
Challenges in Gene Therapy for Somatic Reverted Mosaicism in X-Linked Combined Immunodeficiency by CRISPR/Cas9 and Prime Editing
 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. The Components for Gene-Editing
2.3. Cell Culture
2.4. Genomic Analysis
2.5. In Vitro CRISPR/Cas9 Cutting Assay
2.6. Screening of sgRNAs
2.7. Cell Proliferation Assay
2.8. Transfection of CRISPR/Cas9-ssODN
2.9. Transfection of Prime Editing (PE2)
2.10. Editing Efficiency Analysis
2.10.1. DsRed Expression
2.10.2. GFP Expression
2.10.3. Sanger Sequencing
2.10.4. Restriction Fragment Length Polymorphism (RFLP) Analysis
2.10.5. Droplet Digital PCR (ddPCR) Analysis
2.11. Statistical Analysis
3. Results
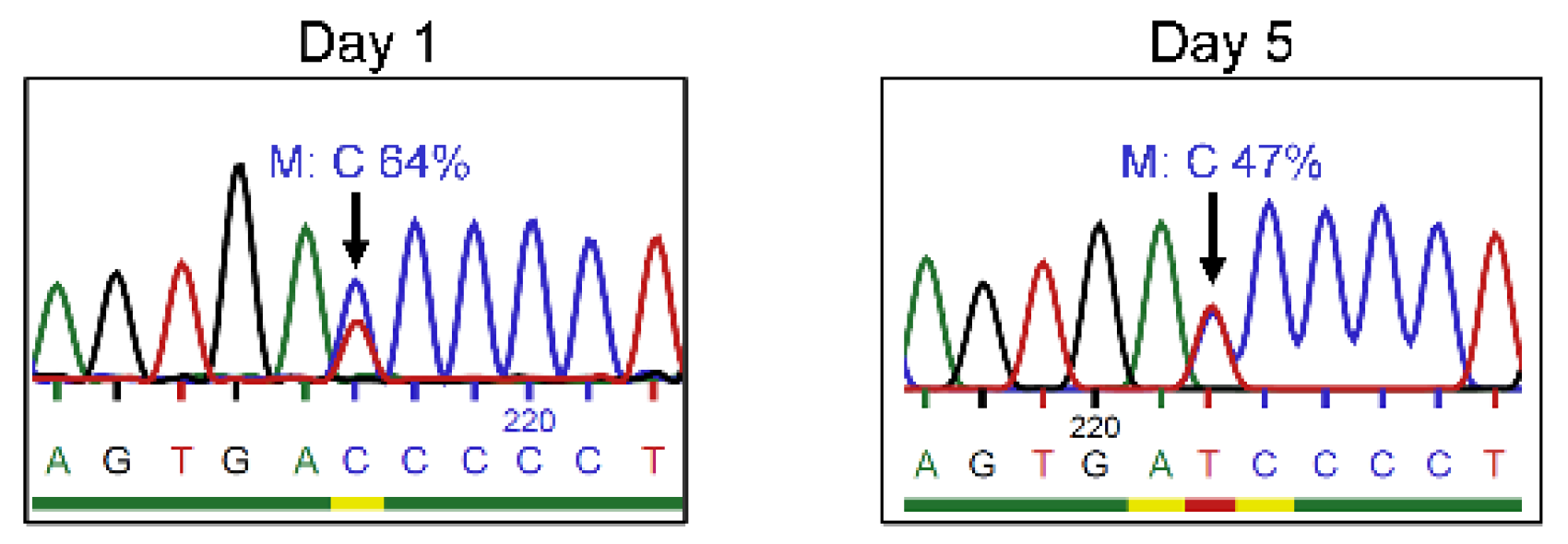
3.1. Reduced Frequency of Mutant c.458T>C IL2RG in Mosaic T Cells after In Vitro Cultivation
3.2. Gene Correction by CRISPR/Cas9-ssODN Approach
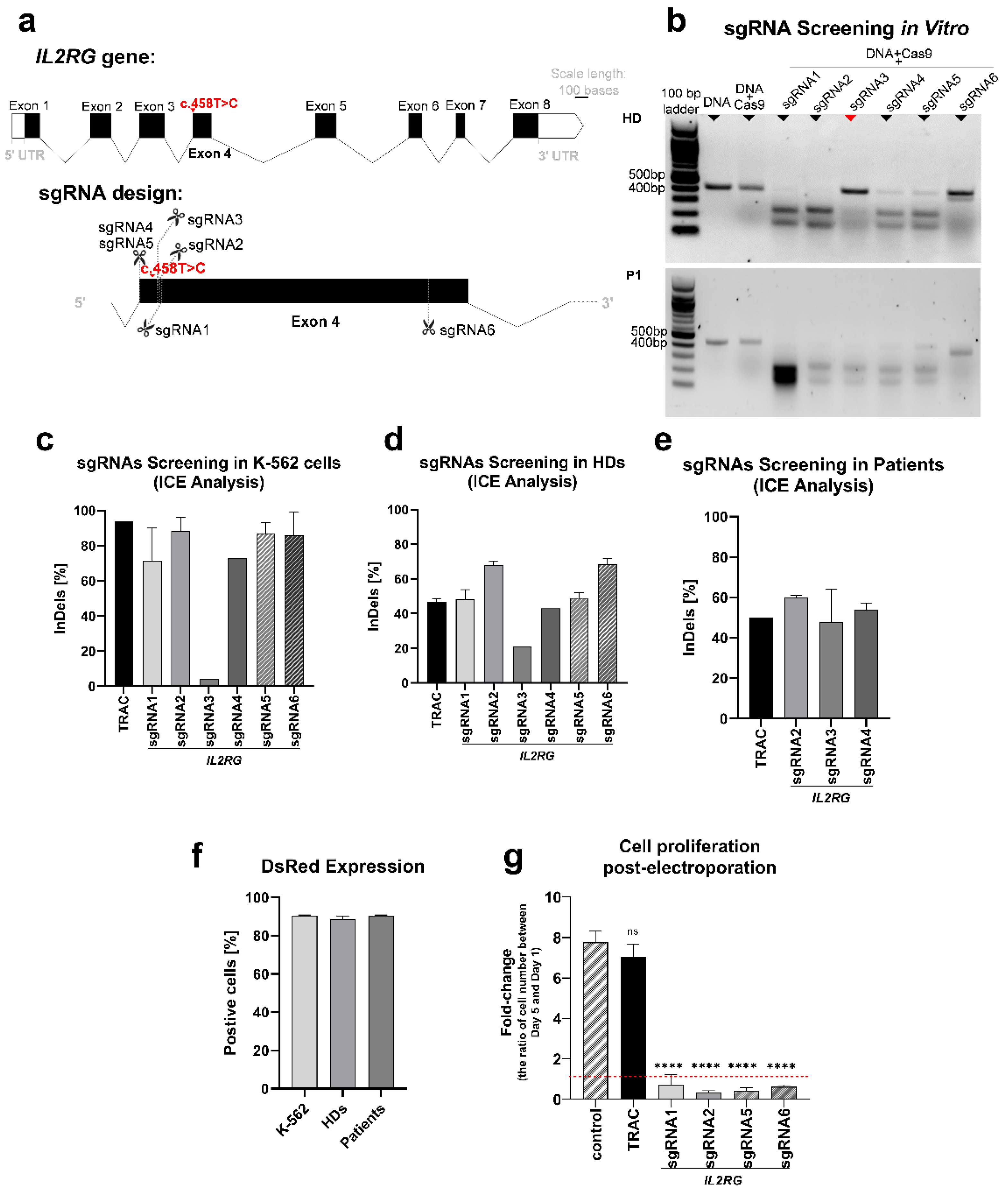
3.2.1. sgRNAs Screening
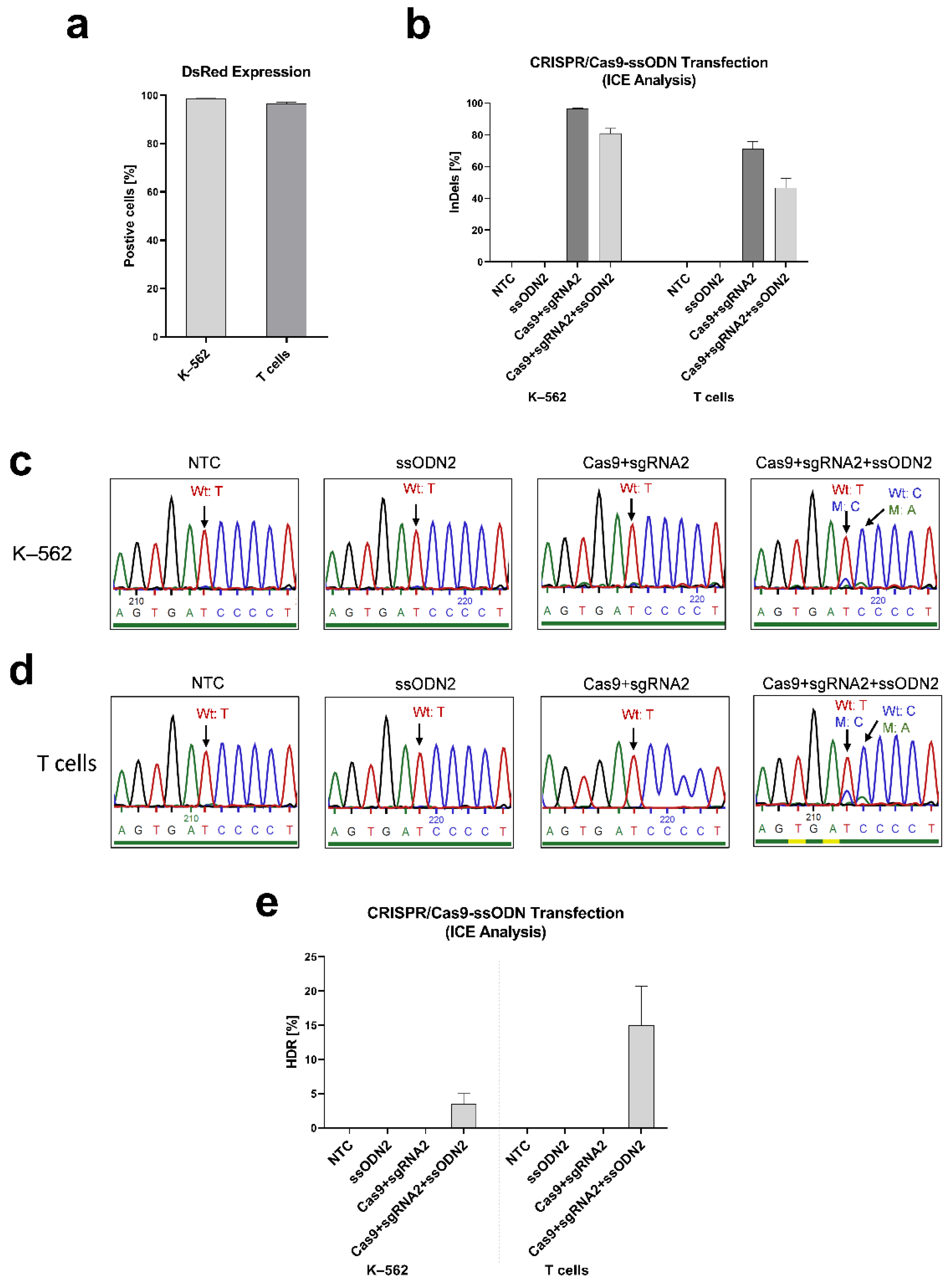
3.2.2. Inducing IL2RG Mutation in K–562 and T cells
3.2.3. Correcting the IL2RG c.458T>C Mutation in Mosaic T Cells
3.3. Gene Editing by Prime Editing
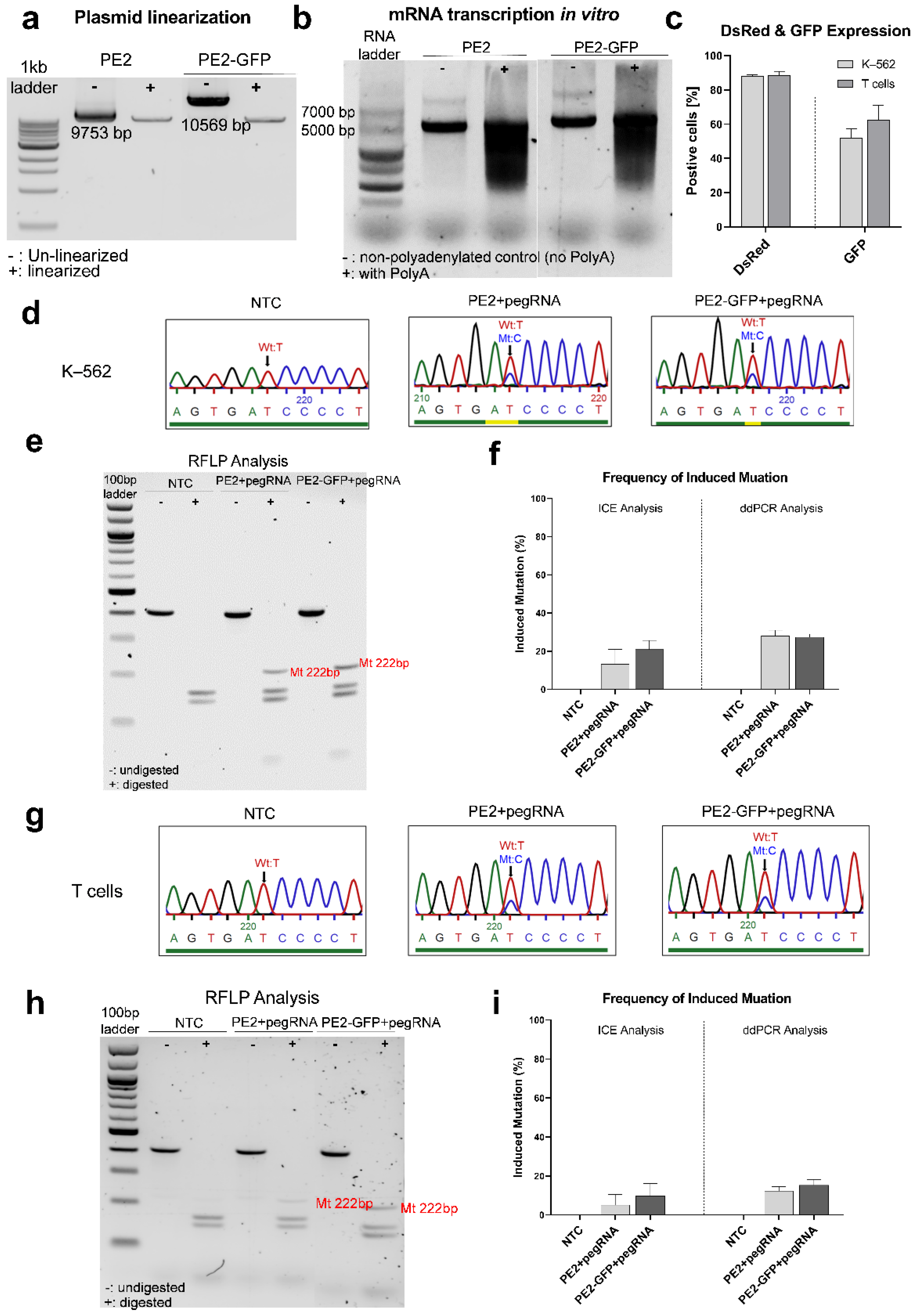
3.3.1. Inducing the IL2RG c.458T>C Mutation in K–562 and T Cells
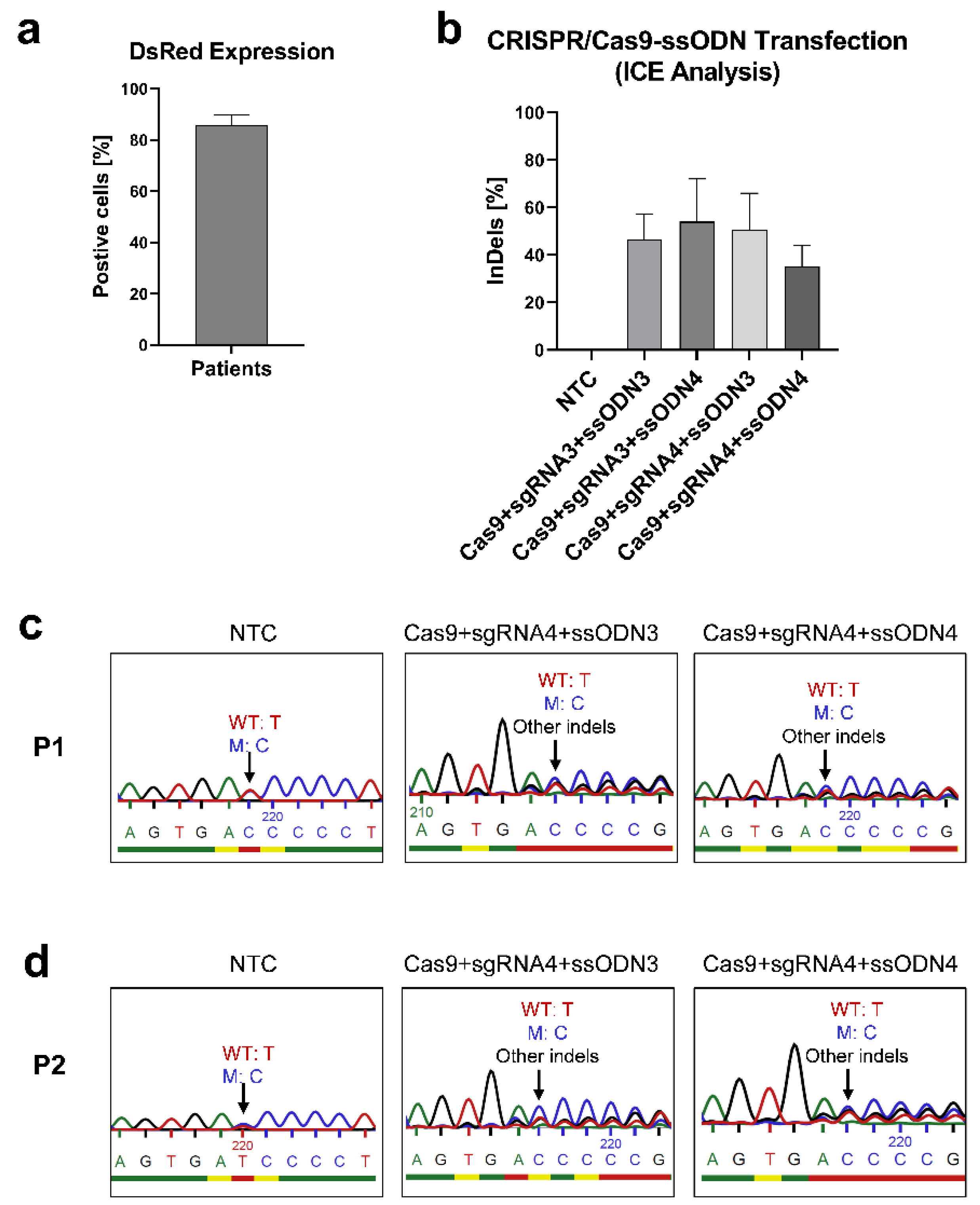
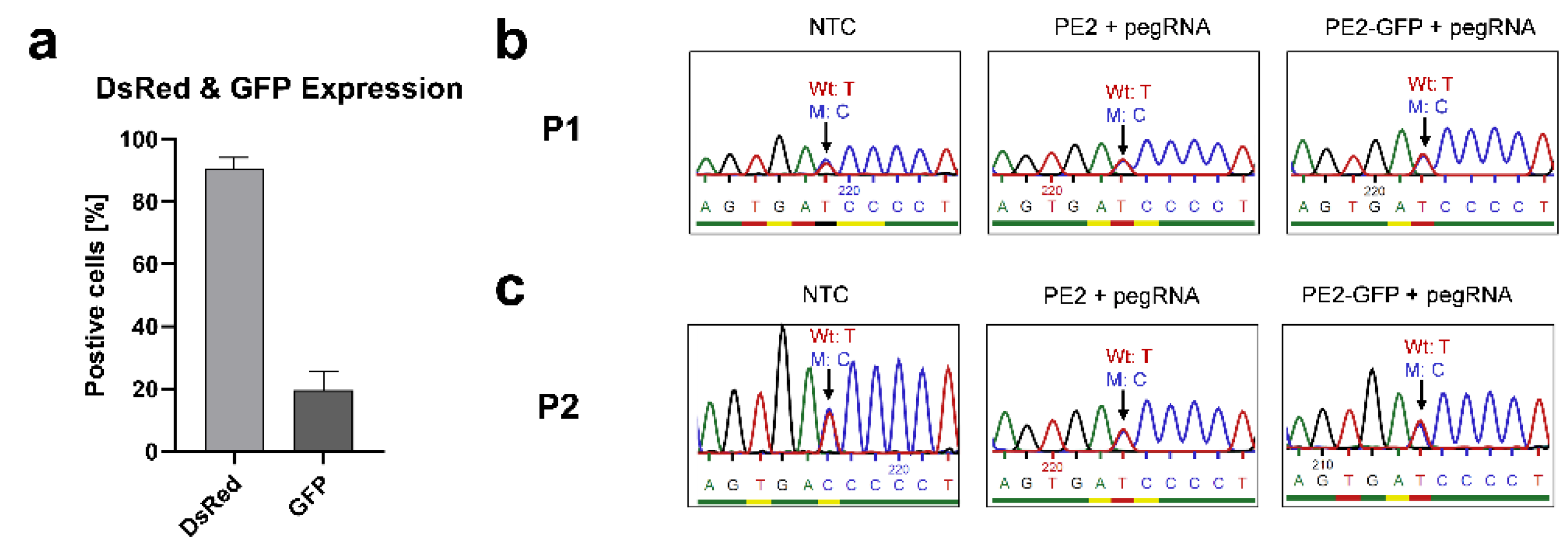
3.3.2. Correcting the Mutant IL2RG in Mosaic T Cells
4. Discussion
- The presence of a revertant mutation reduces the editing efficiency. Because the wild-type and mutant sequences with only one nucleotide variation coexist in the targeted population, this hampers specific targeting in both CRISPR and prime editing systems. Moreover, it leads to more difficulty in detecting the corrected nucleotide when the frequency of gene editing is low. Highly sensitive genotyping techniques such as next-generation sequencing would be required to precisely determine the correction efficiency [54]. Nevertheless, we hypothesize that the correction events for the patients’ treated T cells, if any, would be infrequent at best.
