
Genomic Consequences of Fragmentation in the Endangered Fennoscandian Arctic Fox (Vulpes lagopus)
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
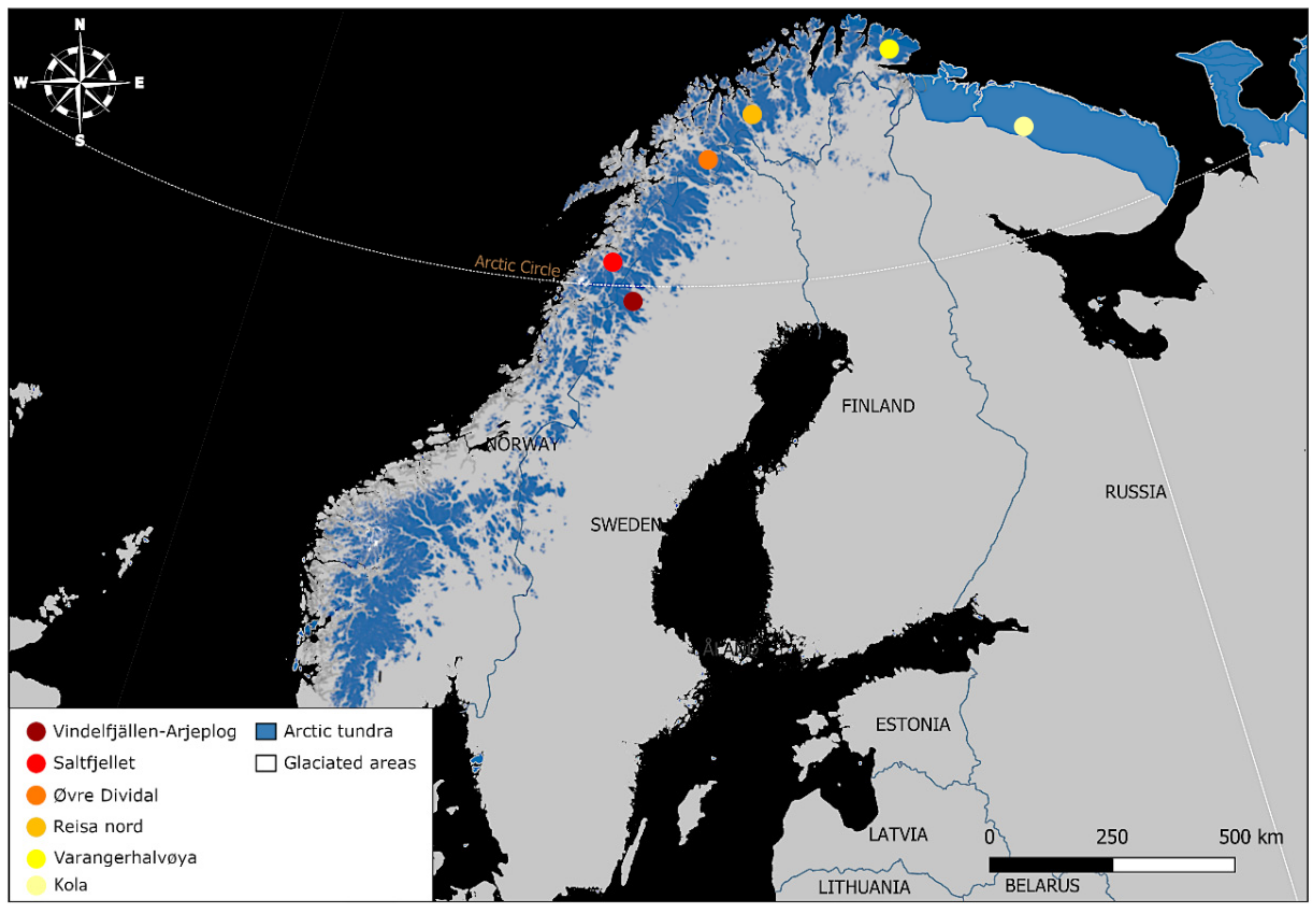
2.1. Study Populations
2.2. DNA Extraction and Resequencing
2.3. Bioinformatics and Variant Calling
2.4. Population Structure
2.5. Genome-Wide Heterozygosity and Genomic Inbreeding Coefficients (FROH)
2.6. Mutational Load
2.7. Statistical Analyses
3. Results
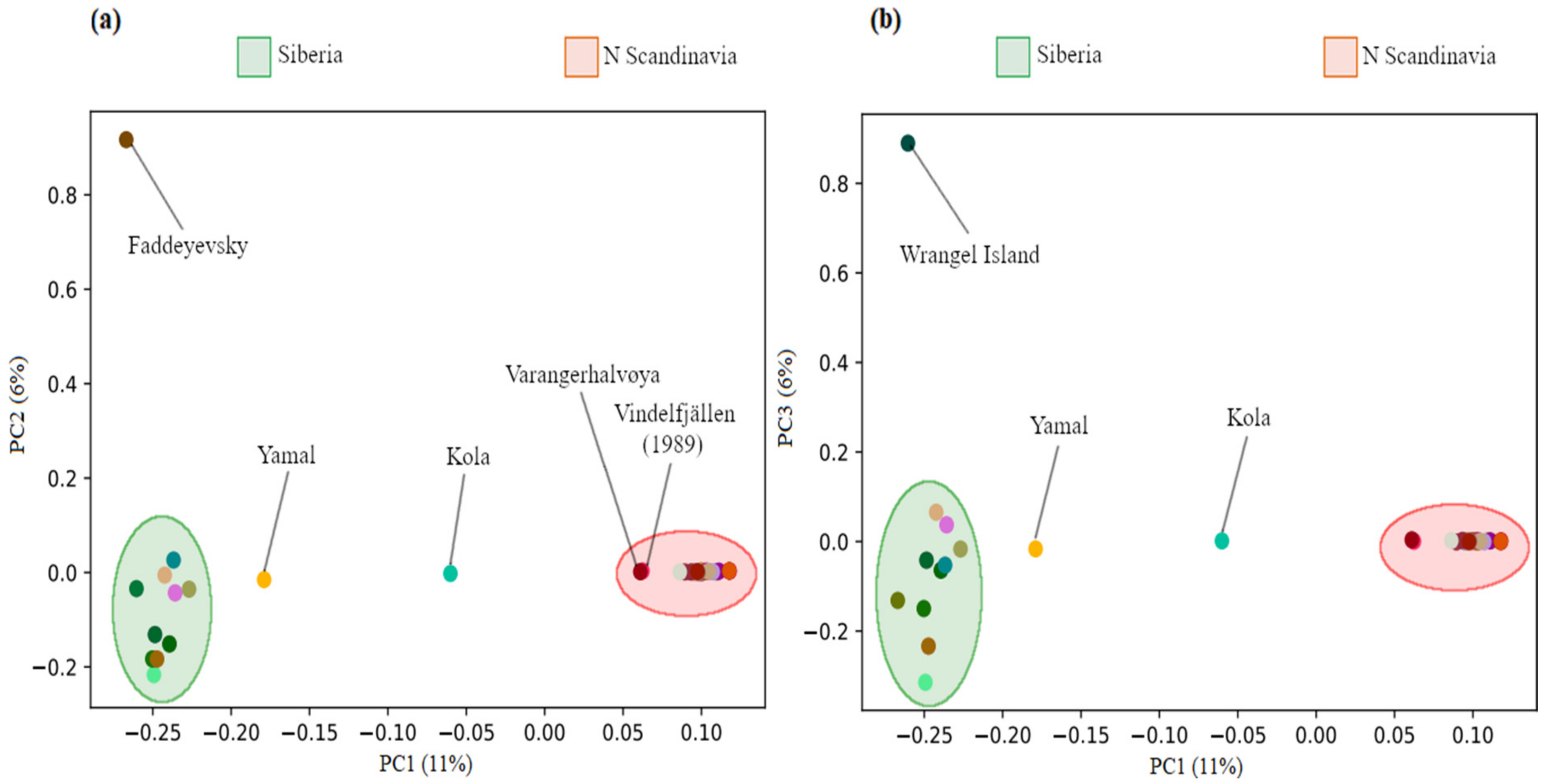
3.1. Population Structure
3.2. Genome-Wide Heterozygosity
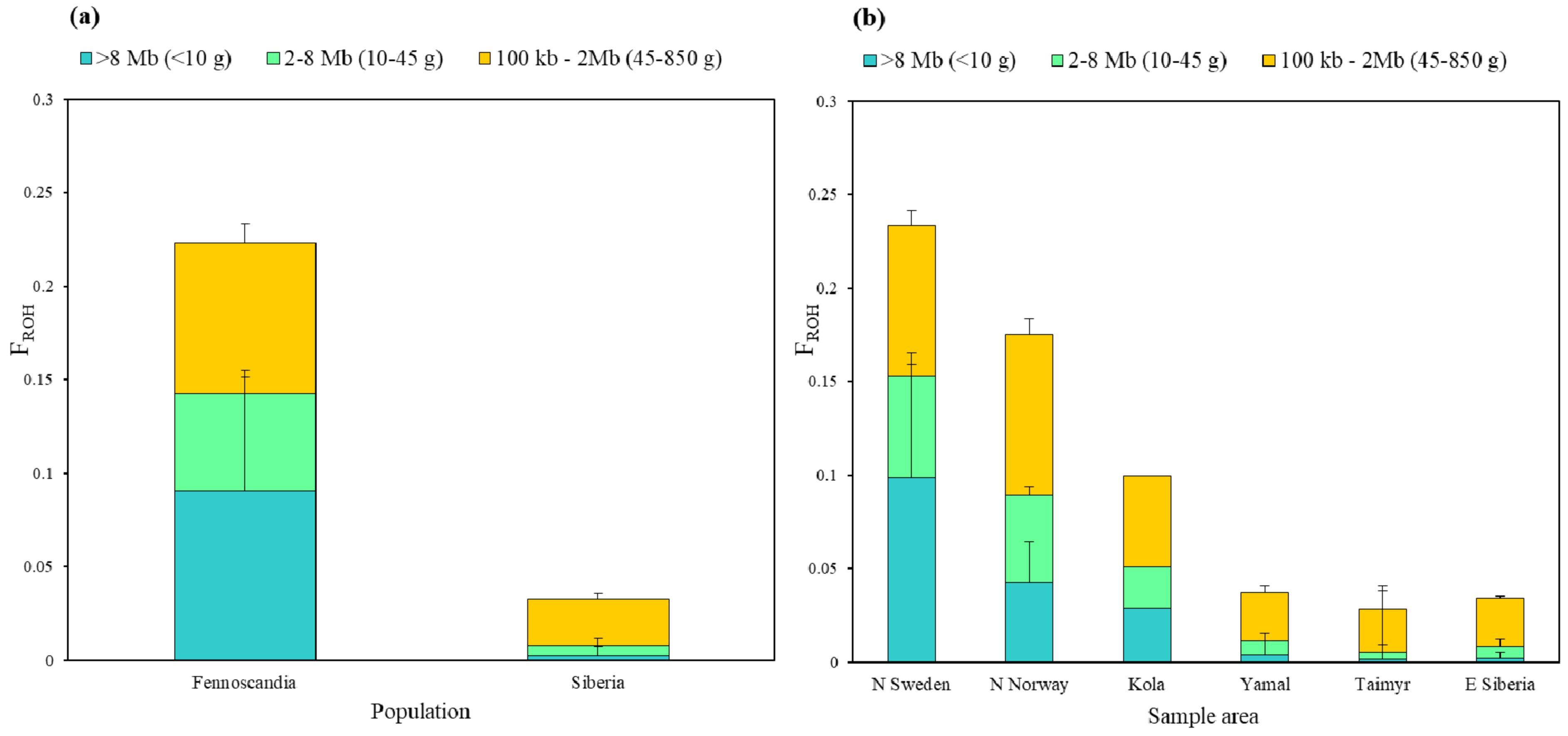
3.3. Genomic Inbreeding Coefficients (FROH)
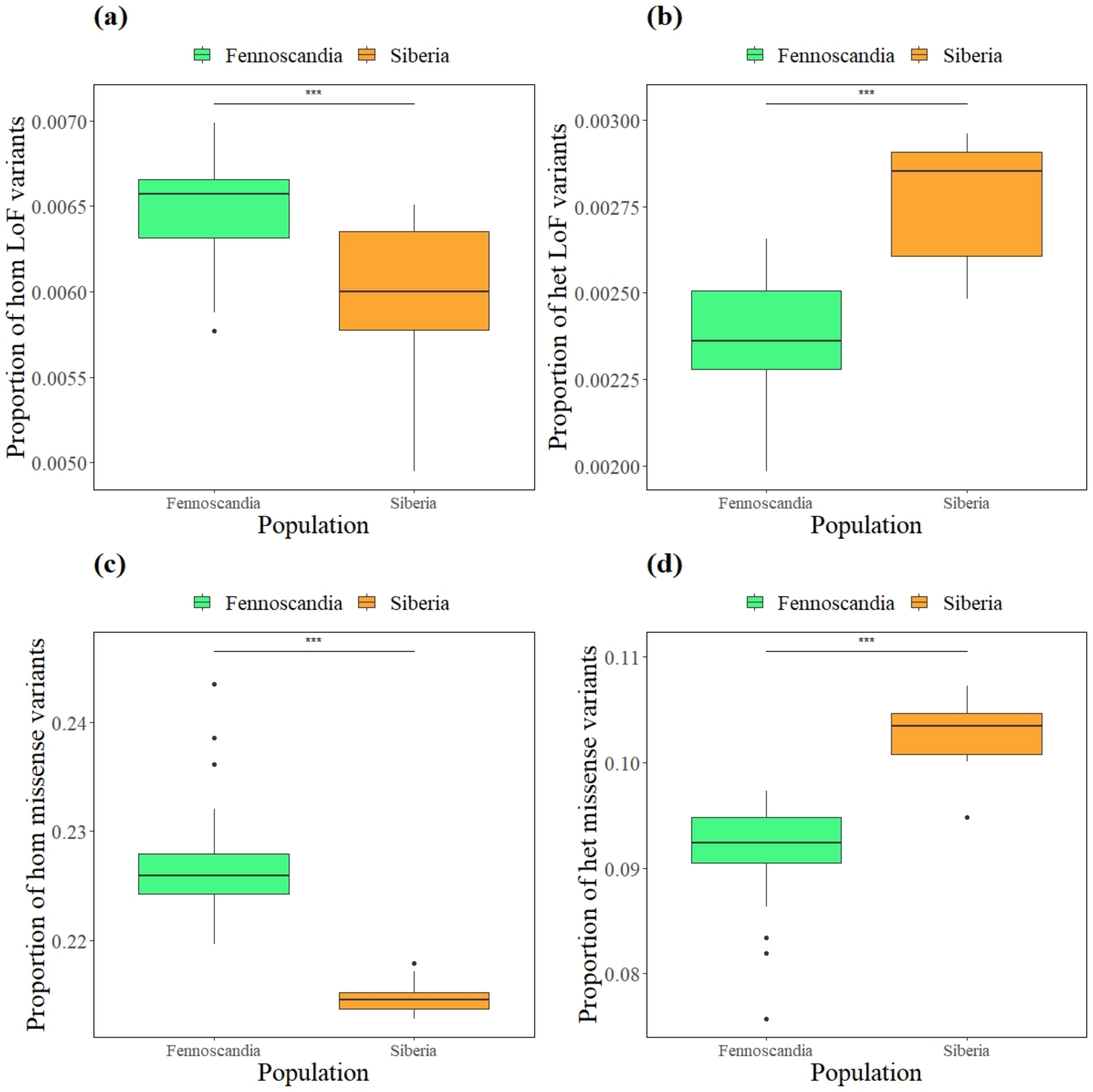
3.4. Mutational Load
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Descamps, S.; Aars, J.; Fuglei, E.; Kovacs, K.M.; Lydersen, C.; Pavlova, O.; Pedersen, Å.Ø.; Ravolainen, V.; Strøm, H. Climate Change Impacts on Wildlife in a High Arctic Archipelago—Svalbard, Norway. Glob. Change Biol. 2017, 23, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Huntington, H.P.; Boyle, M.; Flowers, G.E.; Weatherly, J.W.; Hamilton, L.C.; Hinzman, L.; Gerlach, C.; Zulueta, R.; Nicolson, C.; Overpeck, J. The Influence of Human Activity in the Arctic on Climate and Climate Impacts. Clim. Change 2007, 82, 77. [Google Scholar] [CrossRef]
- Le Moullec, M.; Bender, M. Impacts of Global Warming on Arctic Biota. In Global Arctic: An Introduction to the Multifaceted Dynamics of the Arctic; Springer: Berlin/Heidelberg, Germany, 2021; ISBN 978-3-030-81252-2. [Google Scholar]
- Rundqvist, S.; Hedenås, H.; Sandström, A.; Emanuelsson, U.; Eriksson, H.; Jonasson, C.; Callaghan, T.V. Tree and Shrub Expansion over the Past 34 Years at the Tree-Line near Abisko, Sweden. Ambio 2011, 40, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speed, J.D.M.; Chimal-Ballesteros, J.A.; Martin, M.D.; Barrio, I.C.; Vuorinen, K.E.M.; Soininen, E.M. Will Borealization of Arctic Tundra Herbivore Communities Be Driven by Climate Warming or Vegetation Change? Glob. Change Biol. 2021, 27, 6568–6577. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Evolution in Mendelian Populations. Genetics 1931, 16, 97–159. [Google Scholar] [CrossRef]
- Hanski, I. Metapopulation Dynamics. Nature 1998, 396, 41–49. [Google Scholar] [CrossRef]
