
SPECC1L Mutations Are Not Common in Sporadic Cases of Opitz G/BBB Syndrome

 ,
, 
Abstract
:1. Introduction
2. Patients and Methods
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Opitz, J.M.; Frías, J.L.; Gutenberger, J.E.; Pellet, J.R. The G syndrome of multiple congenital anomalies. Birth Defects Orig. Artic. Ser. 1969, 2, 95–102. [Google Scholar]
- Opitz, J.M.; Summitt, R.L.; Smith, D.W. The BBB syndrome familial telecanthus with associated congenital anomalies. Birth Defects Orig. Artic. Ser. 1969, 2, 86–94. [Google Scholar]
- Robin, N.H.; Opitz, J.M.; Muenke, M. Opitz G/BBB syndrome: Clinical comparisons of families linked to Xp22 and 22q, and a review of the literature. Am. J. Med. Genet. 1996, 62, 305–317. [Google Scholar] [CrossRef]
- Robin, N.H.; Feldman, G.J.; Aronson, A.L.; Mitchell, H.F.; Weksberg, R.; Leonard, C.O.; Burton, B.K.; Josephson, K.D.; Laxova, R.; Aleck, K.A.; et al. Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11.2. Nat. Genet. 1995, 11, 459–461. [Google Scholar] [CrossRef]
- Quaderi, N.A.; Schweiger, S.; Gaudenz, K.; Franco, B.; Rugarli, E.I.; Berger, W.; Feldman, G.J.; Volta, M.; Andolfi, G.; Gilgenkrantz, S.; et al. Opitz G/BBB syndrome, a defect of midline development, is due to mutations in a new RING finger gene on Xp22. Nat. Genet. 1997, 17, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Cainarca, S.; Messali, S.; Ballabio, A.; Meroni, G. Functional characterization of the Opitz syndrome gene product (midin): Evidence for homodimerization and association with microtubules throughout the cell cycle. Hum. Mol. Genet. 1999, 8, 1387–1396. [Google Scholar] [CrossRef] [Green Version]
- Schweiger, S.; Foerster, J.; Lehmann, T.; Suckow, V.; Muller, Y.A.; Walter, G.; Davies, T.; Porter, H.; van Bokhoven, H.; Lunt, P.W.; et al. The Opitz syndrome gene product, MID1, associates with microtubules. Proc. Natl. Acad. Sci. USA 1999, 96, 2794–2799. [Google Scholar] [CrossRef] [Green Version]
- Short, K.M.; Hopwood, B.; Yi, Z.; Cox, T.C. MID1 and MID2 homo- and heterodimerise to tether the rapamycin-sensitive PP2A regulatory subunit, α 4, to microtubules: Implications for the clinical variability of X-linked Opitz GBBB syndrome and other developmental disorders. BMC Cell Biol. 2002, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Winter, J.; Basilicata, M.F.; Stemmler, M.P.; Krauss, S. The MID1 protein is a central player during development and in disease. Front. Biosci. 2016, 21, 664–682. [Google Scholar]
- Baldini, R.; Mascaro, M.; Meroni, G. The MID1 gene product in physiology and disease. Gene 2020, 747, 144655. [Google Scholar] [CrossRef]