- The probability of targeting mutant cells is lower in a mosaic population. Previously, we investigated the levels of γc in patient T cells, finding them to be similar to healthy donors [13]. The similar expression of γc between wild-type/mutant subpopulations and only one amino acid difference in both proteins make fluorescence-activated cell sorter (FACS) inviable; therefore, working with the mosaic population was required. Moreover, the proliferation of patients’ CD3+ T cells was impaired compared with healthy donors [13]. According to the genetic analysis, with the in vitro culturing of CD3+ T cells from patients, the frequency of the mutant base reduced over time (Figure 1), which is consistent with the finding in previous studies that cells containing revertant mutation have a proliferative advantage [8,55]. These observations demonstrate that mutant cells are hardly capable of proliferating in vitro. This technical limitation implies a lower opportunity to target mutant cells in the heterogeneous population. Higher volumes of blood should have been processed to achieve sufficient cellular material and meet the need of in vitro expansion. In this way, the direct genetic manipulation of T cells could have been pursued, but it was not further investigated for ethical reasons. Similarly, hematopoietic stem cells (HSCs) would have been the most suitable candidates for gene modification to achieve durable immune reconstitution, but patient HSCs were not collected nor used in the present study due to medical ethics.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Puck, J.M.; Deschênes, S.M.; Porter, J.C.; Dutra, A.S.; Brown, C.J.; Willard, H.F.; Henthorn, P.S. The interleukin-2 receptor γ chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum. Mol. Genet. 1993, 2, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Qasim, W.; Davies, E.G.; Rao, K.; Amrolia, P.J.; Veys, P. How I treat severe combined immunodeficiency. Blood 2013, 122, 3749–3758. [Google Scholar] [CrossRef] [PubMed]
