
Real-Time Polymerase Chain Reaction: Current Techniques, Applications, and Role in COVID-19 Diagnosis
 , ,
, ,
Abstract
:1. Introduction
2. Basic Principles
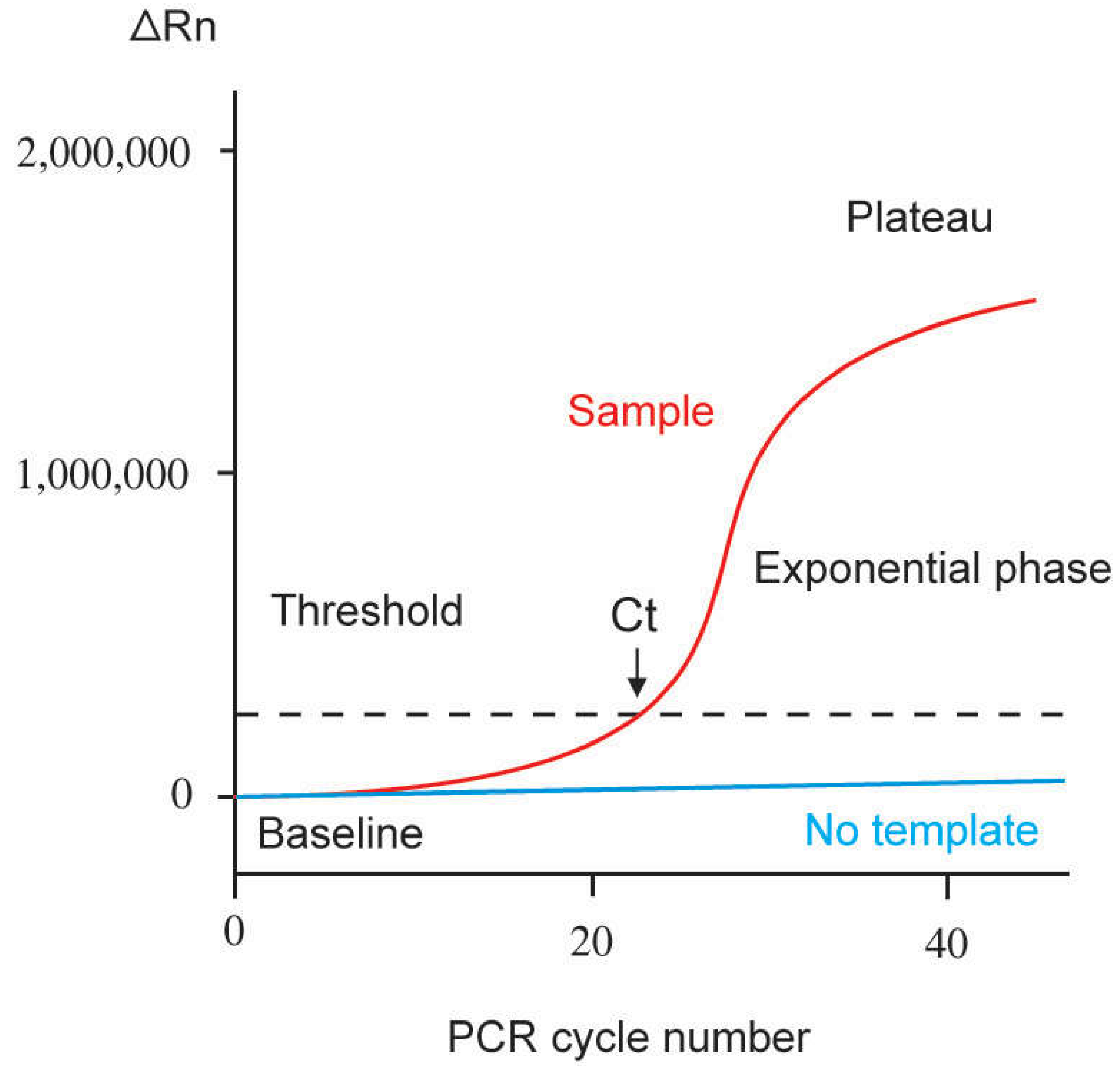
2.1. Quantification
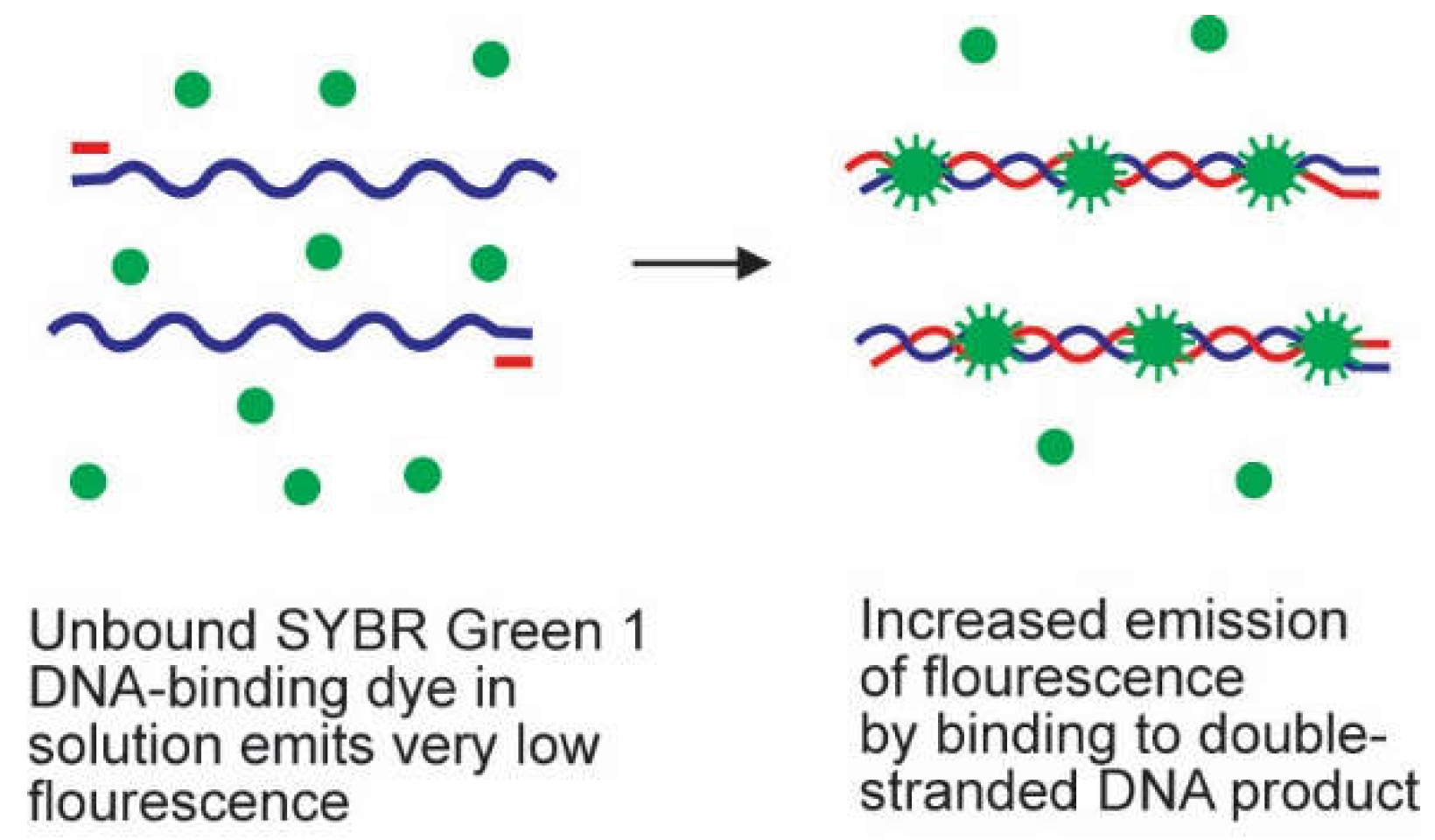
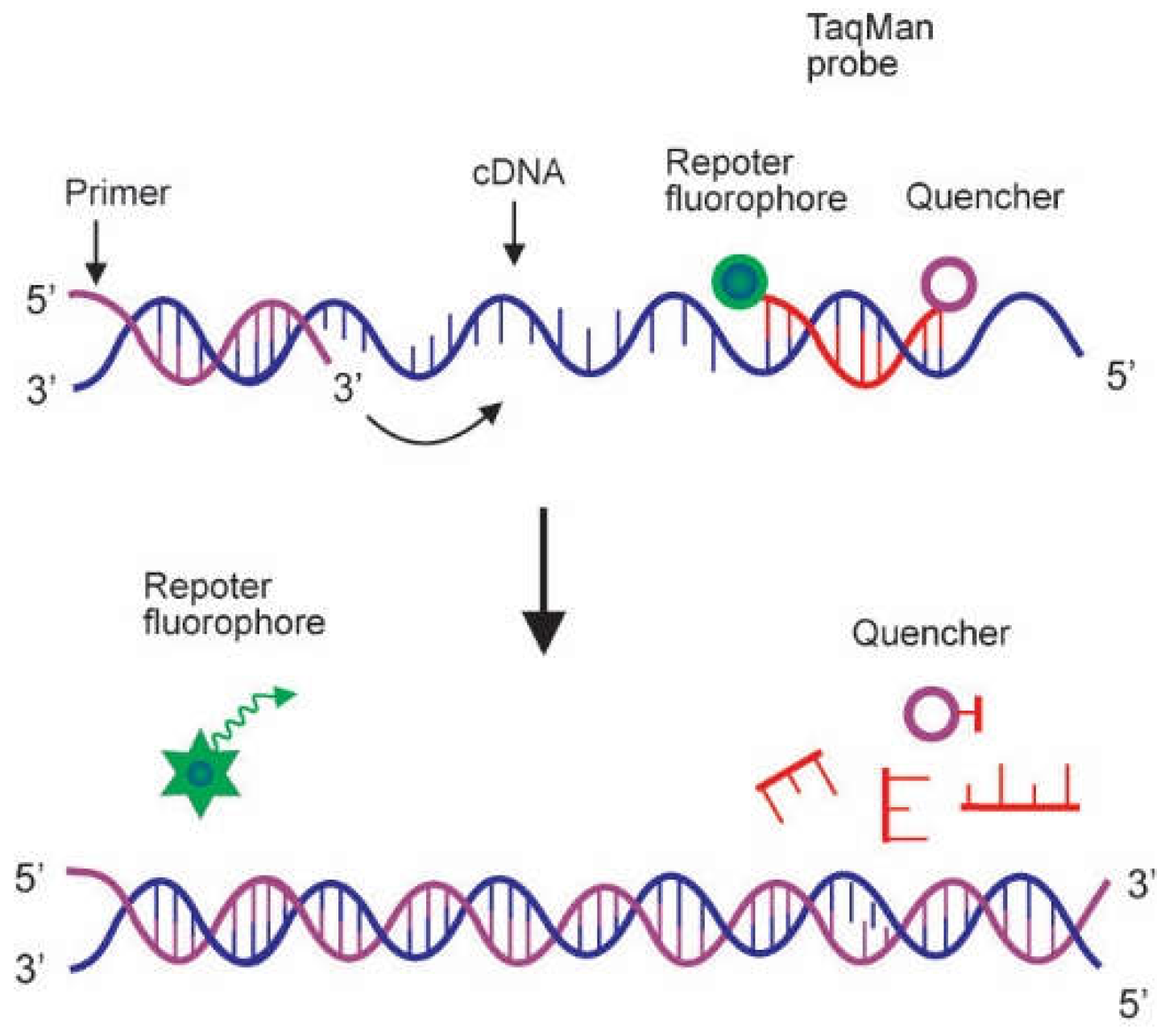
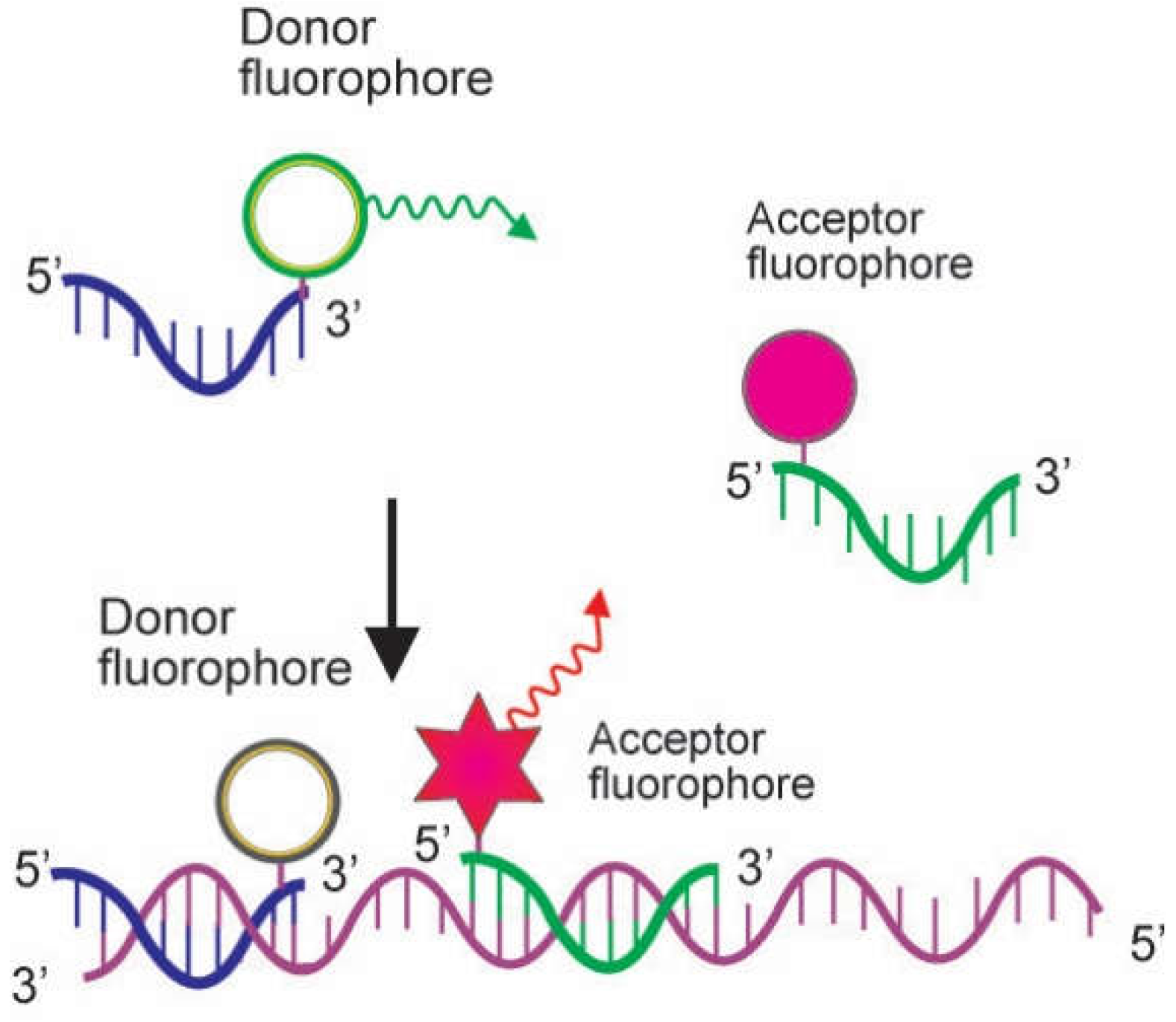
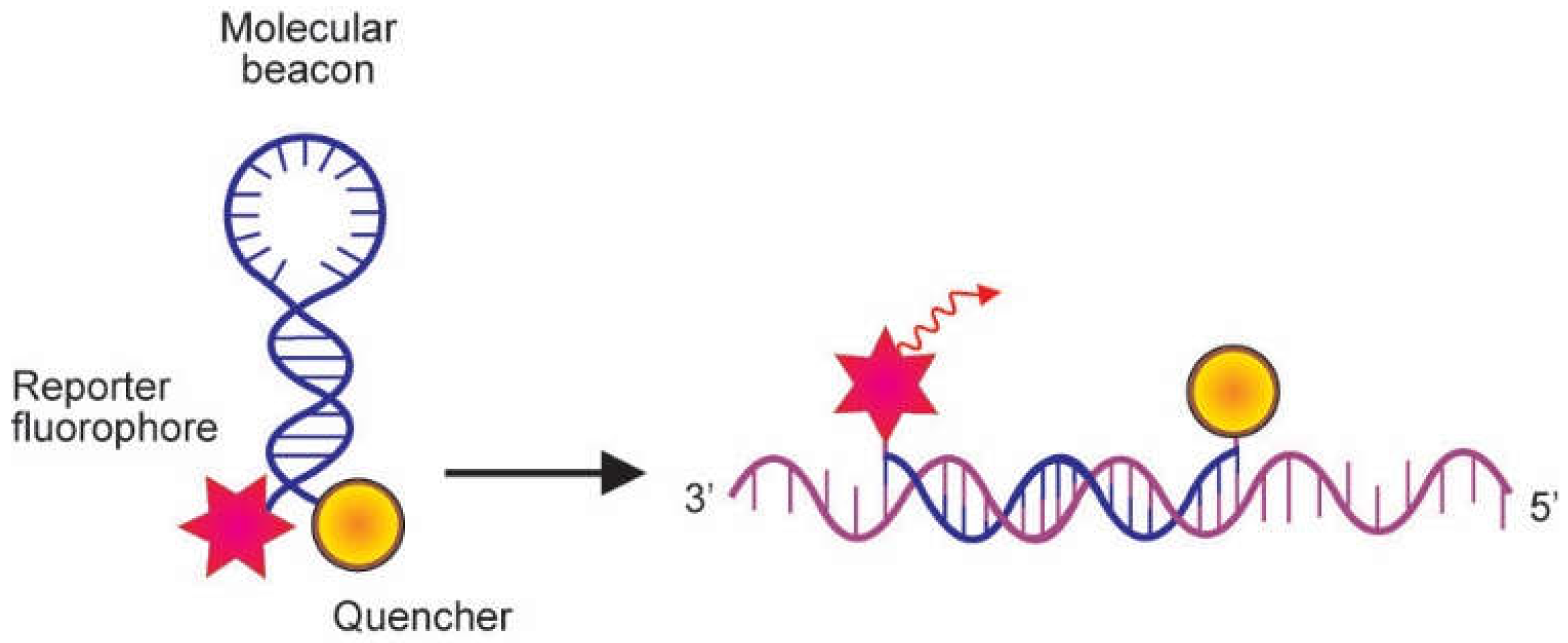
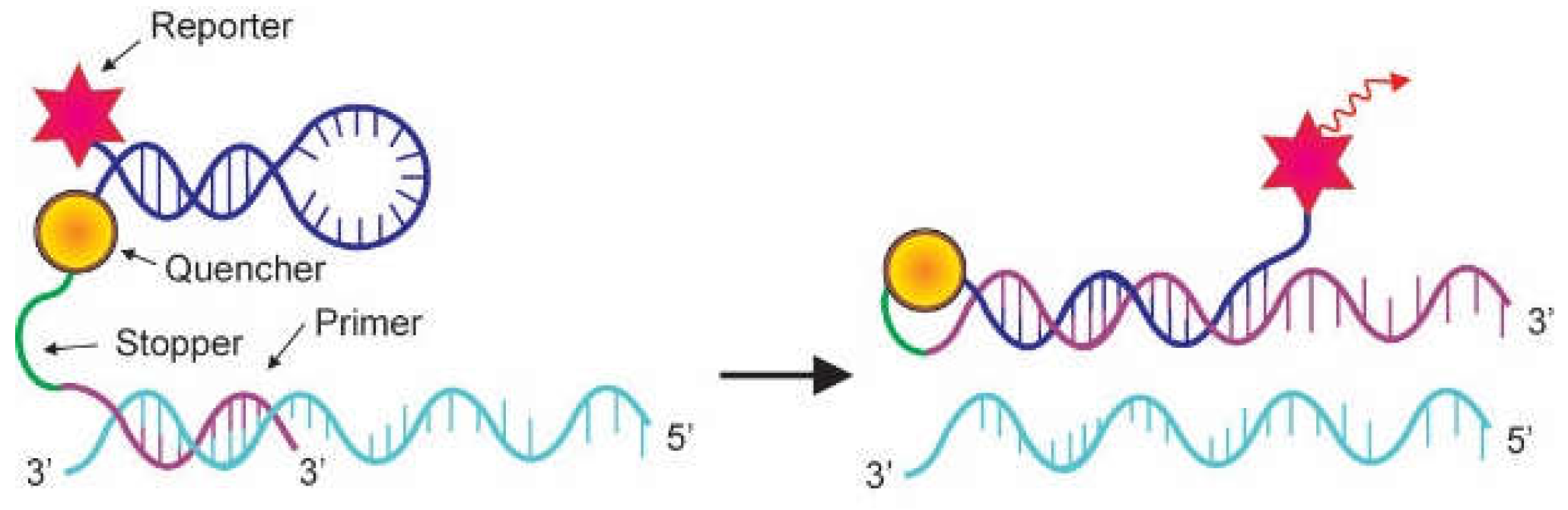
2.2. Probes
3. Applications
3.1. Analysis of Gene Expression
3.2. Detection of Mutation
3.3. Food Analysis
3.4. Bioremediation Monitoring
3.5. Detection and Quantification of Pathogen
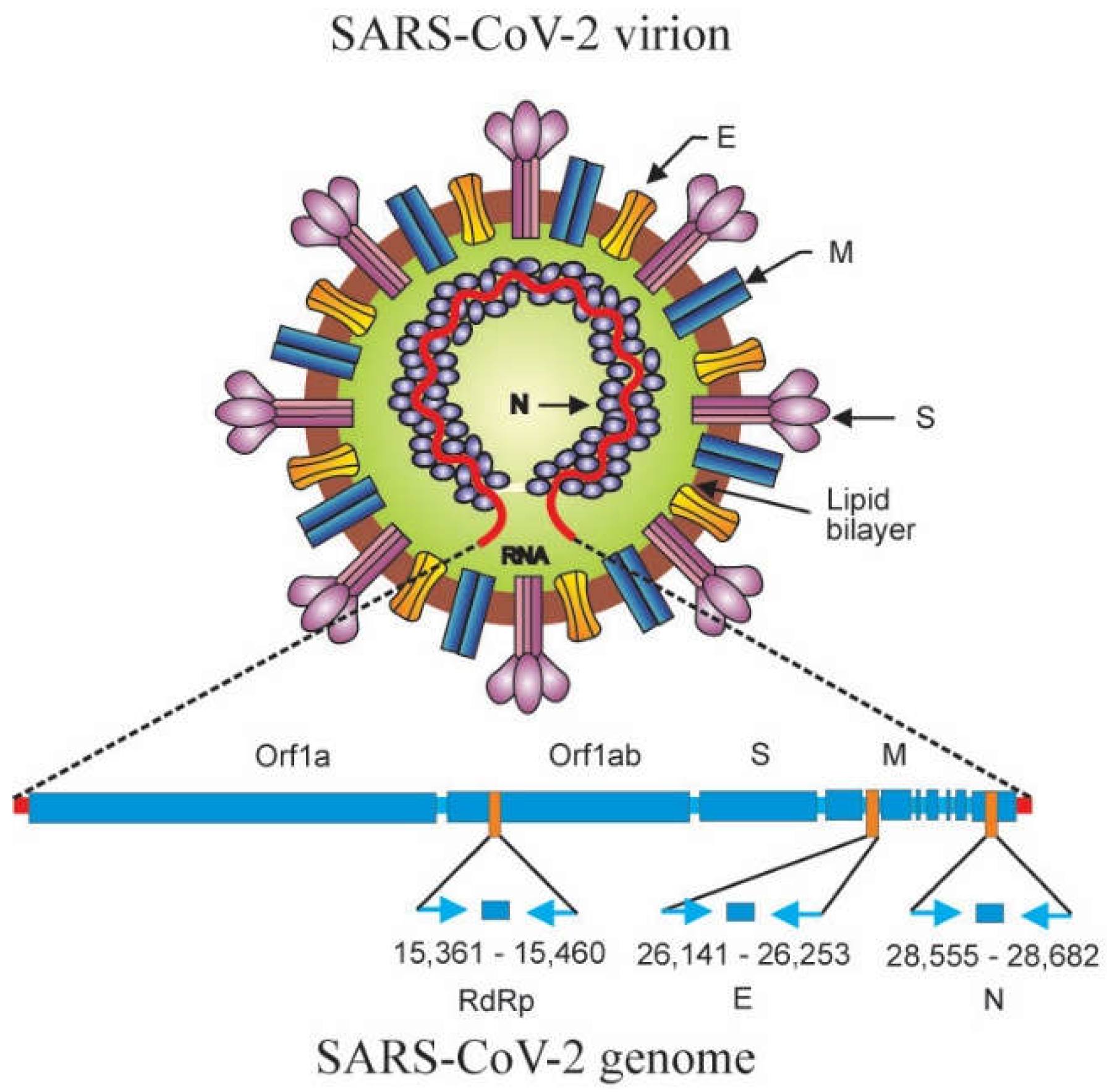
4. Detection and Quantification of SARS-CoV-2
4.1. Detection of SARS-CoV-2 Variants
4.2. Diagnosis of SARS-CoV-2
4.3. Viral Load Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Templeton, N.S. The polymerase chain reaction. History, methods, and applications. Diagn. Mol. Pathol. 1992, 1, 58–72. [Google Scholar] [CrossRef] [PubMed]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L.; et al. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sucahya, P.K. Barriers to COVID-19 RT-PCR testing in Indonesia: A health policy perspective. J. Indones. Health Policy Admin. 2020, 5, 36–42. [Google Scholar] [CrossRef]
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R.H. Basic principles of real-time quantitative PCR. Expert Rev. Mol. Diagn. 2005, 5, 209–219. [Google Scholar] [CrossRef]
- Artika, I.M.; Wiyatno, A.; Ma’roef, C.N. Pathogenic viruses: Molecular detection and characterization. Infect. Genet. Evol. 2020, 81, 1–14. [Google Scholar] [CrossRef]
- Rocha, A.J.; Miranda, R.d.S.; Sousa, A.J.S.; da Silva, A.L.C. Guidelines for successful quantitative gene expression in real-time qPCR assays. In Polymerase Chain Reaction for Biomedical Applications; InTech: London, UK, 2016. [Google Scholar]
- Riswari, S.F.; Ma’roef, C.N.; Djauhari, H.; Kosasih, H.; Perkasa, A.; Yudhaputri, F.A.; Artika, I.M.; Williams, M.; van der Ven, A.; Myint, K.S.; et al. Study of viremic profile in febrile specimens of chikungunya in Bandung, Indonesia. J. Clin. Virol. 2016, 74, 61–65. [Google Scholar] [CrossRef] [Green Version]
- Deepak, S.A.; Kottapalli, K.R.; Rakwal, R.; Oros, G.; Rangappa, K.; Iwahashi, H.; Masuo, Y.; Agrawal, G.K. Real-time PCR: Revolutionizing detection and expression analysis of genes. Curr. Genom. 2007, 8, 234–251. [Google Scholar] [CrossRef]
- Wong, M.L.; Medrano, J.F. Real-time PCR for mRNA quantitation. Biotechniques 2005, 39, 75–85. [Google Scholar] [CrossRef]