- Cho, H.J.; Shin, M.Y.; Ahn, K.M.; Lee, S.I.; Kim, H.J.; Ki, C.S.; Kim, J.W. X-linked Opitz G/BBB syndrome: Identification of a novel mutation and prenatal diagnosis in a Korean family. J. Korean Med. Sci. 2006, 21, 790–793. [Google Scholar] [CrossRef]
- Cox, T.C.; Allen, L.R.; Cox, L.L.; Hopwood, B.; Goodwin, B.; Haan, E.; Suthers, G.K. New mutations in MID1 provide support for loss of function as the cause of X-linked Opitz syndrome. Hum. Mol. Genet. 2000, 9, 2553–2562. [Google Scholar] [CrossRef] [Green Version]
- De Falco, F.; Cainarca, S.; Andolfi, G.; Ferrentino, R.; Berti, C.; Criado, G.R.; Rittinger, O.; Dennis, N.; Odent, S.; Rastogi, A.; et al. X-linked Opitz syndrome: Novel mutations in the MID1 gene and redefinition of the clinical spectrum. Am. J. Med. Genet. 2003, 120, 222–228. [Google Scholar] [CrossRef]
- Ferrentino, R.; Bassi, M.T.; Chitayat, D.; Tabolacci, E.; Meroni, G. MID1 mutation screening in a large cohort of Opitz G/BBB syndrome patients: Twenty-nine novel mutations identified. Hum. Mutat. 2007, 28, 206–207. [Google Scholar] [CrossRef] [Green Version]
- Fontanella, B.; Russolillo, G.; Meroni, G. MID1 mutations in patients with X-linked Opitz G/BBB syndrome. Hum. Mutat. 2008, 29, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Gaudenz, K.; Roessler, E.; Quaderi, N.; Franco, B.; Feldman, G.; Gasser, D.L.; Wittwer, B.; Horst, J.; Montini, E.; Opitz, J.M.; et al. Opitz G/BBB syndrome in Xp22: Mutations in the MID1 gene cluster in the carboxy-terminal domain. Am. J. Hum. Genet. 1998, 63, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.H.; Liu, Y.F.; Yu, J.S.; Ng, Y.Y.; Chen, S.J.; Su, P.H.; Chen, J.Y. A MID1 gene mutation in a patient with Opitz G/BBB syndrome that altered the 3D structure of SPRY domain. Am. J. Med. Genet. A 2012, 158, 726–731. [Google Scholar] [CrossRef]
- Li, B.; Zhou, T.; Zou, Y. Mid1/Mid2 expression in craniofacial development and a literature review of X-linked opitz syndrome. Mol. Genet. Genom. Med. 2016, 4, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Mnayer, L.; Khuri, S.; Merheby, H.A.; Meroni, G.; Elsas, L.J. A structure-function study of MID1 mutations associated with a mild Opitz phenotype. Mol. Genet. Metab. 2006, 87, 198–203. [Google Scholar] [CrossRef]
- Pinson, L.; Auge, J.; Audollent, S.; Mattei, G.; Etchevers, H.; Gigarel, N.; Razavi, F.; Lacombe, D.; Odent, S.; Le Merrer, M.; et al. Embryonic expression of the human MID1 gene and its mutations in Opitz syndrome. J. Med. Genet. 2004, 41, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Ruiter, M.; Kamsteeg, E.J.; Meroni, G.; de Vries, B.B. A MID1 mutation associated with reduced penetrance of X-linked Opitz G/BBB syndrome. Clin. Dysmorphol. 2010, 19, 195–197. [Google Scholar] [CrossRef] [PubMed]