- Kumrah, R.; Vignesh, P.; Patra, P.; Singh, A.; Anjani, G.; Saini, P.; Sharma, M.; Kaur, A.; Rawat, A. Genetics of severe combined immunodeficiency. Genes Dis. 2020, 7, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Abolhassani, H.; Appelberg, S.K.; Sundin, M.; Hammarström, L. hypomorphic mutation: Identification of a novel pathogenic mutation in exon 8 and a review of the literature. Allergy Asthma Clin. Immunol. Off. J. Can. Soc. Allergy Clin. Immunol. 2019, 15, 2. [Google Scholar] [CrossRef] [Green Version]
- Stephan, V.; Wahn, V.; Le Deist, F.; Dirksen, U.; Broker, B.; Müller-Fleckenstein, I.; Horneff, G.; Schroten, H.; Fischer, A.; de Saint Basile, G. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N. Engl. J. Med. 1996, 335, 1563–1567. [Google Scholar] [CrossRef]
- Kawai, T.; Saito, M.; Nishikomori, R.; Yasumi, T.; Izawa, K.; Murakami, T.; Okamoto, S.; Mori, Y.; Nakagawa, N.; Imai, K.; et al. Multiple reversions of an IL2RG mutation restore T cell function in an X-linked severe combined immunodeficiency patient. J. Clin. Immunol. 2012, 32, 690–697. [Google Scholar] [CrossRef]
- Hsu, A.P.; Pittaluga, S.; Martinez, B.; Rump, A.P.; Raffeld, M.; Uzel, G.; Puck, J.M.; Freeman, A.F.; Holland, S.M. IL2RG reversion event in a common lymphoid progenitor leads to delayed diagnosis and milder phenotype. J. Clin. Immunol. 2015, 35, 449–453. [Google Scholar] [CrossRef]
- Kuijpers, T.W.; van Leeuwen, E.M.M.; Barendregt, B.H.; Klarenbeek, P.; aan de Kerk, D.J.; Baars, P.A.; Jansen, M.H.; de Vries, N.; van Lier, R.A.W.; van der Burg, M. A reversion of an IL2RG mutation in combined immunodeficiency providing competitive advantage to the majority of CD8+ T cells. Haematologica 2013, 98, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Kury, P.; Führer, M.; Fuchs, S.; Lorenz, M.R.; Giorgetti, O.B.; Bakhtiar, S.; Frei, A.P.; Fisch, P.; Boehm, T.; Schwarz, K.; et al. Long-term robustness of a T-cell system emerging from somatic rescue of a genetic block in T-cell development. EBioMedicine 2020, 59, 102961. [Google Scholar] [CrossRef]
- Okuno, Y.; Hoshino, A.; Muramatsu, H.; Kawashima, N.; Wang, X.; Yoshida, K.; Wada, T.; Gunji, M.; Toma, T.; Kato, T.; et al. Late-Onset Combined Immunodeficiency with a Novel IL2RG Mutation and Probable Revertant Somatic Mosaicism. J. Clin. Immunol. 2015, 35, 610–614. [Google Scholar] [CrossRef]