- Higuchi, H.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR analysis: Real time monitoring of DNA amplification reactions. Biotechnology 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- Mackay, I.M.; Arden, K.E.; Nitsche, A. Real-time PCR in virology. Nucleic Acids Res. 2002, 30, 1292–1305. [Google Scholar] [CrossRef] [Green Version]
- Wacker, M.J.; Godard, M.P. Analysis of one-step and two-step real-time RT-PCR using SuperScript III. J. Biomol. Tech. 2005, 16, 266–271. [Google Scholar]
- Navarro, E.; Serrano-Heras, G.; Castaño, M.J.; Solera, J. Real-time PCR detection chemistry. Clin. Chim. Acta 2015, 439, 231–250. [Google Scholar] [CrossRef]
- Tajadini, M.; Panjehpour, M.; Javanmard, S.H. Comparison of SYBR Green and TaqMan methods in quantitative real-time polymerase chain reaction analysis of four adenosine receptor subtypes. Adv. Biomed. Res. 2014, 3, 1–6. [Google Scholar] [CrossRef]
- Marinowic, D.R.; Zanirati, G.; Rodrigues, F.V.F.; Grahl, M.V.C.; Alcará, A.M.; Machado, D.C.; Da Costa, J.C. A new SYBR Green real-time PCR to detect SARS-CoV-2. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Pan, Z.; Lu, J.; Wang, N.; He, W.-T.; Zhang, L.; Zhao, W.; Su, S. Development of a TaqMan-probe-based multiplex real-time PCR for the simultaneous detection of emerging and reemerging swine coronaviruses. Virulence 2020, 11, 707–718. [Google Scholar] [CrossRef]
- Caplin, B.E.; Rasmussen, R.P.; Bernard, P.S.; Wittwer, C.T. LightCycler™ hybridization probes: The most direct way to monitor PCR amplification for quantification and mutation detection. Biochemica 1999, 1, 1–7. [Google Scholar]
- Bassy, O.; Jiménez-Mateo, O.; Ortega, M.V.; Granja, C.; Cabria, J.C. Rapid identification of Bacillus anthracis by real-time PCR with dual hybridization probes in environmental swabs. Mol. Cell Probes 2018, 37, 22–27. [Google Scholar] [CrossRef]
- Wang, C.; Yang, C.J. Application of molecular beacons in real-time PCR. In Molecular Beacons; Yang, C.J., Tan, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Chrysostomou, A.C.; Rodosthenous, J.H.; Topcu, C.; Papa, C.; Aristokleous, A.; Stathi, G.; Christodoulou, C.; Eleftheriou, C.; Stylianou, D.C.; Kostrikis, L.G. A multiallelic molecular beacon-based real-time RT-PCR assay for the detection of SARS-CoV-2. Life 2021, 11, 1146. [Google Scholar] [CrossRef]
- Singh, J.; Batish, V.K.; Grover, S. A scorpion probe–based real-time PCR assay for detection of E. coli O157:H7 in dairy products. Foodborne Pathog. Dis. 2009, 6, 395–400. [Google Scholar] [CrossRef]
- Said-Salman, I.H.; Jebaii, F.A.; Yusef, H.H.; Moustafa, M.E. Global gene expression analysis of Escherichia coli K-12 DH5α after exposure to 2.4 GHz wireless fidelity radiation. Sci. Rep. 2019, 9, 14425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Watanabe, A.; Tamura, M.; Tsutsumi, Y. The gene expression analysis of Arabidopsis thaliana ABC transporters by real-time PCR for screening monolignol-transporter candidates. J. Wood Sci. 2018, 64, 477–484. [Google Scholar] [CrossRef]
