
Proteomic and Transcriptomic Landscapes of Alström and Bardet–Biedl Syndromes
 , , , , ,
, , , , ,
Abstract
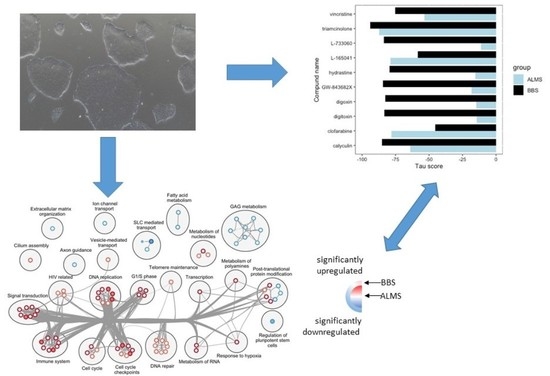
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. RNA Isolation and Microarrays Gene Expression Study—Transcriptomics Analysis
2.3. Proteomic Analysis
2.4. Statistical Analysis
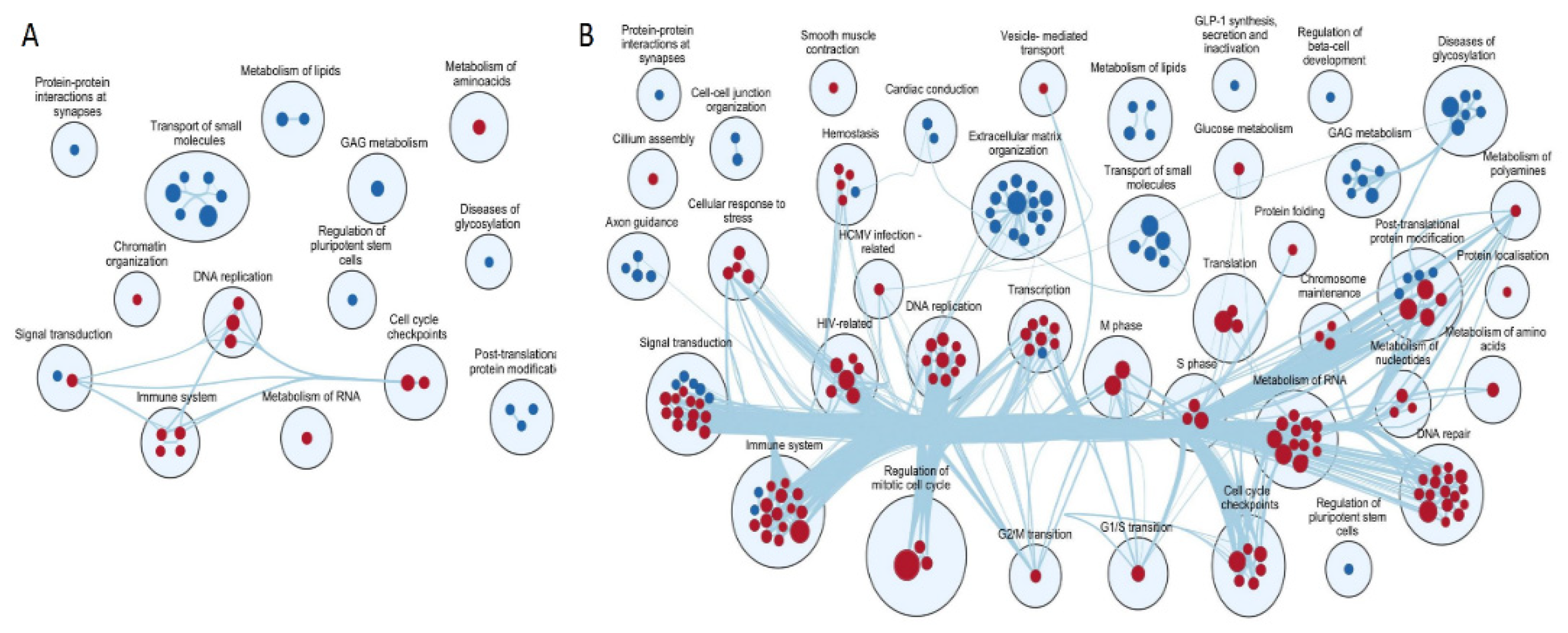
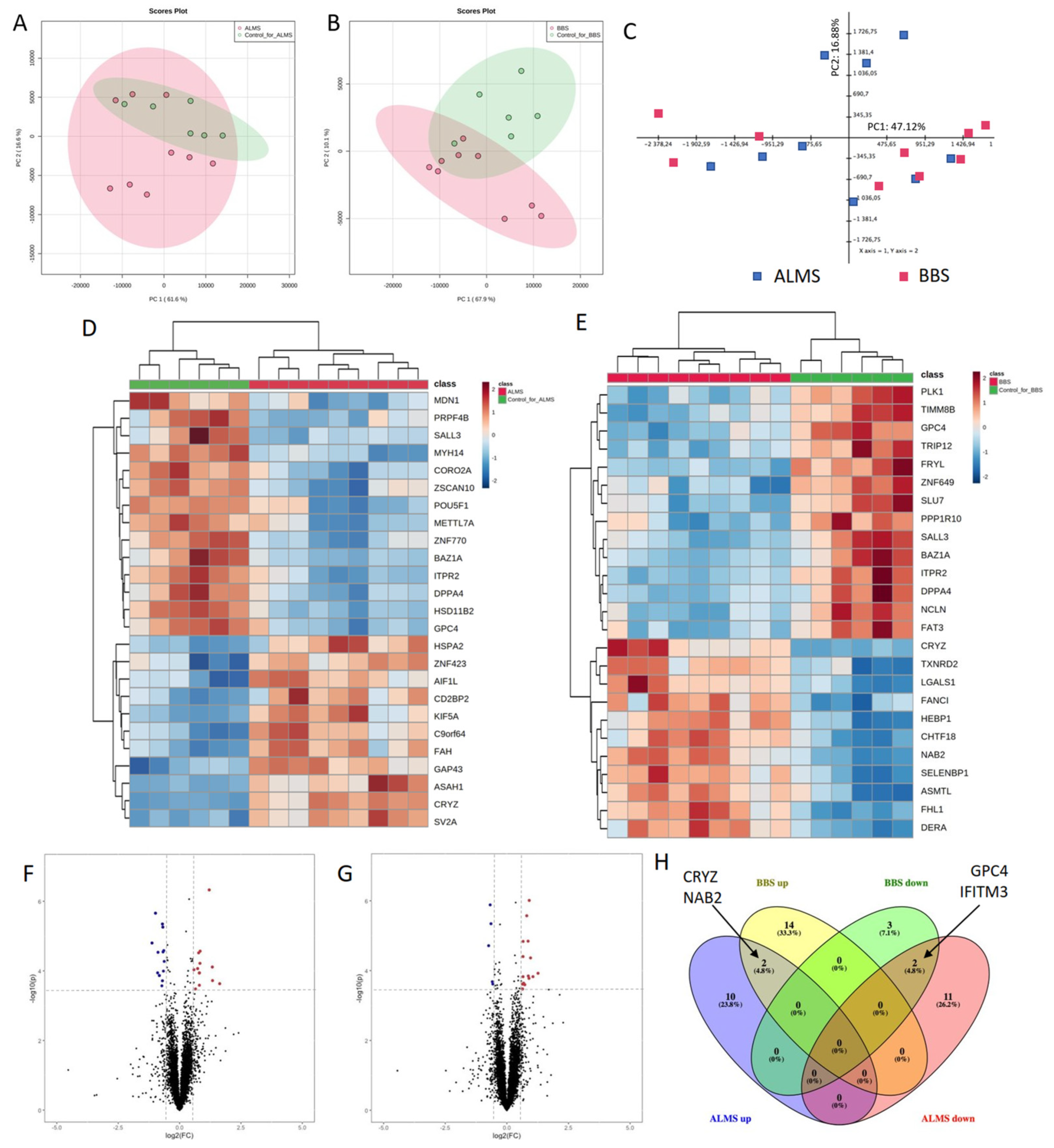
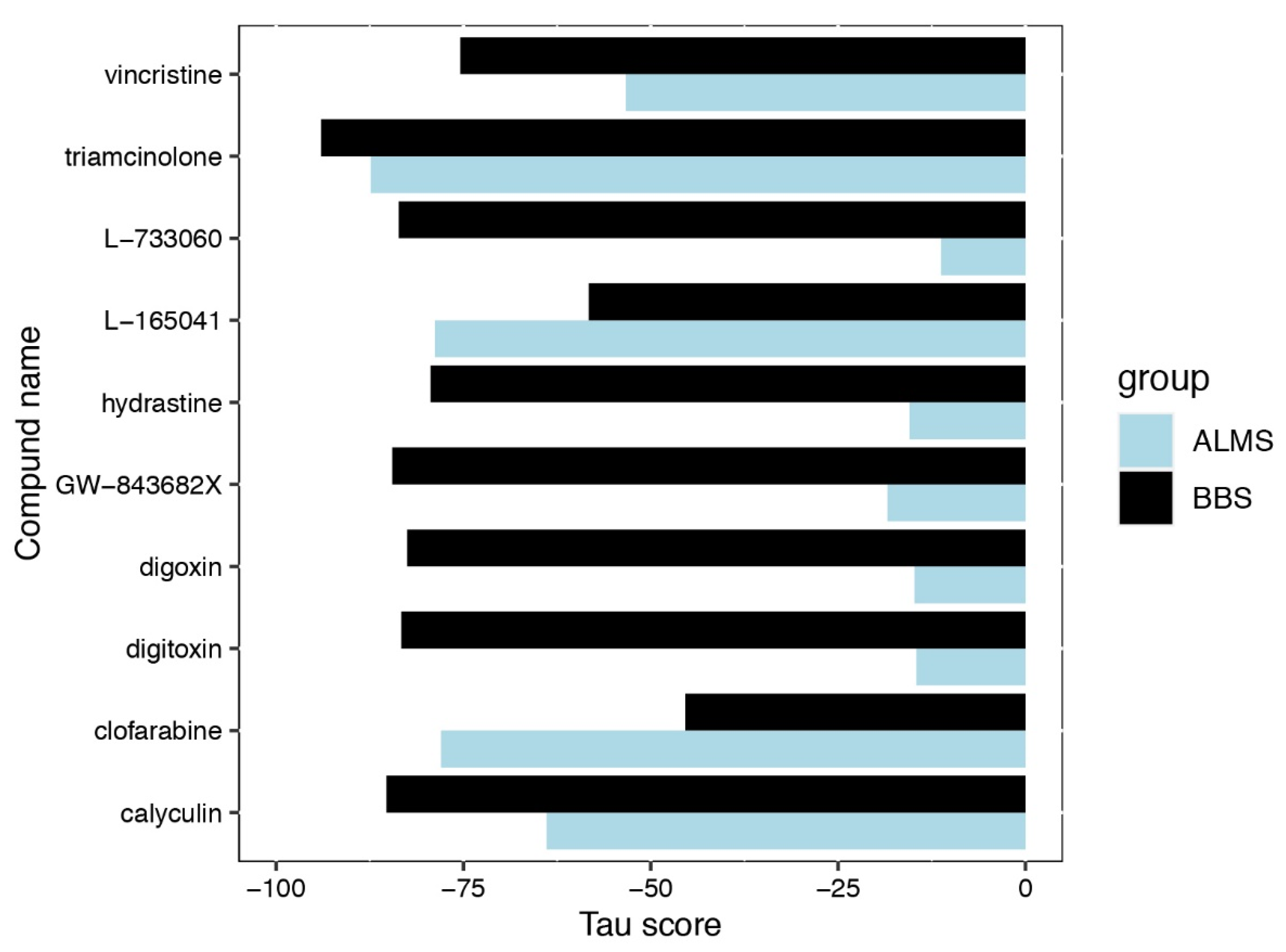
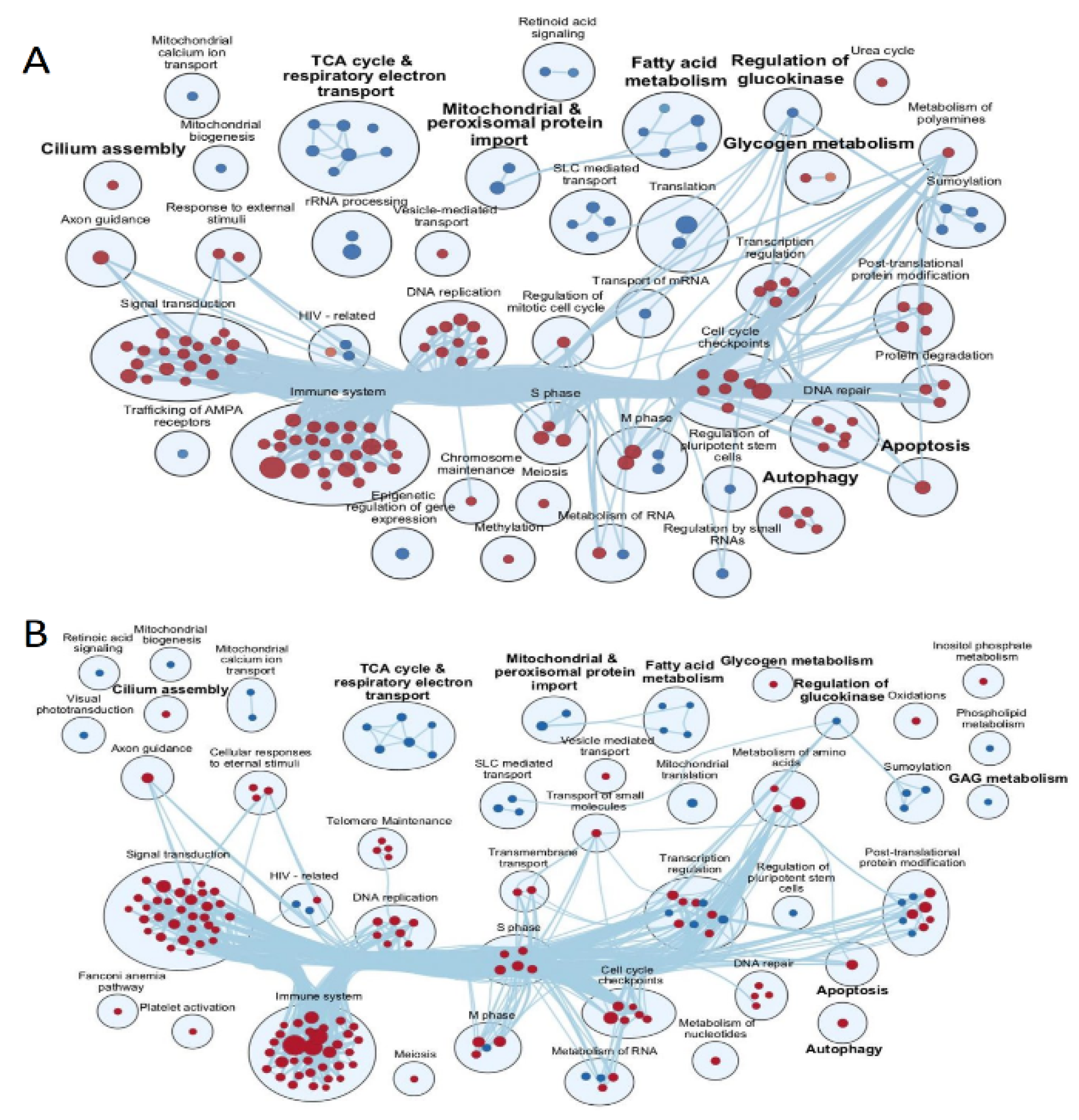
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marshall, J.D.; Bronson, R.T.; Collin, G.B.; Nordstrom, A.D.; Maffei, P.; Paisey, R.B.; Carey, C.; MacDermott, S.; Russell-Eggitt, I.; Shea, S.E.; et al. New Alström Syndrome Phenotypes Based on the Evaluation of 182 Cases. Arch. Intern. Med. 2005, 165, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alstrom Syndrome: Genetics and Clinical Overview. Curr. Genom. 2011, 12, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, D.; Cerutti, M.; Pezzani, L.; Maffei, P.; Milan, G.; Esposito, S. Syndromic obesity: Clinical implications of a correct diagnosis. Ital. J. Pediatr. 2014, 40, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, G.B.; Marshall, J.D.; Ikeda, A.; So, W.V.; Russell-Eggitt, I.; Maffei, P.; Beck, S.; Boerkoel, C.F.; Sicolo, N.; Martin, M.; et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alström syndrome. Nat. Genet. 2002, 31, 74–78. [Google Scholar] [CrossRef]
- Marshall, J.D.; Muller, J.; Collin, G.B.; Milan, G.; Kingsmore, S.F.; Dinwiddie, D.; Farrow, E.G.; Miller, N.A.; Favaretto, F.; Maffei, P.; et al. Alström Syndrome: Mutation Spectrum of ALMS1. Hum. Mutat. 2015, 36, 660–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priya, S.; Nampoothiri, S.; Sen, P.; Sripriya, S. Bardet–Biedl syndrome: Genetics, molecular pathophysiology, and disease management. Indian J. Ophthalmol. 2016, 64, 620–627. [Google Scholar] [CrossRef]