- Speckmann, C.; Pannicke, U.; Wiech, E.; Schwarz, K.; Fisch, P.; Friedrich, W.; Niehues, T.; Gilmour, K.; Buiting, K.; Schlesier, M.; et al. Clinical and immunologic consequences of a somatic reversion in a patient with X-linked severe combined immunodeficiency. Blood 2008, 112, 4090–4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Kuehn, H.S.; Thauland, T.J.; Lee, C.M.; De Ravin, S.S.; Malech, H.L.; Keyes, T.J.; Jager, A.; Davis, K.L.; Garcia-Lloret, M.I.; et al. Progressive B Cell Loss in Revertant X-SCID. J. Clin. Immunol. 2020, 40, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Gratz, H.P.; Ureña-Bailén, G.; Gratz, P.G.; Schilbach-Stückle, K.; Renno, T.; Güngör, D.; Mader, D.A.; Malenke, E.; Antony, J.S.; et al. Somatic Reversion of a Novel Mutation Resulting in Atypical X-Linked Combined Immunodeficiency. Genes 2021, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Buckley, R.H.; Schiff, R.I.; Schiff, S.E.; Markert, M.L.; Williams, L.W.; Harville, T.O.; Roberts, J.L.; Puck, J.M. Human severe combined immunodeficiency: Genetic, phenotypic, and functional diversity in one hundred eight infants. J. Pediatr. 1997, 130, 378–387. [Google Scholar] [CrossRef]
- Blanco, E.; Izotova, N.; Booth, C.; Thrasher, A.J. Immune Reconstitution After Gene Therapy Approaches in Patients With X-Linked Severe Combined Immunodeficiency Disease. Front. Immunol. 2020, 11, 608653. [Google Scholar] [CrossRef] [PubMed]
- Thornhill, S.I.; Schambach, A.; Howe, S.J.; Ulaganathan, M.; Grassman, E.; Williams, D.; Schiedlmeier, B.; Sebire, N.J.; Gaspar, H.B.; Kinnon, C.; et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 590–598. [Google Scholar] [CrossRef]
- Zychlinski, D.; Schambach, A.; Modlich, U.; Maetzig, T.; Meyer, J.; Grassman, E.; Mishra, A.; Baum, C. Physiological promoters reduce the genotoxic risk of integrating gene vectors. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 718–725. [Google Scholar] [CrossRef]
- Pai, S.-Y.; Thrasher, A.J. Gene therapy for X-linked severe combined immunodeficiency: Historical outcomes and current status. J. Allergy Clin. Immunol. 2020, 146, 258–261. [Google Scholar] [CrossRef]