- Söylemez, Z.; Arıkan, E.S.; Solak, M.; Arıkan, Y.; Tokyol, Ç.; Şeker, H. Investigation of the expression levels of CPEB4, APC, TRIP13, EIF2S3, EIF4A1, IFNg, PIK3CA and CTNNB1 genes in different stage colorectal tumors. Turk. J. Med. Sci. 2021, 51, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Masyeni, S.; Hadi, U.; Kuntaman, K.; Dewi, Y.P. Profiling of microRNA expression within the cells of peripheral blood mononuclear after an infection with serotype-2 of dengue virus: Preliminary study. Biomed. Pharmacol. J. 2018, 11, 923–927. [Google Scholar] [CrossRef]
- Alvarez-Garcia, V.; Bartos, C.; Keraite, I.; Brennan, P.M.; Kersaudy-Kerhoas, M.; Gharbi, K.; Oikonomidou, O.; Leslie, N.R. A simple and robust real-time qPCR method for the detection of PIK3CA mutations. Sci. Rep. 2018, 8, 4290. [Google Scholar] [CrossRef] [Green Version]
- Lung, J.; Hung, M.-S.; Lin, Y.-C.; Jiang, Y.Y.; Fang, Y.-H.; Lu, M.-S.; Hsieh, C.-C.; Wang, C.-S.; Kuan, F.-C.; Lu, C.-H.; et al. A highly sensitive and specifc real-time quantitative PCR for BRAF V600E/K mutation screening. Sci. Rep. 2020, 10, 16943. [Google Scholar] [CrossRef]
- Liang, J.; Liang, X.; Ma, H.; Nie, L.; Tian, Y.; Chen, G.; Wang, Y. Detection of hepatitis B virus M204V mutation quantitatively via real-time PCR. J. Clin. Transl. Hepatol. 2021, 9, 143–148. [Google Scholar] [CrossRef]
- Donà, V.; Smid, J.H.; Kasraian, S.; Egli-Gany, D.; Dost, F.; Imeri, F.; Unemo, M.; Low, N.; Endimiani, A. Mismatch amplification mutation assay-based real-time PCR for rapid detection of Neisseria gonorrhoeae and antimicrobial resistance determinants in clinical specimens. J. Clin. Microbiol. 2018, 56, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mano, J.; Hatano, S.; Nagatomi, Y.; Futo, S.; Takabatake, R.; Kitta, K. Highly sensitive GMO detection using real-time PCR with a large amount of DNA template: Single-laboratory validation. J. AOAC Int. 2018, 101, 507–514. [Google Scholar] [CrossRef]
- Park, S.-B.; Kim, J.-Y.; Lee, D.-G.; Kim, J.-H.; Shin, M.-K.; Kim, H.-Y. Development of a systematic qPCR array for screening GM soybeans. Foods 2021, 10, 610. [Google Scholar] [CrossRef]
- Fraiture, M.-A.; Gobbo, A.; Marchesi, U.; Verginelli, D.; Papazova, N.; Roosens, N.H.C. Development of a real-time PCR marker targeting a new unauthorized genetically modified microorganism producing protease identified by DNA walking. Int. J. Food Microbiol. 2021, 354, 1–9. [Google Scholar] [CrossRef]