- Castro-Sánchez, S.; Satta, M.; Corton, M.; Guillén, E.; Ayuso, C.; Valverde, D. Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. J. Med. Genet. 2015, 52, 503–513. [Google Scholar] [CrossRef]
- Forsythe, E.; Beales, P.L. Bardet–Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Forsythe, E.; Sparks, K.; Hoskins, B.; Bagkeris, E.; McGowan, B.; Carroll, P.; Huda, M.; Mujahid, S.; Peters, C.; Barrett, T.; et al. Genetic predictors of cardiovascular morbidity in Bardet–Biedl syndrome. Clin. Genet. 2015, 87, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Testa, F.; Zacchia, M.; Crispo, A.A.; Di Iorio, V.; Capolongo, G.; Rinaldi, L.; D’Antonio, M.; Fioretti, T.; Iadicicco, P.; et al. Genetic characterization of Italian patients with Bardet-Biedl syndrome and correlation to ocular, renal and audio-vestibular phenotype: Identification of eleven novel pathogenic sequence variants. BMC Med. Genet. 2017, 18, 10. [Google Scholar] [CrossRef]
- Forsythe, E.; Kenny, J.; Bacchelli, C.; Beales, P.L. Managing Bardet–Biedl Syndrome—Now and in the Future. Front. Ped. 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hearn, T.; Spalluto, C.; Phillips, V.J.; Renforth, G.L.; Copin, N.; Hanley, N.A.; Wilson, D.I. Subcellular Localization of ALMS1 Supports Involvement of Centrosome and Basal Body Dysfunction in the Pathogenesis of Obesity, Insulin Resistance, and Type 2 Diabetes. Diabetes 2005, 54, 1581–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knorz, V.J.; Spalluto, C.; Lessard, M.; Purvis, T.L.; Adigun, F.F.; Collin, G.B.; Hanley, N.A.; Wilson, D.I.; Hearn, T. Centriolar Association of ALMS1 and Likely Centrosomal Functions of the ALMS Motif–containing Proteins C10orf90 and KIAA1731. Mol. Biol. Cell 2010, 21, 3617–3629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, G.B.; Cyr, E.; Bronson, R.; Marshall, J.D.; Gifford, E.J.; Hicks, W.; Murray, S.A.; Zheng, Q.Y.; Smith, R.S.; Naggert, J.K.; et al. Alms1-disrupted mice recapitulate human Alström syndrome. Hum. Mol. Genet. 