- Booth, C.; Romano, R.; Roncarolo, M.G.; Thrasher, A.J. Gene therapy for primary immunodeficiency. Hum. Mol. Genet. 2019, 28, R15–R23. [Google Scholar] [CrossRef] [Green Version]
- Pavel-Dinu, M.; Wiebking, V.; Dejene, B.T.; Srifa, W.; Mantri, S.; Nicolas, C.E.; Lee, C.; Bao, G.; Kildebeck, E.J.; Punjya, N.; et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019, 10, 1634. [Google Scholar] [CrossRef]
- Rozov, S.M.; Permyakova, N.V.; Deineko, E.V. The Problem of the Low Rates of CRISPR/Cas9-Mediated Knock-ins in Plants: Approaches and Solutions. Int. J. Mol. Sci. 2019, 20, 3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Gao, G. Prime Editing: A Novel Cas9-Reverse Transcriptase Fusion May Revolutionize Genome Editing. Hum. Gene Ther. 2019, 30, 1445–1446. [Google Scholar] [CrossRef] [PubMed]
- Bousso, P.; Wahn, V.; Douagi, I.; Horneff, G.; Pannetier, C.; Le Deist, F.; Zepp, F.; Niehues, T.; Kourilsky, P.; Fischer, A.; et al. Diversity, functionality, and stability of the T cell repertoire derived in vivo from a single human T cell precursor. Proc. Natl. Acad. Sci. USA 2000, 97, 274–278. [Google Scholar] [CrossRef] [Green Version]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [Green Version]
- Osborn, M.J.; Webber, B.R.; Knipping, F.; Lonetree, C.-l.; Tennis, N.; DeFeo, A.P.; McElroy, A.N.; Starker, C.G.; Lee, C.; Merkel, S.; et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Chow, R.D.; Chen, J.S.; Shen, J.; Chen, S. A web tool for the design of prime-editing guide RNAs. Nat. Biomed. Eng. 2021, 5, 190–194. [Google Scholar] [CrossRef]
- Lamsfus-Calle, A.; Daniel-Moreno, A.; Antony, J.S.; Epting, T.; Heumos, L.; Baskaran, P.; Admard, J.; Casadei, N.; Latifi, N.; Siegmund, D.M.; et al. Comparative targeting analysis of KLF1, BCL11A, and HBG1/2 in CD34 HSPCs by CRISPR/Cas9 for the induction of fetal hemoglobin. Sci. Rep. 2020, 10, 10133. [Google Scholar] [CrossRef]
- Antony, J.S.; Daniel-Moreno, A.; Lamsfus-Calle, A.; Raju, J.; Kaftancioglu, M.; Ureña-Bailén, G.; Rottenberger, J.; Hou, Y.; Santhanakumaran, V.; Lee, J.-H.; et al. A Mutation-Agnostic Hematopoietic Stem Cell Gene Therapy for Metachromatic Leukodystrophy. CRISPR J. 2022, 5, 66–79. [Google Scholar] [CrossRef]