- Sanchiz, A.; Sánchez-Enciso, P.; Cuadrado, C.; Linacero, R. Detection of peanut allergen by real-time PCR: Looking for a suitable detection marker as affected by processing. Foods 2021, 10, 1421. [Google Scholar] [CrossRef]
- Orbayinah, S.; Widada, H.; Hermawan, A.; Sudjadi, S.; Rohman, A. Application of real-time polymerase chain reaction using species specific primer targeting on mitochondrial cytochrome-b gene for analysis of pork in meatball products. J. Adv. Vet. Anim. Res. 2019, 6, 260–265. [Google Scholar] [CrossRef]
- Baek, K.-H.; Yoon, B.-D.; Cho, D.-H.; Kim, B.-H.; Oh, H.-M.; Kim, H.S. Monitoring bacterial population dynamics using real-time PCR during the bioremediation of crude-oil-contaminated soil. J. Microbiol. Biotechnol. 2009, 19, 339–345. [Google Scholar] [CrossRef]
- Cao, Y.; Yu, M.; Dong, G.; Chen, B.; Zhang, B. Digital PCR as an emerging tool for monitoring of microbial biodegradation. Molecules 2020, 25, 706. [Google Scholar] [CrossRef] [Green Version]
- Kayashima, T.; Suzuki, H.; Maeda, T.; Ogawa, H.I. Real-time PCR for rapidly detecting aniline-degrading bacteria in activated sludge. Chemosphere 2013, 91, 1338–1343. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, C.; Aranda-Medina, M.; Rodríguez, A.; Hernández, A.; Córdoba, M.G.; Cuadros-Blázquez, F.; Ruiz-Moyano, S. Development of real-time PCR methods for the quantification of Methanoculleus, Methanosarcina and Methanobacterium in anaerobic digestion. J. Microbiol. Methods 2022, 199, 106529. [Google Scholar] [CrossRef]
- Lim, H.J.; Kang, E.-R.; Park, M.Y.; Kim, B.K.; Kim, M.J.; Jung, S.; Roh, K.H.; Sung, N.; Yang, J.-H.; Lee, M.-W.; et al. Development of a multiplex real-time PCR assay for the simultaneous detection of four bacterial pathogens causing pneumonia. PLoS ONE 2021, 16, e0253402. [Google Scholar] [CrossRef]
- Kim, W.-B.; Park, C.; Cho, S.-Y.; Chun, H.-S.; Lee, D.G. Development of multiplex real-time PCR for rapid identification and quantitative analysis of Aspergillus species. PLoS ONE 2020, 15, e0229561. [Google Scholar] [CrossRef] [Green Version]
- Sabiiti, W.; Mtafya, B.; De Lima, D.A.; Dombay, E.; Baron, V.O.; Azam, K.; Oravcova, K.; Sloan, D.J.; Gillespie, S.H. A tuberculosis molecular bacterial load assay (TB-MBLA). J. Vis. Exp. 2020, 158, e60460. [Google Scholar] [CrossRef]
- Nuralitha, S.; Murdiyarso, L.S.; Siregar, J.E.; Syafruddin, D.; Roelands, J.; Verhoef, J.; Hoepelman, A.I.M.; Marzuki, S. Within-host selection of drug resistance in a mouse model reveals dosedependent selection of atovaquone resistance mutations. Antimicrob. Agents Chemother. 2017, 61, e01867-16. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.-C.; Tan, L.-K.; Tan, C.-H.; Tan, S.S.; Hapuarachchi, H.C.; Pok, K.Y.; Lai, Y.-L.; Lam-Phua, S.-G.; Bucht, G.; Lin, R.T.P.; et al. Entomologic and virologic investigation of chikungunya, Singapore. Emerg. Infect. Dis. 2009, 15, 1243–1249. [Google Scholar] [CrossRef]