2005, 14, 2323–2333. [Google Scholar] [CrossRef]
- Collin, G.B.; Marshall, J.D.; King, B.L.; Milan, G.; Maffei, P.; Jagger, D.J.; Naggert, J.K. The Alström Syndrome Protein, ALMS1, Interacts with α-Actinin and Components of the Endosome Recycling Pathway. PLoS ONE 2012, 7, e37925. [Google Scholar] [CrossRef] [Green Version]
- Starks, R.D.; Beyer, A.M.; Guo, D.F.; Boland, L.; Zhang, Q.; Sheffield, V.C.; Rahmouni, K. Regulation of Insulin Receptor Trafficking by Bardet Biedl Syndrome Proteins. PLoS Genet. 2015, 11, e1005311. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, S.; Katoh, Y.; Kobayashi, T.; Nakayama, K. BBS1 is involved in retrograde trafficking of ciliary GPCRs in the context of the BBSome complex. PLoS ONE 2018, 13, e0195005. [Google Scholar] [CrossRef]
- Klink, B.U.; Gatsogiannis, C.; Hofnagel, O.; Wittinghofer, A.; Raunser, S. Structure of the human BBSome core complex. eLife 2020, 9, e53910. [Google Scholar] [CrossRef]
- Song, D.K.; Choi, J.H.; Kim, M.-S. Primary Cilia as a Signaling Platform for Control of Energy Metabolism. Diabetes Metab. J. 2018, 42, 117–127. [Google Scholar] [CrossRef]
- Mariman, E.C.M.; Vink, R.G.; Roumans, N.J.T.; Bouwman, F.G.; Stumpel, C.T.R.M.; Aller, E.E.J.G.; van Baak, M.; Wang, P. The cilium: A cellular antenna with an influence on obesity risk. Br. J. Nutr. 2016, 116, 576–592. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchese, E.; Ruoppolo, M.; Perna, A.; Capasso, G.; Zacchia, M. Exploring Key Challenges of Understanding the Pathogenesis of Kidney Disease in Bardet–Biedl Syndrome. Kidney Int. Rep. 2020, 5, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Scesa, G.; Adami, R.; Bottai, D. iPSC Preparation and Epigenetic Memory: Does the Tissue Origin Matter? Cells 2021, 10, 1470. [Google Scholar] [CrossRef] [PubMed]