- Nei, M.; Maruyama, T.; Chakraborty, R. The Bottleneck Effect and Genetic Variability in Populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Wright, S. Evolution and the Genetics of Populations, Volume 3: Experimental Results and Evolutionary Deductions; University of Chicago Press: Chicago, IL, USA, 1984; ISBN 978-0-226-91040-6. [Google Scholar]
- Stephens, P.A.; Sutherland, W.J.; Freckleton, R.P. What Is the Allee Effect? Oikos 1999, 87, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Caughley, G. Directions in Conservation Biology. J. Anim. Ecol. 1994, 63, 215–244. [Google Scholar] [CrossRef]
- Gilpin, M.E.; Soulé, M.E. Minimum Viable Populations: Processes of Species Extinction. In Conservation Biology. The Science of Scarcity and Diversity; Sinauer Assoc.: Sunderland, MA, USA, 1986. [Google Scholar]
- Saccheri, I.; Kuussaari, M.; Kankare, M.; Vikman, P.; Fortelius, W.; Hanski, I. Inbreeding and Extinction in a Butterfly Metapopulation. Nature 1998, 392, 491–494. [Google Scholar] [CrossRef]
- Blomqvist, D.; Pauliny, A.; Larsson, M.; Flodin, L.-Å. Trapped in the Extinction Vortex? Strong Genetic Effects in a Declining Vertebrate Population. BMC Evol. Biol. 2010, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Palomares, F.; Godoy, J.A.; López-Bao, J.V.; Rodríguez, A.; Roques, S.; Casas-Marce, M.; Revilla, E.; Delibes, M. Possible Extinction Vortex for a Population of Iberian Lynx on the Verge of Extirpation. Conserv. Biol. 2012, 26, 689–697. [Google Scholar] [CrossRef]
- Haldane, J.B.S. The Effect of Variation of Fitness. Am. Nat. 1937, 71, 337–349. [Google Scholar] [CrossRef]
- Charlesworth, B.; Charlesworth, D. The Genetic Basis of Inbreeding Depression. Genet. Res. 1999, 74, 329–340. [Google Scholar] [CrossRef]
- Keller, L.F.; Waller, D.M. Inbreeding Effects in Wild Populations. Trends Ecol. Evol. 2002, 17, 230–241. [Google Scholar] [CrossRef]
- Lynch, M.; Walsh, B. Genetics and Analysis of Quantitative Traits; Sinauer: Sunderland, MA, USA, 1998; Volume 1. [Google Scholar]
- Darwin, C. Sexual Selection in Relation to Monkeys. Nature 1876, 15, 18–19. [Google Scholar] [CrossRef] [Green Version]
- Whiteley, A.R.; Fitzpatrick, S.W.; Funk, W.C.; Tallmon, D.A. Genetic Rescue to the Rescue. Trends Ecol. Evol. 2015, 30, 42–49. [Google Scholar] [CrossRef]
- Hedrick, P.W.; Garcia-Dorado, A. Understanding Inbreeding Depression, Purging, and Genetic Rescue. Trends Ecol. Evol. 2016, 31, 940–952. [Google Scholar] [CrossRef]