- Sari, K.; Myint, K.S.A.; Andayani, A.R.; Adi, P.D.; Dheni, R.; Perkasa, A.; Ma’roef, C.N.; Witari, N.P.D.; Megawati, D.; Powers, A.M.; et al. Chikungunya fever outbreak identified in North Bali, Indonesia. Trans. R. Soc. Trop. Med. Hyg. 2017, 111, 325–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Siyabi, T.; Binkhamis, K.; Wilcox, M.; Wong, S.; Pabbaraju, K.; Tellier, R.; Hatchette, T.F.; LeBlanc, J.J. A cost effective real-time PCR for the detection of adenovirus from viral swabs. Virol. J. 2013, 10, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.K.; Kabra, S.K.; Lodha, R.; Ratageri, V.H.; Ray, P. Virus load and clinical features during the acute phase of Chikungunya infection in children. PLoS ONE 2019, 14, e0211036. [Google Scholar]
- Liu, R.; Yi, S.; Zhang, J.; Lv, Z.; Zhu, C.; Zhang, Y. Viral load dynamics in sputum and nasopharyngeal swab in patients with COVID-19. J. Dent. Res. 2020, 99, 1239–1244. [Google Scholar] [CrossRef]
- Adeola, F. Normalization of gene expression by quantitative RT-PCR in human cell line: Comparison of 12 endogenous reference genes. Ethiop. J. Health Sci. 2018, 28, 741–748. [Google Scholar] [CrossRef]
- Kuang, J.; Yan, X.; Genders, A.J.; Granata, C.; Bishop, D.J. An overview of technical considerations when using quantitative real-time PCR analysis of gene expression in human exercise research. PLoS ONE 2018, 10, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Freitas, F.C.P.; Depintor, T.S.; Agostini, L.T.; Luna-Lucena, D.; Nunes, F.M.F.; Bitondi, M.M.G.; Simões, Z.L.P.; Lourenço, A.P. Evaluation of reference genes for gene expression analysis by real-time quantitative PCR (qPCR) in three stingless bee species (Hymenoptera: Apidae: Meliponini). Sci. Rep. 2019, 9, 17692. [Google Scholar] [CrossRef] [Green Version]
- He, J.-Q.; Sandford, A.J.; Wang, I.-M.; Stepaniants, S.; Knight, D.A.; Kicic, A.; Stick, S.M.; Pare, P.D. Selection of housekeeping genes for real-time PCR in atopic human bronchial epithelial cells. Eur. Respir. J. 2008, 32, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Turabelidze, A.; Guo, S.; DiPietro, L.A. Importance of housekeeping gene selection for accurate reverse transcription-quantitative polymerase chain reaction in a wound healing model. Wound Repair Regen. 2010, 18, 460–466. [Google Scholar] [CrossRef]
- Kozera, B.; Rapacz, M. Reference genes in real-time PCR. J. Appl. Genet. 2013, 54, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Panina, Y.; Germond, A.; Masui, S.; Watanabe, T.M. Validation of common housekeeping genes as reference for qPCR gene expression analysis during iPS reprogramming process. Sci. Rep. 2018, 8, 8716. [Google Scholar] [CrossRef]