- Drozd, A.M.; Walczak, M.P.; Piaskowski, S.; Stoczynska-Fidelus, E.; Rieske, P.; Grzela, D.P. Generation of human iPSCs from cells of fibroblastic and epithelial origin by means of the oriP/EBNA-1 episomal reprogramming system. Stem Cell Res. Ther. 2015, 6, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmyslowska, A.; Borowiec, M.; Antosik, K.; Ploski, R.; Ciechanowska, M.; Iwaniszewska, B.; Jakubiuk-Tomaszuk, A.; Janczyk, W.; Krawczynski, M.; Salmonowicz, B.; et al. Genetic evaluation of patients with Alström syndrome in the Polish population. Clin. Genet. 2016, 89, 448–453. [Google Scholar] [CrossRef]
- Jeziorny, K.; Antosik, K.; Jakiel, P.; Młynarski, W.; Borowiec, M.; Zmysłowska, A. Next-Generation Sequencing in the Diagnosis of Patients with Bardet–Biedl Syndrome—New Variants and Relationship with Hyperglycemia and Insulin Resistance. Genes 2020, 11, 1283. [Google Scholar] [CrossRef]
- Zhou, T.; Benda, C.; Dunzinger, S.; Huang, Y.; Ho, J.C.; Yang, J.; Wang, Y.; Zhang, Y.; Zhuang, Q.; Li, Y.; et al. Generation of human induced pluripotent stem cells from urine samples. Nat. Protoc. 2012, 7, 2080–2089. [Google Scholar] [CrossRef]
- Grot, D.; Wasiak, K.; Tyszkowski, J.; Stoczynska-Fidelus, E.; Ochedalski, T.P.; Rieske, P. Regeneration difficulties in patients with FQAD can limit the use of iPSc-based cell therapy. Stem Cell Res. Ther. 2022, 13, 210. [Google Scholar] [CrossRef]