- Hasselgren, M.; Norén, K. Inbreeding in Natural Mammal Populations: Historical Perspectives and Future Challenges. Mammal. Rev. 2019, 49, 369–383. [Google Scholar] [CrossRef]
- Roelke, M.E.; Martenson, J.S.; O’Brien, S.J. The Consequences of Demographic Reduction and Genetic Depletion in the Endangered Florida Panther. Curr. Biol. 1993, 3, 340–350. [Google Scholar] [CrossRef]
- Liberg, O.; Andrén, H.; Pedersen, H.-C.; Sand, H.; Sejberg, D.; Wabakken, P.; Åkesson, M.; Bensch, S. Severe Inbreeding Depression in a Wild Wolf Canis Lupus Population. Biol. Lett. 2005, 1, 17–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Räikkönen, J.; Vucetich, J.A.; Peterson, R.O.; Nelson, M.P. Congenital Bone Deformities and the Inbred Wolves (Canis lupus) of Isle Royale. Biol. Conserv. 2009, 142, 1025–1031. [Google Scholar] [CrossRef]
- Pemberton, J.M.; Ellis, P.E.; Pilkington, J.G.; Bérénos, C. Inbreeding Depression by Environment Interactions in a Free-Living Mammal Population. Heredity 2017, 118, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åkesson, M.; Liberg, O.; Sand, H.; Wabakken, P.; Bensch, S.; Flagstad, Ø. Genetic Rescue in a Severely Inbred Wolf Population. Mol. Ecol. 2016, 25, 4745–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselgren, M.; Angerbjörn, A.; Eide, N.E.; Erlandsson, R.; Flagstad, Ø.; Landa, A.; Wallén, J.; Norén, K. Genetic Rescue in an Inbred Arctic Fox (Vulpes lagopus) Population. Proc. R. Soc. B Biol. Sci. 2018, 285, 20172814. [Google Scholar] [CrossRef] [Green Version]
- Kardos, M.; Taylor, H.R.; Ellegren, H.; Luikart, G.; Allendorf, F.W. Genomics Advances the Study of Inbreeding Depression in the Wild. Evol. Appl. 2016, 9, 1205–1218. [Google Scholar] [CrossRef]
- Broman, K.W.; Weber, J.L. Long Homozygous Chromosomal Segments in Reference Families from the Centre d’Étude Du Polymorphisme Humain. Am. J. Hum. Genet. 1999, 65, 1493–1500. [Google Scholar] [CrossRef] [Green Version]
- Allendorf, F.W. Genetics and the Conservation of Natural Populations: Allozymes to Genomes. Mol. Ecol. 2017, 26, 420–430. [Google Scholar] [CrossRef] [Green Version]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of Homozygosity in European Populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.A.; Räikkönen, J.; Vucetich, L.M.; Vucetich, J.A.; Peterson, R.O.; Lohmueller, K.E.; Wayne, R.K. Genomic Signatures of Extensive Inbreeding in Isle Royale Wolves, a Population on the Threshold of Extinction. Sci. Adv. 2019, 5, eaau0757. [Google Scholar] [CrossRef]