- So, J.; Suckow, V.; Kijas, Z.; Kalscheuer, V.; Moser, B.; Winter, J.; Baars, M.; Firth, H.; Lunt, P.; Hamel, B.; et al. Mild phenotypes in a series of patients with Opitz GBBB syndrome with MID1 mutations. Am. J. Med. Genet. A 2005, 132, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Lehmann, T.; Suckow, V.; Kijas, Z.; Kulozik, A.; Kalscheuer, V.; Hamel, B.; Devriendt, K.; Opitz, J.; Lenzner, S. Duplication of the MID1 first exon in a patient with Opitz G/BBB syndrome. Hum. Genet. 2003, 112, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Longman, C.; Irving, M.; Splitt, M. Neonatal teeth in X-linked Opitz (G/BBB) syndrome. Clin. Dysmorphol. 2006, 15, 185–186. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Zhao, S.; Markljung, E.; Nordenskjold, A. Hypospadias associated with hypertelorism, the mildest phenotype of Opitz syndrome. J. Hum. Genet. 2011, 56, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Kruszka, P.; Li, D.; Harr, M.H.; Wilson, N.R.; Swarr, D.; McCormick, E.M.; Chiavacci, R.M.; Li, M.; Martinez, A.F.; Hart, R.A.; et al. Mutations in SPECC1L, encoding sperm antigen with calponin homology and coiled-coil domains 1-like, are found in some cases of autosomal dominant Opitz G/BBB syndrome. J. Med. Genet. 2015, 52, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Bhoj, E.J.; Haye, D.; Toutain, A.; Bonneau, D.; Nielsen, I.K.; Lund, I.B.; Bogaard, P.; Leenskjold, S.; Karaer, K.; Wild, K.T.; et al. Phenotypic spectrum associated with SPECC1L pathogenic variants: New families and critical review of the nosology of Teebi, Opitz GBBB, and Baraitser-Winter syndromes. Eur. J. Med. Genet. 2019, 62, 103588. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A.; van Dyke, M.; Stock, J. Predicting coiled coils from protein sequences. Science 1991, 252, 1162–1164. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Bhoj, E.J.; Li, D.; Harr, M.H.; Tian, L.; Wang, T.; Zhao, Y.; Qiu, H.; Kim, C.; Hoffman, J.D.; Hakonarson, H.; et al. Expanding the SPECC1L mutation phenotypic spectrum to include Teebi hypertelorism syndrome. Am. J. Med. Genet. A 2015, 167, 2497–2502. [Google Scholar] [CrossRef]