- Zmyslowska, A.; Kuljanin, M.; Malachowska, B.; Stanczak, M.; Michalek, D.; Wlodarczyk, A.; Grot, D.; Taha, J.; Pawlik, B.; Lebiedzińska-Arciszewska, M.; et al. Multiomic analysis on human cell model of wolfram syndrome reveals changes in mitochondrial morphology and function. Cell Commun. Signal. 2021, 19, 1–16. [Google Scholar] [CrossRef]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the Multiplexing Capacity of TMTs Using Reporter Ion Isotopologues with Isobaric Masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Yoav, B.Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment Map: A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef] [Green Version]
- Kucera, M.; Isserlin, R.; Arkhangorodsky, A.; Bader, G.D. AutoAnnotate: A Cytoscape app for summarizing networks with semantic annotations. F1000Research 2016, 5, 1717. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Ávalos, Y.; Peña-Oyarzun, D.; Budini, M.; Morselli, E.; Criollo, A. New Roles of the Primary Cilium in Autophagy. BioMed Res. Int. 2017, 2017, 4367019. [Google Scholar] [CrossRef] [PubMed]
- Wiegering, A.; Rüther, U.; Gerhardt, C. The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy. Cells 2019, 8, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostelley, T.L.; Lodh, S.; Zaghloul, N.A. Whole organism transcriptome analysis of zebrafish models of Bardet-Biedl Syndrome and Alström Syndrome provides mechanistic insight into shared and divergent phenotypes. BMC Genom. 2016, 17, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, R.F.; George, A.M.; Roussel, E.M.; Faux, M.C.; Wigle, J.T.; Eisenstat, D.D. Genetic Regulation of Vertebrate Forebrain Development by Homeobox Genes. Front. Neurosci. 2022, 16, 843794. [Google Scholar] [CrossRef]
- Li, W.; Zhu, Z.; He, K.; Ma, X.; Pignolo, R.J.; Sieck, G.C.; Hu, J.; Wang, H. Primary cilia in satellite cells are the mechanical sensors for muscle hypertrophy. Proc. Natl. Acad. Sci. USA 2022, 119, e2103615119. [Google Scholar] [CrossRef]
- Aruga, J.; Millen, K.J. ZIC1 Function in Normal Cerebellar Development and Human Developmental Pathology. Adv. Exp. Med. Biol. 2018, 1046, 249–268. [Google Scholar] [CrossRef]