- Robinson, J.A.; Ortega-Del Vecchyo, D.; Fan, Z.; Kim, B.Y.; von Holdt, B.M.; Marsden, C.D.; Lohmueller, K.E.; Wayne, R.K. Genomic Flatlining in the Endangered Island Fox. Curr. Biol. 2016, 26, 1183–1189. [Google Scholar] [CrossRef] [Green Version]
- Saremi, N.F.; Supple, M.A.; Byrne, A.; Cahill, J.A.; Coutinho, L.L.; Dalén, L.; Figueiró, H.V.; Johnson, W.E.; Milne, H.J.; O’Brien, S.J.; et al. Puma Genomes from North and South America Provide Insights into the Genomic Consequences of Inbreeding. Nat. Commun. 2019, 10, 4769. [Google Scholar] [CrossRef] [Green Version]
- Loison, A.; Strand, O.; Linnell, J.D.C. Effect of Temporal Variation in Reproduction on Models of Population Viability: A Case Study for Remnant Arctic Fox (Alopex lagopus) Populations in Scandinavia. Biol. Conserv. 2001, 97, 347–359. [Google Scholar] [CrossRef]
- Lönnberg, E. Fjällrävstammen i Sverige 1926; Royal Swedish Academy of Sciences: Uppsala, Sweden, 1927. [Google Scholar]
- Nyström, V.; Angerbjörn, A.; Dalén, L. Genetic Consequences of a Demographic Bottleneck in the Scandinavian Arctic Fox. Oikos 2006, 114, 84–94. [Google Scholar] [CrossRef]
- Larsson, P.; von Seth, J.; Hagen, I.J.; Götherström, A.; Androsov, S.; Germonpré, M.; Bergfeldt, N.; Fedorov, S.; Eide, N.E.; Sokolova, N.; et al. Consequences of Past Climate Change and Recent Human Persecution on Mitogenomic Diversity in the Arctic Fox. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190212. [Google Scholar] [CrossRef] [Green Version]
- Hersteinsson, P.; MacDonald, D.W. Interspecific Competition and the Geographical Distribution of Red and Arctic Foxes Vulpes vulpes and Alopex lagopus. Oikos 1992, 64, 505. [Google Scholar] [CrossRef]
- Tannerfeldt, M.; Elmhagen, B.; Angerbjörn, A. Exclusion by Interference Competition? The Relationship between Red and Arctic Foxes. Oecologia 2002, 132, 213–220. [Google Scholar] [CrossRef]
- Dalén, L.; Kvaløy, K.; Linnell, J.D.C.; Elmhagen, B.; Strand, O.; Tannerfeldt, M.; Henttonen, H.; Fuglei, E.; Landa, A.; Angerbjörn, A. Population Structure in a Critically Endangered Arctic Fox Population: Does Genetics Matter? Mol. Ecol. 2006, 15, 2809–2819. [Google Scholar] [CrossRef]