- Saadi, I.; Alkuraya, F.S.; Gisselbrecht, S.S.; Goessling, W.; Cavallesco, R.; Turbe-Doan, A.; Petrin, A.L.; Harris, J.; Siddiqui, U.; Grix, A.W., Jr.; et al. Deficiency of the cytoskeletal protein SPECC1L leads to oblique facial clefting. Am. J. Hum. Genet. 2011, 89, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Geng, Y.; Duan, H.; Xu, L.; Yu, Z.; Yuan, J.; Wei, M. A novel p.Pro871Leu missense mutation in SPECC1L gene causing craniosynostosis in a patient. Orthod. Craniofac. Res. 2021, 24, 480–485. [Google Scholar]
- Hall, E.G.; Wenger, L.W.; Wilson, N.R.; Undurty-Akella, S.S.; Standley, J.; Augustine-Akpan, E.A.; Kousa, Y.A.; Acevedo, D.S.; Goering, J.P.; Pitstick, L.; et al. SPECC1L regulates palate development downstream of IRF6. Hum. Mol. Genet. 2020, 29, 845–858. [Google Scholar] [CrossRef]
- Wild, K.T.; Gordon, T.; Bhoj, E.J.; Du, H.; Jhangiani, S.N.; Posey, J.E.; Lupski, J.R.; Scott, D.A.; Zackai, E.H. Congenital diaphragmatic hernia as a prominent feature of a SPECC1L-related syndrome. Am. J. Med. Genet. A 2020, 182, 2919–2925. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhu, L.; Wu, D.; Yang, R.; Qi, M.; Huang, X. A novel SPECC1L mutation causing Teebi hypertelorism syndrome: Expanding phenotypic and genetic spectrum. Eur. J. Med. Genet. 2020, 63, 103851. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Wilson, N.R.; Olm-Shipman, A.J.; Acevedo, D.S.; Palaniyandi, K.; Hall, E.G.; Kosa, E.; Stumpff, K.M.; Smith, G.J.; Pitstick, L.; Liao, E.C.; et al. SPECC1L deficiency results in increased adherens junction stability and reduced cranial neural crest cell delamination. Sci. Rep. 2016, 6, 17735. [Google Scholar] [CrossRef] [Green Version]
- Gfrerer, L.; Shubinets, V.; Hoyos, T.; Kong, Y.; Nguyen, C.; Pietschmann, P.; Morton, C.C.; Maas, R.L.; Liao, E.C. Functional analysis of SPECC1L in craniofacial development and oblique facial cleft pathogenesis. Plast. Reconstr. Surg. 2014, 134, 748–759. [Google Scholar] [CrossRef] [Green Version]



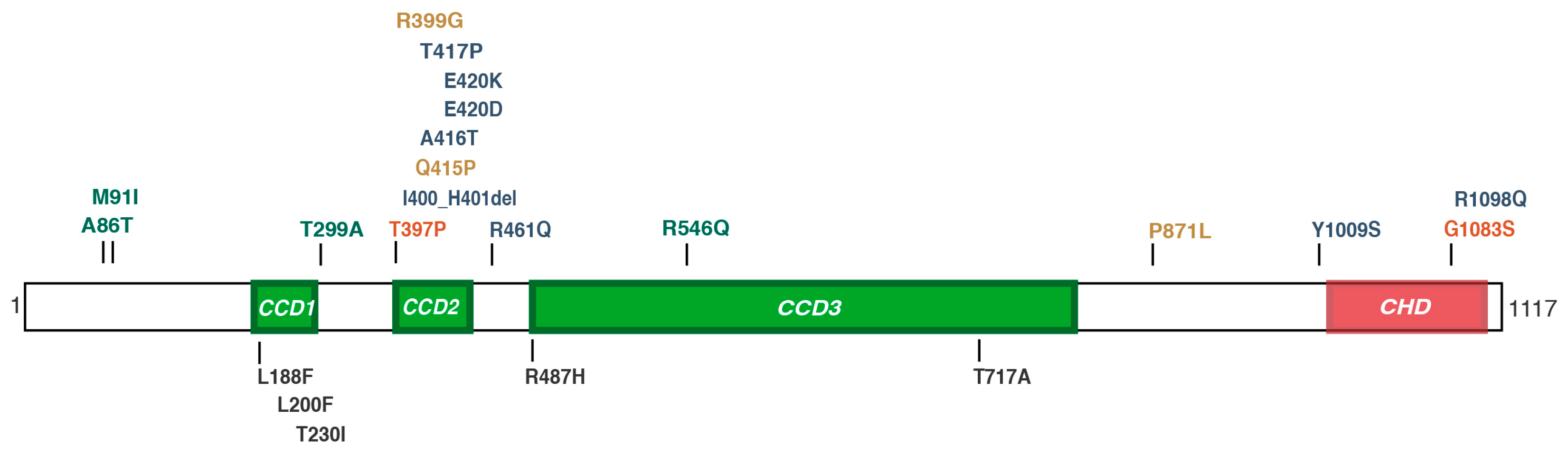{kind=link}
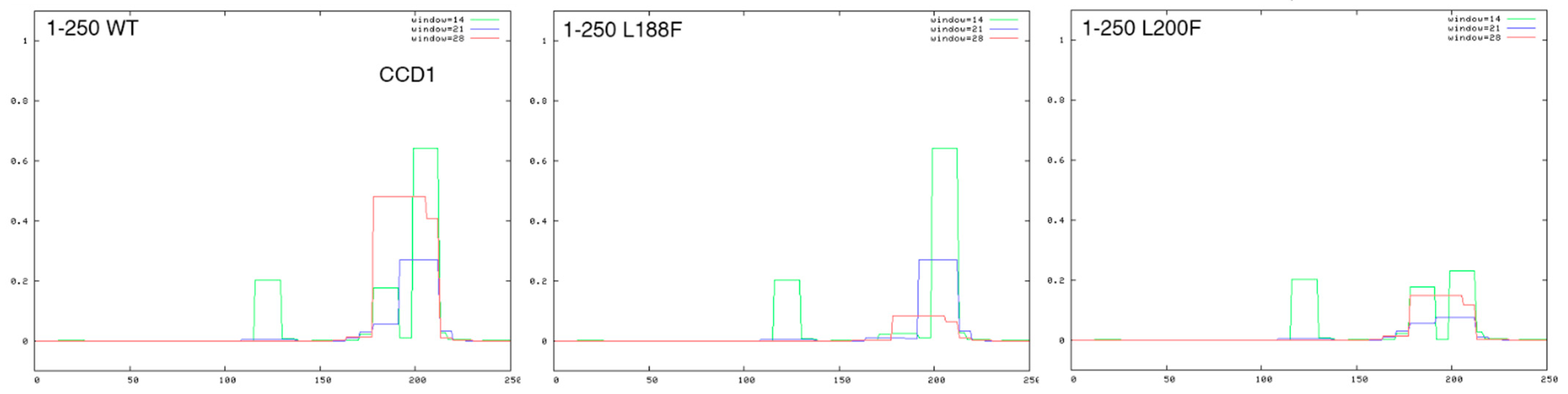{kind=link}
| Patient | Hypertelorsim and Other Facial Features | Laryngo–Tracheo–Esophageal Abnormalities | Hypospadias (Other External Genitalia Abnormalities) | Other Midline Signs a |
|---|---|---|---|---|
| OS212 | + | + | + | |
| OS269 * | + | + | + | |
| OS286 | + | + | + | |
| OS293 | + | + | + | |
| OS299 | + | (+) | + | |
| OS302 | + | + | + | |
| OS303 | + | + | + | |
| OS306 | + | (+) | + | |
| OS310 * | + | + | ||
| OS311 (F) | + | + | ||
| OS312 | + | + | + | |
| OS315 | + | + | ||
| OS316 (F) | + | + | ||
| OS320 (F) | + | + | + | |
| OS324 | + | + | (+) | |
| OS325 * | + | + | ||
| OS326 | + | + | + | + |
| OS327 | + | |||
| OS330 | + | + | ||