- Markan, K.R.; Boland, L.K.; King-McAlpin, A.Q.; Claflin, K.E.; Leaman, M.P.; Kemerling, M.K.; Stonewall, M.M.; Amendt, B.A.; Ankrum, J.A.; Potthoff, M.J. Adipose TBX1 regulates β-adrenergic sensitivity in subcutaneous adipose tissue and thermogenic capacity in vivo. Mol. Metab. 2020, 36, 100965. [Google Scholar] [CrossRef]
- Geusz, R.J.; Wang, A.; Lam, D.K.; Vinckier, N.K.; Alysandratos, K.-D.; Roberts, D.A.; Wang, J.; Kefalopoulou, S.; Ramirez, A.; Qiu, Y.; et al. Sequence logic at enhancers governs a dual mechanism of endodermal organ fate induction by FOXA pioneer factors. Nat. Commun. 2021, 12, 6636. [Google Scholar] [CrossRef]
- Lowenstein, E.D.; Rusanova, A.; Stelzer, J.; Hernaiz-Llorens, M.; Schroer, A.E.; Epifanova, E.; Bladt, F.; Isik, E.G.; Buchert, S.; Jia, S.; et al. Olig3 regulates early cerebellar development. eLife 2021, 10, e64684. [Google Scholar] [CrossRef]
- Alam, G.; Luan, Z.; Gul, A.; Lu, H.; Zhou, Y.; Huo, X.; Li, Y.; Du, C.; Luo, Z.; Zhang, H.; et al. Activation of farnesoid X receptor (FXR) induces crystallin zeta expression in mouse medullary collecting duct cells. Pflug. Arch. 2020, 472, 1631–1641. [Google Scholar] [CrossRef]
- Lucerna, M.; Mechtcheriakova, D.; Kadl, A.; Schabbauer, G.; Schäfer, R.; Gruber, F.; Koshelnick, Y.; Müller, H.-D.; Issbrücker, K.; Clauss, M.; et al. NAB2, a Corepressor of EGR-1, Inhibits Vascular Endothelial Growth Factor-mediated Gene Induction and Angiogenic Responses of Endothelial Cells. J. Biol. Chem. 2003, 278, 11433–11440. [Google Scholar] [CrossRef] [PubMed]
- Oikari, L.; Yu, C.; Okolicsanyi, R.K.; Avgan, N.; Peall, I.; Griffiths, L.R.; Haupt, L.M. HSPGs glypican-1 and glypican-4 are human neuronal proteins characteristic of different neural phenotypes. J. Neurosci. Res. 2020, 98, 1619–1645. [Google Scholar] [CrossRef] [PubMed]