- Hemphill-Keeling, E.J.; Flagstad, Ø.; Jensen, H.; Norén, K.; Wallén, J.F.; Landa, A.; Angerbjörn, A.; Eide, N.E. Genetic Consequences of Conservation Action: Restoring the Arctic Fox (Vulpes lagopus) Population in Scandinavia. Biol. Conserv. 2020, 248, 108534. [Google Scholar] [CrossRef]
- Tarroux, A.; Berteaux, D.; Bêty, J. Northern Nomads: Ability for Extensive Movements in Adult Arctic Foxes. Polar Biol. 2010, 33, 1021–1026. [Google Scholar] [CrossRef]
- Herfindal, I.; Linnell, J.D.C.; Elmhagen, B.; Andersen, R.; Eide, N.E.; Frafjord, K.; Henttonen, H.; Kaikusalo, A.; Mela, M.; Tannerfeldt, M.; et al. Population Persistence in a Landscape Context: The Case of Endangered Arctic Fox Populations in Fennoscandia. Ecography 2010, 33, 932–941. [Google Scholar] [CrossRef]
- Norén, K.; Dalén, L.; Flagstad, Ø.; Berteaux, D.; Wallén, J.; Angerbjörn, A. Evolution, Ecology and Conservation—Revisiting Three Decades of Arctic Fox Population Genetic Research. Polar Res. 2017, 36, 4. [Google Scholar] [CrossRef] [Green Version]
- Dalén, L.; Götherström, A.; Tannerfeldt, M.; Angerbjörn, A. Is the Endangered Fennoscandian Arctic Fox (Alopex lagopus) Population Genetically Isolated? Biol. Conserv. 2002, 105, 171–178. [Google Scholar] [CrossRef]
- Tirronen, K.; Ehrich, D.; Panchenko, D.; Dalén, L.; Angerbjörn, A. The Arctic Fox (Vulpes lagopus L.) on the Kola Peninsula (Russia): Silently Disappearing in the Mist of Data Deficiency? Polar Biol. 2021, 44, 913–925. [Google Scholar] [CrossRef]
- Strand, O.; Landa, A.; Linnell, J.D.C.; Zimmermann, B.; Skogland, T. Social Organization and Parental Behavior in the Arctic Fox. J. Mammal. 2000, 81, 223–233. [Google Scholar] [CrossRef]
- Ims, R.A.; Killengreen, S.T.; Ehrich, D.; Flagstad, Ø.; Hamel, S.; Henden, J.-A.; Jensvoll, I.; Yoccoz, N.G. Ecosystem Drivers of an Arctic Fox Population at the Western Fringe of the Eurasian Arctic. Polar Res. 2017, 36, 8. [Google Scholar] [CrossRef] [Green Version]
- Norén, K.; Godoy, E.; Dalén, L.; Meijer, T.; Angerbjörn, A. Inbreeding Depression in a Critically Endangered Carnivore. Mol. Ecol. 2016, 25, 3309–3318. [Google Scholar] [CrossRef]
- Hasselgren, M.; Dussex, N.; von Seth, J.; Angerbjörn, A.; Olsen, R.-A.; Dalén, L.; Norén, K. Genomic and Fitness Consequences of Inbreeding in an Endangered Carnivore. Mol. Ecol. 2021, 30, 2790–2799. [Google Scholar] [CrossRef]