- Conant, D.; Hsiau, T.; Rossi, N.; Oki, J.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; et al. Inference of CRISPR Edits from Sanger Trace Data. CRISPR J. 2022, 5, 123–130. [Google Scholar] [CrossRef]
- Paquet, D.; Kwart, D.; Chen, A.; Sproul, A.; Jacob, S.; Teo, S.; Olsen, K.M.; Gregg, A.; Noggle, S.; Tessier-Lavigne, M. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 2016, 533, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Medley, J.C.; Hebbar, S.; Sydzyik, J.T.; Zinovyeva, A.Y. Single nucleotide substitutions effectively block Cas9 and allow for scarless genome editing in Caenorhabditis elegans. Genetics 2022, 220, iyab199. [Google Scholar] [CrossRef] [PubMed]
- Hiramoto, T.; Li, L.B.; Funk, S.E.; Hirata, R.K.; Russell, D.W. Nuclease-free Adeno-Associated Virus-Mediated Il2rg Gene Editing in X-SCID Mice. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1255–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacein-Bey-Abina, S.; Von Kalle, C.; Schmidt, M.; McCormack, M.P.; Wulffraat, N.; Leboulch, P.; Lim, A.; Osborne, C.S.; Pawliuk, R.; Morillon, E.; et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; Le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.-L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [Green Version]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Mamcarz, E.; Zhou, S.; Lockey, T.; Abdelsamed, H.; Cross, S.J.; Kang, G.; Ma, Z.; Condori, J.; Dowdy, J.; Triplett, B.; et al. Lentiviral Gene Therapy Combined with Low-Dose Busulfan in Infants with SCID-X1. N. Engl. J. Med. 2019, 380, 1525–1534. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S.; Pai, S.-Y.; Gaspar, H.B.; Armant, M.; Berry, C.C.; Blanche, S.; Bleesing, J.; Blondeau, J.; de Boer, H.; Buckland, K.F.; et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2014, 371, 1407–1417. [Google Scholar] [CrossRef] [Green Version]
- De Ravin, S.S.; Wu, X.; Moir, S.; Anaya-O’Brien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Su, L.; Marquesen, M.; et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra357. [Google Scholar] [CrossRef] [Green Version]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Schiroli, G.; Ferrari, S.; Conway, A.; Jacob, A.; Capo, V.; Albano, L.; Plati, T.; Castiello, M.C.; Sanvito, F.; Gennery, A.R.; et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017, 9, eaan0820. [Google Scholar] [CrossRef]