- Morlan, J.; Baker, J.; Sinicropi, D. Mutation detection by real-time PCR: A simple, robust and highly selective method. PLoS ONE 2009, 4, e4584. [Google Scholar] [CrossRef]
- Chhalliyil, P.; Ilves, H.; Kazakov, S.A.; Howard, S.J.; Johnston, B.H.; Fagan, J. A real-time quantitative PCR method specific for detection and quantification of the first commercialized genome-edited plant. Foods 2020, 9, 1245. [Google Scholar] [CrossRef]
- Fraiture, M.-A.; Herman, P.; Taverniers, I.; De Loose, M.; Deforce, D.; Roosens, N.H. Current and new approaches in GMO detection: Challenges and solutions. Biomed. Res. Int. 2015, 2015, 392872. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Zhang, B.; Greer, C.W.; Lee, K.; Cai, Q.; Song, X.; Tremblay, J.; Zhu, Z.; Dong, G.; Chen, B. Metagenomic and metatranscriptomic responses of chemical dispersant application during a marine dilbit spill. Appl. Environ. Microbiol. 2022, 88, e02151-21. [Google Scholar] [CrossRef]
- Qin, N.; Zhao, P.; Ho, E.A.; Xin, G.; Ren, C.L. Microfluidic technology for antibacterial resistance study and antibiotic susceptibility testing: Review and perspective. ACS Sens. 2022, 6, 3–21. [Google Scholar] [CrossRef]
- Putri, N.D.; Dhenni, R.; Handryastuti, S.; Johar, E.; Ma’roef, C.N.; Fadhilah, A.; Perma Iskandar, A.T.; Prayitno, A.; Karyanti, M.R.; Satari, H.I.; et al. Absence of evidence of Zika virus infection in cord blood and urine from newborns with congenital abnormalities, Indonesia. Am. J. Trop. Med. Hyg. 2020, 102, 876–879. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef]
- LeBlanc, J.J.; Gubbay, J.B.; Li, Y.; Needle, R.; Arneson, S.R.; Marcino, D.; Charest, H.; Desnoyers, G.; Dust, K.; Fattouh, R.; et al. Real-time PCR-based SARS-CoV-2 detection in Canadian laboratories. J. Clin. Virol. 2020, 128, 104433. [Google Scholar] [CrossRef] [PubMed]
- Setiadi, W.; Rozi, I.E.; Safari, D.; Daningrat, W.O.D.; Johar, E.; Yohan, B.; Yudhaputri, F.A.; Lestari, K.D.; Oktavianthi, S.; Myint, K.S.A.; et al. Prevalence and epidemiological characteristics of COVID-19 after one year of pandemic in Jakarta and neighbouring areas, Indonesia: A single center study. PLoS ONE 2022, 17, e0268241. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). The species Severe acute respiratory syndromerelated coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Coronavirus Disease (COVID-19) Pandemic. COVID-19 Weekly Epidemiological Update. 2022. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---12-october-2022. (accessed on 18 October 2022).