- Lotsander, A.; Hasselgren, M.; Larm, M.; Wallén, J.; Angerbjörn, A.; Norén, K. Low Persistence of Genetic Rescue across Generations in the Arctic Fox (Vulpes lagopus). J. Hered. 2021, 112, 276–285. [Google Scholar] [CrossRef]
- Xue, Y.; Prado-Martinez, J.; Sudmant, P.H.; Narasimhan, V.; Ayub, Q.; Szpak, M.; Frandsen, P.; Chen, Y.; Yngvadottir, B.; Cooper, D.N.; et al. Mountain Gorilla Genomes Reveal the Impact of Long-Term Population Decline and Inbreeding. Science 2015, 348, 242–245. [Google Scholar] [CrossRef]
- Grossen, C.; Guillaume, F.; Keller, L.F.; Croll, D. Purging of Highly Deleterious Mutations through Severe Bottlenecks in Alpine Ibex. Nat. Commun. 2020, 11, 1001. [Google Scholar] [CrossRef] [Green Version]
- Dussex, N.; Valk, T.V.D.; Morales, H.E.; Wheat, C.W.; Díez-del-Molino, D.; Seth, J.V.; Foster, Y.; Kutschera, V.E.; Guschanski, K.; Rhie, A.; et al. Population Genomics of the Critically Endangered Kākāpō. Cell Genom. 2021, 10, e1–e9. [Google Scholar] [CrossRef]
- von Seth, J.; Dussex, N.; Díez-del-Molino, D.; van der Valk, T.; Kutschera, V.E.; Kierczak, M.; Steiner, C.C.; Liu, S.; Gilbert, M.T.P.; Sinding, M.-H.S.; et al. Genomic Insights into the Conservation Status of the World’s Last Remaining Sumatran Rhinoceros Populations. Nat. Commun. 2021, 12, 2393. [Google Scholar] [CrossRef]
- Rovbase 3.0. Available online: https://rovbase30.miljodirektoratet.no/ (accessed on 5 September 2022).
- Walker, D.; Raynolds, M.; Daniëls, F.; Einarsson, E.; Elvebakk, A.; Gould, W.; Katenin, A.; Kholod, S.; Markon, C.; Melnikov, E.; et al. The Circumpolar Arctic Vegetation Map. J. Veg. Sci. 2005, 16, 267–282. [Google Scholar] [CrossRef]
- Angerbjörn, A.; Eide, N.E.; Dalén, L.; Elmhagen, B.; Hellström, P.; Ims, R.A.; Killengreen, S.; Landa, A.; Meijer, T.; Mela, M.; et al. Carnivore Conservation in Practice: Replicated Management Actions on a Large Spatial Scale. J. Appl. Ecol. 2013, 50, 59–67. [Google Scholar] [CrossRef]
- Kutschera, V.; Kierczak, M.; Valk, T.; von Seth, J.; Dussex, N.; Lord, E.; Dehasque, M.; Stanton, D.; Khoonsari, P.; Nystedt, B.; et al. GenErode: A Bioinformatics Pipeline to Investigate Genome Erosion in Endangered and Extinct Species. BMC Bioinform. 2022, 23, 228. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.W. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Picard Tools—By Broad Institute. Available online: https://broadinstitute.github.io/picard/ (accessed on 4 October 2022).