- Urnov, F.D.; Miller, J.C.; Lee, Y.-L.; Beausejour, C.M.; Rock, J.M.; Augustus, S.; Jamieson, A.C.; Porteus, M.H.; Gregory, P.D.; Holmes, M.C. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 2005, 435, 646–651. [Google Scholar] [CrossRef]
- Lombardo, A.; Genovese, P.; Beausejour, C.M.; Colleoni, S.; Lee, Y.-L.; Kim, K.A.; Ando, D.; Urnov, F.D.; Galli, C.; Gregory, P.D.; et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat. Biotechnol. 2007, 25, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, T.; Firth, A.L.; Scripture-Adams, D.D.; Galic, Z.; Qualls, S.J.; Gilmore, W.B.; Ke, E.; Singer, O.; Anderson, L.S.; Bornzin, A.R.; et al. Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell 2015, 16, 367–372. [Google Scholar] [CrossRef] [Green Version]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef]
- Genovese, P.; Schiroli, G.; Escobar, G.; Tomaso, T.D.; Firrito, C.; Calabria, A.; Moi, D.; Mazzieri, R.; Bonini, C.; Holmes, M.C.; et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature 2014, 510, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azhagiri, M.K.K.; Babu, P.; Venkatesan, V.; Thangavel, S. Homology-directed gene-editing approaches for hematopoietic stem and progenitor cell gene therapy. Stem Cell Res. Ther. 2021, 12, 500. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, S.; Amaishi, Y.; Maki, I.; Enoki, T.; Mineno, J. Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci. Rep. 2019, 9, 4811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antony, J.S.; Latifi, N.; Haque, A.; Lamsfus-Calle, A.; Daniel-Moreno, A.; Graeter, S.; Baskaran, P.; Weinmann, P.; Mezger, M.; Handgretinger, R.; et al. Gene correction of HBB mutations in CD34+ hematopoietic stem cells using Cas9 mRNA and ssODN donors. Mol. Cell. Pediatr. 2018, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Busquets, O.; Verma, Y.; Syed, K.M.; Kutnowski, N.; Pangilinan, G.R.; Gilbert, L.A.; Bateup, H.S.; Rio, D.C.; Hockemeyer, D.; et al. Highly efficient generation of isogenic pluripotent stem cell models using prime editing. eLife 2022, 11, e79208. [Google Scholar] [CrossRef] [PubMed]