- Artika, I.M.; Dewantari, A.K.; Wiyatno, A. Molecular biology of coronaviruses: Current knowledge. Heliyon 2020, 6, e04743. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Recommendations for National SARS-CoV-2 Testing Strategies and Diagnostic Capacities: Interim Guidance. 2021. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-lab-testing-2021.1-eng (accessed on 18 October 2022).
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in different types of clinical specimens. JAMA 2020, 323, 1843–1844. [Google Scholar] [CrossRef] [Green Version]
- Coil, D.A.; Albertson, T.; Banerjee, S.; Brennan, G.; Campbell, A.J.; Cohen, S.H.; Dandekar, S.; Díaz-Muñoz, S.L.; Eisen, J.A.; Goldstein, T.; et al. SARS-CoV-2 detection and genomic sequencing from hospital surface samples collected at UC Davis. PLoS ONE 2021, 16, e0253578. [Google Scholar] [CrossRef]
- Casabianca, A.; Orlandi, C.; Amagliani, G.; Magnani, M.; Brandi, G.; Schiavano, G.F. SARS-CoV-2 RNA detection on environmental surfaces in a university setting of Central Italy. Int. J. Environ. Res. Public Health 2022, 19, 5560. [Google Scholar] [CrossRef]
- Randazzo, W.; Truchado, P.; Cuevas-Ferrando, E.; Simon, P.; Allende, A.; Sanchez, G. SARS-CoV-2 RNA in wastewater anticipated COVID-19 occurrence in a low prevalence area. Water Res. 2020, 181, 115942. [Google Scholar] [CrossRef]
- Janik, E.; Niemcewicz, M.; Podogrocki, M.; Majsterek, I.; Bijak, M. The emerging concern and interest SARS-CoV-2 variants. Pathogens 2021, 10, 633. [Google Scholar] [CrossRef]
- Fibriani, A.; Stephanie, R.; Alfiantie, A.A.; Siregar, A.L.F.; Pradani, G.A.P.; Yamahoki, N.; Purba, W.S.; Alamanda, C.N.C.; Rahmawati, E.; Rachman, R.W.; et al. Analysis of SARS-CoV-2 genomes from West Java, Indonesia. Viruses 2021, 13, 2097. [Google Scholar] [CrossRef]
- Vega-Magaña, N.; Sánchez-Sánchez, R.; Hernández-Bello, J.; Venancio-Landeros, A.A.; Peña-Rodríguez, M.; Vega-Zepeda, R.A.; Galindo-Ornelas, B.; Díaz-Sánchez, M.; García-Chagollán, M.; Macedo-Ojeda, G.; et al. RT-qPCR assays for rapid detection of the N501Y, 69-70del, K417N, and E484K SARS-CoV-2 mutations: A screening strategy to identify variants with clinical impact. Front. Cell Infect. Microbiol. 2021, 11, 672562. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.-D.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- Setiabudi, D.; Sribudiani, Y.; Hermawan, K.; Andriyoko, B.; Nataprawira, H.M. The Omicron variant of concern: The genomics, diagnostics, and clinical characteristics in children. Front. Pediatr. 2022, 10, 898463. [Google Scholar] [CrossRef]
- Chaintoutis, S.C.; Chassalevris, T.; Tsiolas, G.; Balaska, S.; Vlatakis, I.; Mouchtaropoulou, E.; Siarkou, V.I.; Tychala, A.; Koutsioulis, D.; Skoura, L.; et al. A one-step real-time RT-PCR assay for simultaneous typing of SARS-CoV-2 mutations associated with the E484K and N501Y spike protein amino-acid substitutions. J. Virol. Methods 2021, 296, 114242. [Google Scholar] [CrossRef]
- Gomes, L.; Jeewandara, C.; Jayadas, T.P.; Dissanayake, O.; Harvie, M.; Guruge, D.; Withanage, V.; Mahesh, P.K.B.; Rajapakse, W.; Ramachandran, R.; et al. Surveillance of SARS-CoV-2 variants of concern by identification of single nucleotide polymorphisms in the spike protein by a multiplex real-time PCR. J. Virol. Methods 2022, 300, 114374. [Google Scholar] [CrossRef]
- Tahamtan, A.; Ardebili, A. Real-time RT-PCR in COVID-19 detection: Issues affecting the results. Expert Rev. Mol. Diagn. 2020, 20, 453–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 lineage—United States, December 29, 2020–January 12, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Kanji, J.N.; Zelyas, N.; MacDonald, C.; Pabbaraju, K.; Khan, M.N.; Prasad, A.; Hu, J.; Diggle, M.; Berenger, B.M.; Tipples, G. False negative rate of COVID-19 PCR testing: A discordant testing analysis. Virol. J. 2021, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Woloshin, S.; Patel, N.; Kesselheim, A.S. False negative tests for SARS-CoV-2 infection—Challenges and implications. N. Engl. J. Med. 2020, 383, e38. [Google Scholar] [CrossRef]
- Yip, C.C.-Y.; Sridhar, S.; Leung, K.-H.; Ng, A.C.-K.; Chan, K.-H.; Chan, J.F.-W.; Tsang, O.T.-Y.; Hung, I.F.-N.; Cheng, V.C.-C.; Yuen, K.-Y.; et al. Development and evaluation of novel and highly sensitive single-tube nested real-time RT-PCR assays for SARS-CoV-2 detection. Int. J. Mol. Sci. 2020, 21, 5674. [Google Scholar] [CrossRef]
- Huggett, J.F.; Benes, V.; Bustin, S.A.; Garson, J.A.; Harris, K.; Kammel, M.; Kubista, M.; McHugh, T.D.; Moran-Gilad, J.; Nolan, T.; et al. Cautionary note on contamination of reagents used for molecular detection of SARS-CoV-2. Clin. Chem. 2020, 66, 1369–1372. [Google Scholar] [CrossRef]
- Surkova, E.; Nikolayevskyy, V.; Drobniewski, F. False-positive COVID-19 results: Hidden problems and costs. Lancet Respir. Med. 2020, 8, 1167–1168. [Google Scholar] [CrossRef]
- Healy, B.; Khan, A.; Metezai, H.; Blyth, I.; Asad, H. The impact of false positive COVID-19 results in an area of low prevalence. Clin. Med. 2021, 21, e54–e56. [Google Scholar] [CrossRef]
- Layfield, L.J.; Camp, S.; Bowers, K.; Miller, D.C. SARS-CoV-2 detection by reverse transcriptase polymerase chain reaction testing: Analysis of false positive results and recommendations for quality control measures. Pathol. Res. Pract. 2021, 225, 153579. [Google Scholar] [CrossRef]
- Han, M.S.; Byun, J.-H.; Cho, Y.; Rim, J.H. RT-PCR for SARS-CoV-2: Quantitative versus qualitative. Lancet Infect. Dis. 2020, 21, 165. [Google Scholar] [CrossRef]
- Whale, A.S.; von der Heide, E.K.; Kohlenberg, M.; Brinckmann, A.; Baedker, S.; Karalay, D.; Fernandez-Gonzalez, A.; Busby, E.J.; Bustin, S.A.; Hauser, H.; et al. Digital PCR can augment the interpretation of RT-qPCR Cq values for SARS-CoV-2 diagnostics. Methods 2022, 201, 5–14. [Google Scholar] [CrossRef]
- Ibrahim, F.; Natasha, A.; Saharman, Y.R.; Yasmon, A.; Fithriyah, F.; Karuniawati, A.; Ganiesa, S.; Sudarmono, P. Consideration of the cycle threshold values from real time RT-PCR SARS-CoV-2 interpretation for the clinicians: Analysis of 339 positive cases from a referral laboratory in Jakarta, Indonesia. Acta Med. Indones. 2021, 53, 13–17. [Google Scholar] [PubMed]