- Leelalertlauw, C.; Korwutthikulrangsri, M.; Mahachoklertwattana, P.; Chanprasertyothin, S.; Khlairit, P.; Pongratanakul, S.; Poomthavorn, P. Serum glypican 4 level in obese children and its relation to degree of obesity. Clin. Endocrinol. 2017, 87, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Hur, J.-Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The innate immunity protein IFITM3 modulates γ-secretase in Alzheimer’s disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef]
- Hearn, T. ALMS1 and Alström syndrome: A recessive form of metabolic, neurosensory and cardiac deficits. J. Mol. Med. 2019, 97, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Nachury, M.V.; Loktev, A.V.; Zhang, Q.; Westlake, C.J.; Peränen, J.; Merdes, A.; Slusarski, D.C.; Scheller, R.H.; Bazan, J.F.; Sheffield, V.C.; et al. A Core Complex of BBS Proteins Cooperates with the GTPase Rab8 to Promote Ciliary Membrane Biogenesis. Cell 2007, 129, 1201–1213. [Google Scholar] [CrossRef] [Green Version]
- Breslow, D.K.; Hoogendoorn, S.; Kopp, A.R.; Morgens, D.W.; Vu, B.K.; Kennedy, M.C.; Han, K.; Li, A.; Hess, G.T.; Bassik, M.C.; et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat. Genet. 2018, 50, 460–471. [Google Scholar] [CrossRef]
- Antonellis, P.J.; Engle, S.E.; Brewer, K.M.; Berbari, N.F. The Hedgehog Signaling Pathway is Expressed in the Adult Mouse Hypothalamus and Modulated by Fasting. eNeuro 2021, 8, ENEURO.0276-21.2021. [Google Scholar] [CrossRef]
- Hey, C.; Larsen, L.; Tümer, Z.; Brøndum-Nielsen, K.; Grønskov, K.; Hjortshøj, T.; Møller, L. BBS Proteins Affect Ciliogenesis and Are Essential for Hedgehog Signaling, but Not for Formation of iPSC-Derived RPE-65 Expressing RPE-Like Cells. Int. J. Mol. Sci. 2021, 22, 1345. [Google Scholar] [CrossRef]
