterior fossa | sloping forehead, highly arched eyebrow, exotropia, epicanthus, large ears; hyperactivity | Pakistani (northeast) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caraffi, S.G.; Pollazzon, M.; Farooq, M.; Fatima, A.; Larsen, L.A.; Zuntini, R.; Napoli, M.; Garavelli, L. MCPH1: A Novel Case Report and a Review of the Literature. Genes 2022, 13, 634. https://doi.org/10.3390/genes13040634
Caraffi SG, Pollazzon M, Farooq M, Fatima A, Larsen LA, Zuntini R, Napoli M, Garavelli L. MCPH1: A Novel Case Report and a Review of the Literature. Genes. 2022; 13(4):634. https://doi.org/10.3390/genes13040634
Chicago/Turabian StyleCaraffi, Stefano Giuseppe, Marzia Pollazzon, Muhammad Farooq, Ambrin Fatima, Lars Allan Larsen, Roberta Zuntini, Manuela Napoli, and Livia Garavelli. 2022. "MCPH1: A Novel Case Report and a Review of the Literature" Genes 13, no. 4: 634. https://doi.org/10.3390/genes13040634