- Soeroto, A.Y.; Antartika, R.; Asriputri, N.N.; Suryadinata, H.; Andriyoko, B. Real-time RT-PCR Ct value is not associated with COVID-19 disease severity: An observational study in tertiary COVID-19 referral hospital of West Java, Indonesia. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 4893–4901. [Google Scholar]
- Vasudevan, H.N.; Xu, P.; Servellita, V.; Miller, S.; Liu, L.; Gopez, A.; Chiu, C.Y.; Abate, A.R. Digital droplet PCR accurately quantifes SARS-CoV-2 viral load from crude lysate without nucleic acid purification. Sci. Rep. 2021, 11, 780. [Google Scholar] [CrossRef]
- de Kock, R.; Baselmans, M.; Scharnhorst, V.; Deiman, B. Sensitive detection and quantification of SARS-CoV-2 by multiplex droplet digital RT-PCR. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Purohit, S.; Rao, P.K.; Rawtani, D. Sampling and analytical techniques for COVID-19. In COVID-19 in the Environment: Impact, Concern, and Management of Coronavirus; Rawtani, D., Hussain, C.M., Khatri, N., Eds.; Candice Janco: New Delhi, India, 2022. [Google Scholar]
- Sit, T.H.C.; Brackman, C.J.; Ip, S.M.; Tam, K.W.S.; Law, P.Y.T.; To, E.M.W.; Yu, V.Y.T.; Sims, L.D.; Tsang, D.N.C.; Chu, D.K.W.; et al. Infection of dogs with SARS-CoV-2. Nature 2020, 586, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Chaimayo, C.; Kaewnaphan, B.; Tanlieng, N.; Athipanyasilp, N.; Sirijatuphat, R.; Chayakulkeeree, M.; Angkasekwinai, N.; Sutthent, R.; Puangpunngam, N.; Tharmviboonsri, T.; et al. Rapid SARS-CoV-2 antigen detection assay in comparison with real-time RT-PCR assay for laboratory diagnosis of COVID-19 in Thailand. Virol. J. 2020, 17, 177. [Google Scholar] [CrossRef]
- Peto, T. COVID-19: Rapid antigen detection for SARS-CoV-2 by lateral flow assay: A national systematic evaluation of sensitivity and specificity for mass-testing. EClinicalMedicine 2021, 36, 100924. [Google Scholar] [CrossRef]
- Lee, C.Y.-P.; Lin, R.T.P.; Renia, L.; Ng, L.F.P. Serological approaches for COVID-19: Epidemiologic perspective on surveillance and control. Front. Immunol. 2020, 11, 879. [Google Scholar] [CrossRef]
- Mistry, D.A.; Wang, J.Y.; Moeser, M.-E.; Starkey, T.; Lee, L.Y.W. A systematic review of the sensitivity and specificity of lateral flow devices in the detection of SARS-CoV-2. BMC Infect. Dis. 2021, 21, 879. [Google Scholar] [CrossRef]
- Albert, E.; Torres, I.; Bueno, F.; Huntley, D.; Molla, E.; Fernández-Fuentes, M.Á.; Martínez, M.; Poujois, S.; Forqué, L.; Valdivia, A.; et al. Field evaluation of a rapid antigen test (Panbio™ COVID-19 Ag Rapid Test Device) for COVID-19 diagnosis in primary healthcare centres. Clin. Microbiol. Infect. 2021, 27, 472.e7–472.e10. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Field | Application | References |
|---|---|---|
| Gene expression analysis | Analysis of wireless fidelity radiofrequency radiation on the expression of E. coli genes that potentially alter its pathogenic traits | [23] |
| Examination of plant gene expression impacting lignin synthesis for plant cell wall structure | [24] | |
| Analysis of gene expression as a potential biomarker for early-stage diagnosis in colorectal tumor and cancer patients | [25] | |
| Examination of microRNA expression profile in response to viral infection | [26] | |
| Detection of mutation | Detection of mutation patterns in human cancer cells | [27,28] |
| Detection and quantitative analysis of mutation for monitoring drug resistance | [29,30] | |
| Food Analysis | Detection of genetically modified organisms (GMO) | [31,32,33] |
| Detection of allergens in food | [34] | |
| Detection of pork in food products | [35] | |
| Bioremediation monitoring | Monitoring microbial degradation | [36,37,38,39] |
| Detection and quantification of pathogens | Detection of pathogenic bacteria | [40] |
| Identification of microbial species as etiology of a disease | [41] | |
| Molecular bacterial load assay (i.e., Mycobacterium tuberculosis) | [42] | |
| Determination of growth fitness of plasmodium parasites | [43] | |
| Detection of pathogenic RNA viruses | [3,44,45] | |
| Diagnosis of pathogenic DNA viruses | [46] | |
| Analysis of viral load associated with clinical features of the disease | [8,47,48] |
| Gene Name | Abbreviation | Application | Reference |
|---|---|---|---|
| Glyceraldehide-3-phosphate dehydrogenase | GADPH | Analysis of gene expression in human cell lines, human airway epithelial cells, wound healing model, human skeletal muscle tissue, human breast cells, induced pluripotent stem cell reprogramming | [49,52,53,54,55] |
| Actin, beta | ACTB | Analysis of gene expression in wound healing model, human skeletal muscle tissue, human breast cells | [53,54] |
| Ribosomal RNA 18S | 18S | Analysis of gene expression in | [53,54] |
| wound healing model, human skeletal muscle tissue, human breast cells | |||
| β-2-microglobulin | β-2M | Analysis of gene expression in wound healing model, human skeletal muscle tissue, human breast cells | [53,54] |
| Phosphoglycerate kinase 1 | PGK1 | Analysis of gene expression in induced pluripotent stem cell reprogramming | [55] |
| Polyubiquitin C | UBC | Determination of gene expression in human cell lines | [49] |
| DNA topoisomerase 1 | TOP1 | Study of gene expression in human cell lines | [49] |
| ATP synthase subunit beta, mitochondrial | ATP5B | Elucidation of gene expression in human cell lines, induced pluripotent stem cell reprogramming | [49,55] |
| Cyclophilin A | PPIA | Examination of gene expression in human airway epithelial cells | [52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artika, I.M.; Dewi, Y.P.; Nainggolan, I.M.; Siregar, J.E.; Antonjaya, U. Real-Time Polymerase Chain Reaction: Current Techniques, Applications, and Role in COVID-19 Diagnosis. Genes 2022, 13, 2387. https://doi.org/10.3390/genes13122387
Artika IM, Dewi YP, Nainggolan IM, Siregar JE, Antonjaya U. Real-Time Polymerase Chain Reaction: Current Techniques, Applications, and Role in COVID-19 Diagnosis. Genes. 2022; 13(12):2387. https://doi.org/10.3390/genes13122387
Chicago/Turabian StyleArtika, I Made, Yora Permata Dewi, Ita Margaretha Nainggolan, Josephine Elizabeth Siregar, and Ungke Antonjaya. 2022. "Real-Time Polymerase Chain Reaction: Current Techniques, Applications, and Role in COVID-19 Diagnosis" Genes 13, no. 12: 2387. https://doi.org/10.3390/genes13122387