- Wu, H.-T.; Chen, C.-T.; Cheng, K.-C.; Li, Y.-X.; Yeh, C.-H.; Cheng, J.-T. Pharmacological Activation of Peroxisome Proliferator-activated Receptor δ Improves Insulin Resistance and Hepatic Steatosis in High Fat Diet-induced Diabetic Mice. Horm. Metab. Res. 2011, 43, 631–635. [Google Scholar] [CrossRef]
- Leibowitz, M.D.; Fiévet, C.; Hennuyer, N.; Peinado-Onsurbe, J.; Duez, H.; Berger, J.; Cullinan, C.A.; Sparrow, C.P.; Baffic, J.; Berger, G.D.; et al. Activation of PPARδ alters lipid metabolism in db/db mice. FEBS Lett. 2000, 473, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, A.; Muramatsu, Y.; Yamazaki, T.; Muramoto, M.; Kita, Y.; Yamazaki, S.; Mihara, K.; Moriguchi, A.; Matsuoka, N. Neuroprotective Efficacy of the Peroxisome Proliferator-Activated Receptor δ-Selective Agonists in Vitro and in Vivo. J. Pharmacol. Exp. Ther. 2006, 320, 1087–1096. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyczynska, U.; Stanczak, M.; Kuljanin, M.; Włodarczyk, A.; Stoczynska-Fidelus, E.; Taha, J.; Pawlik, B.; Borowiec, M.; Mancias, J.D.; Mlynarski, W.; et al. Proteomic and Transcriptomic Landscapes of Alström and Bardet–Biedl Syndromes. Genes 2022, 13, 2370. https://doi.org/10.3390/genes13122370
Smyczynska U, Stanczak M, Kuljanin M, Włodarczyk A, Stoczynska-Fidelus E, Taha J, Pawlik B, Borowiec M, Mancias JD, Mlynarski W, et al. Proteomic and Transcriptomic Landscapes of Alström and Bardet–Biedl Syndromes. Genes. 2022; 13(12):2370. https://doi.org/10.3390/genes13122370
Chicago/Turabian StyleSmyczynska, Urszula, Marcin Stanczak, Miljan Kuljanin, Aneta Włodarczyk, Ewelina Stoczynska-Fidelus, Joanna Taha, Bartłomiej Pawlik, Maciej Borowiec, Joseph D. Mancias, Wojciech Mlynarski, and et al. 2022. "Proteomic and Transcriptomic Landscapes of Alström and Bardet–Biedl Syndromes" Genes 13, no. 12: 2370. https://doi.org/10.3390/genes13122370