- GATK. Available online: https://gatk.broadinstitute.org/hc/en-us (accessed on 11 January 2022).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Johnson, P.L.; Slatkin, M. Inference of Population Genetic Parameters in Metagenomics: A Clean Look at Messy Data. Genome Res. 2006, 16, 1320–1327. [Google Scholar] [CrossRef] [Green Version]
- Haubold, B.; Pfaffelhuber, P.; Lynch, M. MlRho—A Program for Estimating the Population Mutation and Recombination Rates from Shotgun-Sequenced Diploid Genomes. Mol. Ecol. 2010, 19, 277–284. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Korneliussen, T.; Albrechtsen, A.; Li, Y.; Wang, J. SNP Calling, Genotype Calling, and Sample Allele Frequency Estimation from New-Generation Sequencing Data. PLoS ONE 2012, 7, e37558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmann, I.; Mang, Y.; Gu, Z.; Li, P.; Francisco, M.; Clark, A.G.; Nielsen, R. Population Genetic Analysis of Shotgun Assemblies of Genomic Sequences from Multiple Individuals. Genome Res. 2008, 18, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukekova, A.V.; Trut, L.N.; Oskina, I.N.; Johnson, J.L.; Temnykh, S.V.; Kharlamova, A.V.; Shepeleva, D.V.; Gulievich, R.G.; Shikhevich, S.G.; Graphodatsky, A.S.; et al. A Meiotic Linkage Map of the Silver Fox, Aligned and Compared to the Canine Genome. Genome Res. 2007, 17, 387–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukekova, A.V.; Johnson, J.L.; Xiang, X.; Feng, S.; Liu, S.; Rando, H.M.; Kharlamova, A.V.; Herbeck, Y.; Serdyukova, N.A.; Xiong, Z.; et al. Red Fox Genome Assembly Identifies Genomic Regions Associated with Tame and Aggressive Behaviours. Nat. Ecol. Evol. 2018, 2, 1479–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassambara, A. Rstatix: Pipe-Friendly Framework for Basic Statistical Tests 2021. R Package Rstatix Version 0.6.0. Available online: https://cran.r-project.org/web/packages/spacyr/index.html (accessed on 13 October 2022).
- Kardos, M.; Åkesson, M.; Fountain, T.; Flagstad, Ø.; Liberg, O.; Olason, P.; Sand, H.; Wabakken, P.; Wikenros, C.; Ellegren, H. Genomic Consequences of Intensive Inbreeding in an Isolated Wolf Population. Nat. Ecol. Evol. 2018, 2, 124–131. [Google Scholar] [CrossRef]
- Buffalo, V.; Coop, G. Estimating the Genome-Wide Contribution of Selection to Temporal Allele Frequency Change. Proc. Natl. Acad. Sci. USA 2020, 117, 20672–20680. [Google Scholar] [CrossRef]
- Brüniche-Olsen, A.; Kellner, K.F.; Anderson, C.J.; DeWoody, J.A. Runs of Homozygosity Have Utility in Mammalian Conservation and Evolutionary Studies. Conserv. Genet. 2018, 19, 1295–1307. [Google Scholar] [CrossRef]
- Kimura, M.; Weiss, G.H. The Stepping Stone Model of Population Structure and the Decrease of Genetic Correlation with Distance. Genetics 1964, 49, 561–576. [Google Scholar] [CrossRef]
- Nesje, A. Latest Pleistocene and Holocene Alpine Glacier Fluctuations in Scandinavia. Quat. Sci. Rev. 2009, 28, 2119–2136. [Google Scholar] [CrossRef]
- Hasselgren, M.; Cockerill, C.A.; Wallén, J.F.; Angerbjorn, A.; Dalén, L.; Dussex, N.D.; Eide, N.E.; Flagstad, Ø.; Landa, A.; von Seth, J.; et al. Genomic Consequences of Reintroductions from a Captive Breeding Station—A Replicated Evaluation. In prep.