| OS331 * | + | + | + | |
| OS332 | + | + | + | |
| OS336 * | + | + | + | + |
| OS337 (F) | + | + | ||
| OS338 | + | + | + | |
| OS345 | + | + |
| Patient | Genomic Location a | Exon b | cDNA Alteration c | Protein Alteration | dbSNP d | Global MAF | SIFT | Mutation Taster | Polyphen2 |
|---|---|---|---|---|---|---|---|---|---|
| OS310 OS325 | 22:24321542 | 5 | c.562C>T | p.Leu188Phe | rs56168869 | 0.0034 | Deleterious | Benign | Probably Damaging—score 0.998 |
| OS296 | 22:24321580 | 5 | c.600A>T | p.Leu200Phe | rs56112030 | 0.0094 | Tolerated | Benign | Benign—score 0 |
| OS331 | 22:24321669 | 5 | c.689C>T | p.Thr230Ile | rs117220882 | 0.0058 | Tolerated | Benign | Benign—score 0.002 |
| OS269 | 22:24322440 | 5 | c.1460G>A | p.Arg487His | rs55723436 | 0.0036 | Tolerated | Benign | Probably Damaging—score 0.999 |
| OS336 | 22:24328848 | 7 | c.2149A>G | p.Thr717Ala | rs6004132 | 0.0006 | Tolerated | Benign | Benign—score 0.001 |
| Patient | cDNA Alteration | Protein Alteration | Global MAF | MAF GO- ESP | MAF ExAC | GnomAD | TOP Med | EAS | AMR | AFR | EUR | SAS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OS310 OS325 | c.562C>T | p.Leu188Phe | 0.0034 | 0.0057 | 0.0072 | 0.0085 | 0.005 | 0 | 0.011 | 0 | 0.009 | 0 |
| OS296 | c.600A>T | p.Leu200Phe | 0.0094 | 0.0071 | 0.0116 | 0.0072 | 0.0056 | 0.002 | 0 | 0 | 0.013 | 0.033 |
| OS331 | c.689C>T | p.Thr230Ile | 0.0058 | - | 0.0022 | 0.0023 | 0.0022 | 0.029 | 0 | 0 | 0 | 0 |
| OS269 | c.1460G>A | p.Arg487His | 0.0036 | 0.0068 | 0.0057 | 0.0055 | 0.0058 | 0 | 0.006 | 0.001 | 0.01 | 0.003 |
| OS336 | c.2149A>G | p.Thr717Ala | 0.0006 | 0.0015 | 0.0005 | 0.0012 | 0.0016 | 0 | 0 | 0.002 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Migliore, C.; Vendramin, A.; McKee, S.; Prontera, P.; Faravelli, F.; Sachdev, R.; Dias, P.; Mascaro, M.; Licastro, D.; Meroni, G. SPECC1L Mutations Are Not Common in Sporadic Cases of Opitz G/BBB Syndrome. Genes 2022, 13, 252. https://doi.org/10.3390/genes13020252
Migliore C, Vendramin A, McKee S, Prontera P, Faravelli F, Sachdev R, Dias P, Mascaro M, Licastro D, Meroni G. SPECC1L Mutations Are Not Common in Sporadic Cases of Opitz G/BBB Syndrome. Genes. 2022; 13(2):252. https://doi.org/10.3390/genes13020252
Chicago/Turabian StyleMigliore, Chiara, Anna Vendramin, Shane McKee, Paolo Prontera, Francesca Faravelli, Rani Sachdev, Patricia Dias, Martina Mascaro, Danilo Licastro, and Germana Meroni. 2022. "SPECC1L Mutations Are Not Common in Sporadic Cases of Opitz G/BBB Syndrome" Genes 13, no. 2: 252. https://doi.org/10.3390/genes13020252