- Tsiatis, A.C.; Norris-Kirby, A.; Rich, R.G.; Hafez, M.J.; Gocke, C.D.; Eshleman, J.R.; Murphy, K.M. Comparison of Sanger sequencing, pyrosequencing, and melting curve analysis for the detection of KRAS mutations: Diagnostic and clinical implications. J. Mol. Diagn. JMD 2010, 12, 425–432. [Google Scholar] [CrossRef]
- Miyazawa, H.; Wada, T. Reversion Mosaicism in Primary Immunodeficiency Diseases. Front. Immunol. 2021, 12, 783022. [Google Scholar] [CrossRef]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M.; et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 2021, 184, 5635–5652.e5629. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef]
- Aluri, J.; Cooper, M.A. Genetic Mosaicism as a Cause of Inborn Errors of Immunity. J. Clin. Immunol. 2021, 41, 718–728. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Y.; Ureña-Bailén, G.; Mohammadian Gol, T.; Gratz, P.G.; Gratz, H.P.; Roig-Merino, A.; Antony, J.S.; Lamsfus-Calle, A.; Daniel-Moreno, A.; Handgretinger, R.; et al. Challenges in Gene Therapy for Somatic Reverted Mosaicism in X-Linked Combined Immunodeficiency by CRISPR/Cas9 and Prime Editing. Genes 2022, 13, 2348. https://doi.org/10.3390/genes13122348
Hou Y, Ureña-Bailén G, Mohammadian Gol T, Gratz PG, Gratz HP, Roig-Merino A, Antony JS, Lamsfus-Calle A, Daniel-Moreno A, Handgretinger R, et al. Challenges in Gene Therapy for Somatic Reverted Mosaicism in X-Linked Combined Immunodeficiency by CRISPR/Cas9 and Prime Editing. Genes. 2022; 13(12):2348. https://doi.org/10.3390/genes13122348
Chicago/Turabian StyleHou, Yujuan, Guillermo Ureña-Bailén, Tahereh Mohammadian Gol, Paul Gerhard Gratz, Hans Peter Gratz, Alicia Roig-Merino, Justin S. Antony, Andrés Lamsfus-Calle, Alberto Daniel-Moreno, Rupert Handgretinger, and et al. 2022. "Challenges in Gene Therapy for Somatic Reverted Mosaicism in X-Linked Combined Immunodeficiency by CRISPR/Cas9 and Prime Editing" Genes 13, no. 12: 2348. https://doi.org/10.3390/genes13122348