- Van der Valk, T.; de Manuel, M.; Marques-Bonet, T.; Guschanski, K. Estimates of Genetic Load in Small Populations Suggest Extensive Purging of Deleterious Alleles. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Kyriazis, C.C.; Wayne, R.K.; Lohmueller, K.E. Strongly Deleterious Mutations Are a Primary Determinant of Extinction Risk Due to Inbreeding Depression. Evol. Lett. 2021, 5, 33–47. [Google Scholar] [CrossRef]
- Sams, A.J.; Boyko, A.R. Fine-Scale Resolution of Runs of Homozygosity Reveal Patterns of Inbreeding and Substantial Overlap with Recessive Disease Genotypes in Domestic Dogs. G3 Genes Genomes Genet. 2019, 9, 117–123. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, I.M.; Larkin, D.M.; Lewin, H.A.; Hayes, B.J.; Goddard, M.E. Inferring Demography from Runs of Homozygosity in Whole-Genome Sequence, with Correction for Sequence Errors. Mol. Biol. Evol. 2013, 30, 2209–2223. [Google Scholar] [CrossRef] [Green Version]
- Díez-del-Molino, D.; Sánchez-Barreiro, F.; Barnes, I.; Gilbert, M.T.P.; Dalén, L. Quantifying Temporal Genomic Erosion in Endangered Species. Trends Ecol. Evol. 2018, 33, 176–185. [Google Scholar] [CrossRef]
- Palkopoulou, E.; Mallick, S.; Skoglund, P.; Enk, J.; Rohland, N.; Li, H.; Omrak, A.; Vartanyan, S.; Poinar, H.; Götherström, A.; et al. Complete Genomes Reveal Signatures of Demographic and Genetic Declines in the Woolly Mammoth. Curr. Biol. 2015, 25, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Fang, Q.; Barnett, R.; Li, C.; Han, S.; Kuhlwilm, M.; Zhou, L.; Pan, H.; Deng, Y.; Chen, G.; et al. The Genomic Footprints of the Fall and Recovery of the Crested Ibis. Curr. Biol. 2019, 29, 340–349.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmhagen, B.; Berteaux, D.; Burgess, R.M.; Ehrich, D.; Gallant, D.; Henttonen, H.; Ims, R.A.; Killengreend, S.T.; Niemimaa, J.; Norén, K.; et al. Homage to Hersteinsson and Macdonald: Climate Warming and Resource Subsidies Cause Red Fox Range Expansion and Arctic Fox Decline. Polar Res. 2017, 36 (Suppl. S1). [Google Scholar] [CrossRef]
- Sokolov, A.A.; Sokolova, N.A.; Ims, R.A.; Brucker, L.; Ehrich, D. Emergent Rainy Winter Warm Spells May Promote Boreal Predator Expansion into the Arctic. ARCTIC 2016, 69, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Kiener, T.V.; Zaitsev, V.A. Range Structure in the Red Fox (Vulpes vulpes L.) in the Forest Zone of Eastern Europe. Contemp. Probl. Ecol. 2010, 3, 119–126. [Google Scholar] [CrossRef]
- Dalén, L.; Nyström, V.; Valdiosera, C.; Germonpré, M.; Sablin, M.; Turner, E.; Angerbjörn, A.; Arsuaga, J.L.; Götherström, A. Ancient DNA Reveals Lack of Postglacial Habitat Tracking in the Arctic Fox. Proc. Natl. Acad. Sci. USA 2007, 104, 6726–6729. [Google Scholar] [CrossRef] [Green Version]
- Dalén, L.; Fuglei, E.; Hersteinsson, P.; Kapel, C.M.O.; Roth, J.D.; Samelius, G.; Tannerfeldt, M.; Angerbjörn, A. Population History and Genetic Structure of a Circumpolar Species: The Arctic Fox. Biol. J. Linn. Soc. 2005, 84, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Kutschera, V.E.; Nilsson, M.A.; Janke, A. Genetic Signatures of Adaptation Revealed from Transcriptome Sequencing of Arctic and Red Foxes. BMC Genom. 2015, 16, 585. [Google Scholar] [CrossRef] [Green Version]
- Gilg, O.; Kovacs, K.M.; Aars, J.; Fort, J.; Gauthier, G.; Grémillet, D.; Ims, R.A.; Meltofte, H.; Moreau, J.; Post, E.; et al. Climate Change and the Ecology and Evolution of Arctic Vertebrates. Ann. N. Y. Acad. Sci. 2012, 1249, 166–190. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Country | Sample Area | Year | Tissue Type | Sampling Method | n |
|---|---|---|---|---|---|---|
| Fennoscandia | Northern Sweden | Vindelfjällen | 1989–2019 | Ear tissue | Ear tagging | 15 |
| Arjeplog | 2008–2019 | Ear tissue | Ear tagging | 11(5) | ||
| Northern Norway | Saltfjellet | 2007 | Ear tissue | Ear tagging | 1 | |
| Reisa nord | 2007 | Ear tissue | Ear tagging | 1 | ||
| Varangerhalvøya | 2011 | Muscle tissue | Injured fox | 1 | ||
| Øvre Dividal | 2005 | Ear tissue | Ear tagging | 1 | ||
| Russia | Kola | 1990s | Muscle tissue | Carcass sampling | 1 | |
| Siberia | Russia | Indigirka | 1994 | Muscle tissue | Carcass sampling | 1(1) |
| Yamal | 1994–2011 | Skin/muscle tissue | Carcass sampling | 4 | ||
| Taymyr | 1994 | Skin/muscle tissue | Carcass sampling | 5(4) | ||
| Faddeyevsky Island | 1994 | Muscle tissue | Carcass sampling | 1(1) | ||
| Wrangel Island | 2008 | Skin tissue | Carcass sampling | 1(1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cockerill, C.A.; Hasselgren, M.; Dussex, N.; Dalén, L.; von Seth, J.; Angerbjörn, A.; Wallén, J.F.; Landa, A.; Eide, N.E.; Flagstad, Ø.; et al. Genomic Consequences of Fragmentation in the Endangered Fennoscandian Arctic Fox (Vulpes lagopus). Genes 2022, 13, 2124. https://doi.org/10.3390/genes13112124
Cockerill CA, Hasselgren M, Dussex N, Dalén L, von Seth J, Angerbjörn A, Wallén JF, Landa A, Eide NE, Flagstad Ø, et al. Genomic Consequences of Fragmentation in the Endangered Fennoscandian Arctic Fox (Vulpes lagopus). Genes. 2022; 13(11):2124. https://doi.org/10.3390/genes13112124
Chicago/Turabian StyleCockerill, Christopher A., Malin Hasselgren, Nicolas Dussex, Love Dalén, Johanna von Seth, Anders Angerbjörn, Johan F. Wallén, Arild Landa, Nina E. Eide, Øystein Flagstad, and et al. 2022. "Genomic Consequences of Fragmentation in the Endangered Fennoscandian Arctic Fox (Vulpes lagopus)" Genes 13, no. 11: 2124. https://doi.org/10.3390/genes13112124