
Epilepsy and BRAF Mutations: Phenotypes, Natural History and Genotype-Phenotype Correlations

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Study Design
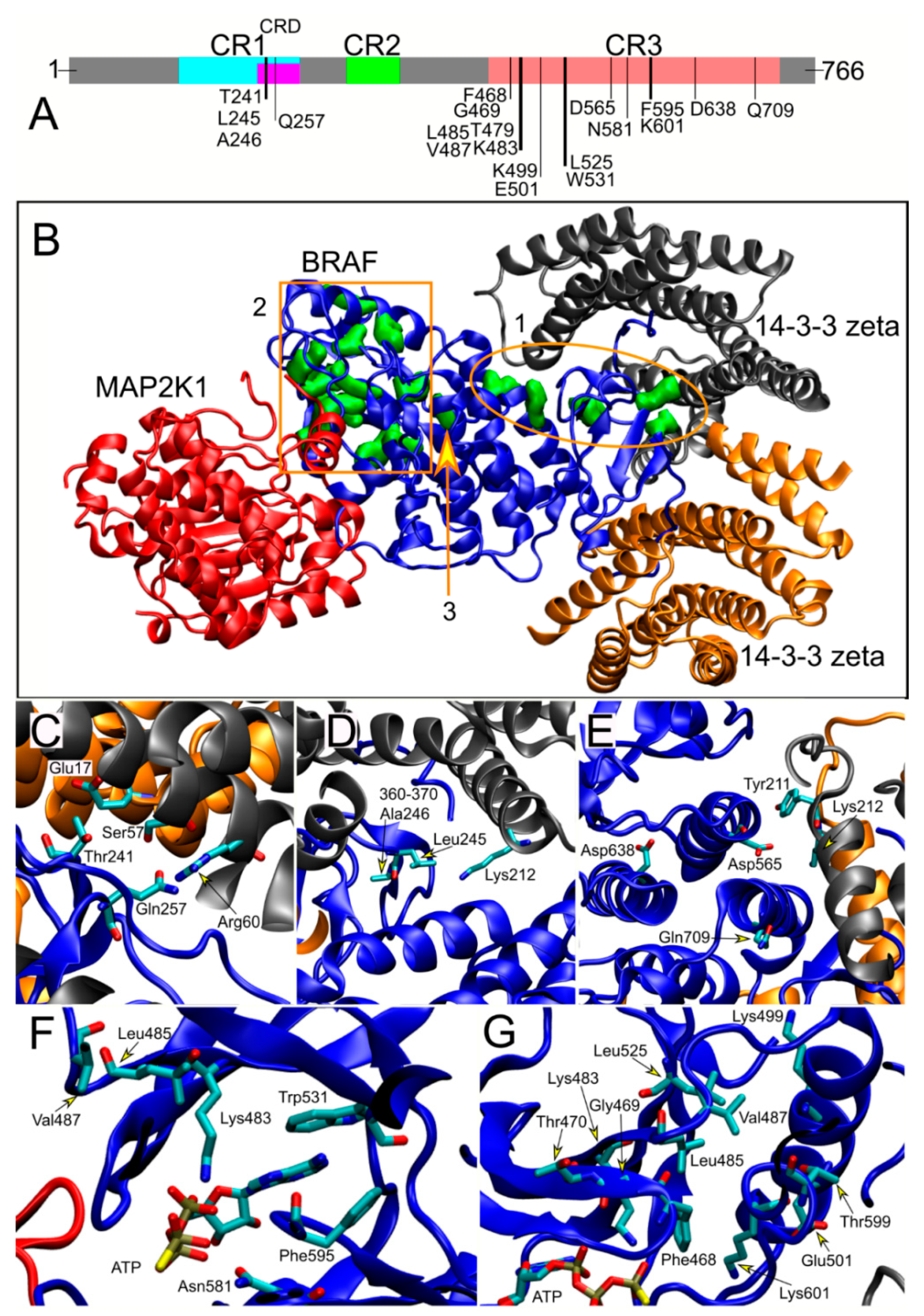
2.3. Structural Analysis
2.4. Statistical Analysis
3. Results
3.1. Neurological Data
3.2. Electroclinical Data
3.2.1. Clinical Epilepsy Data
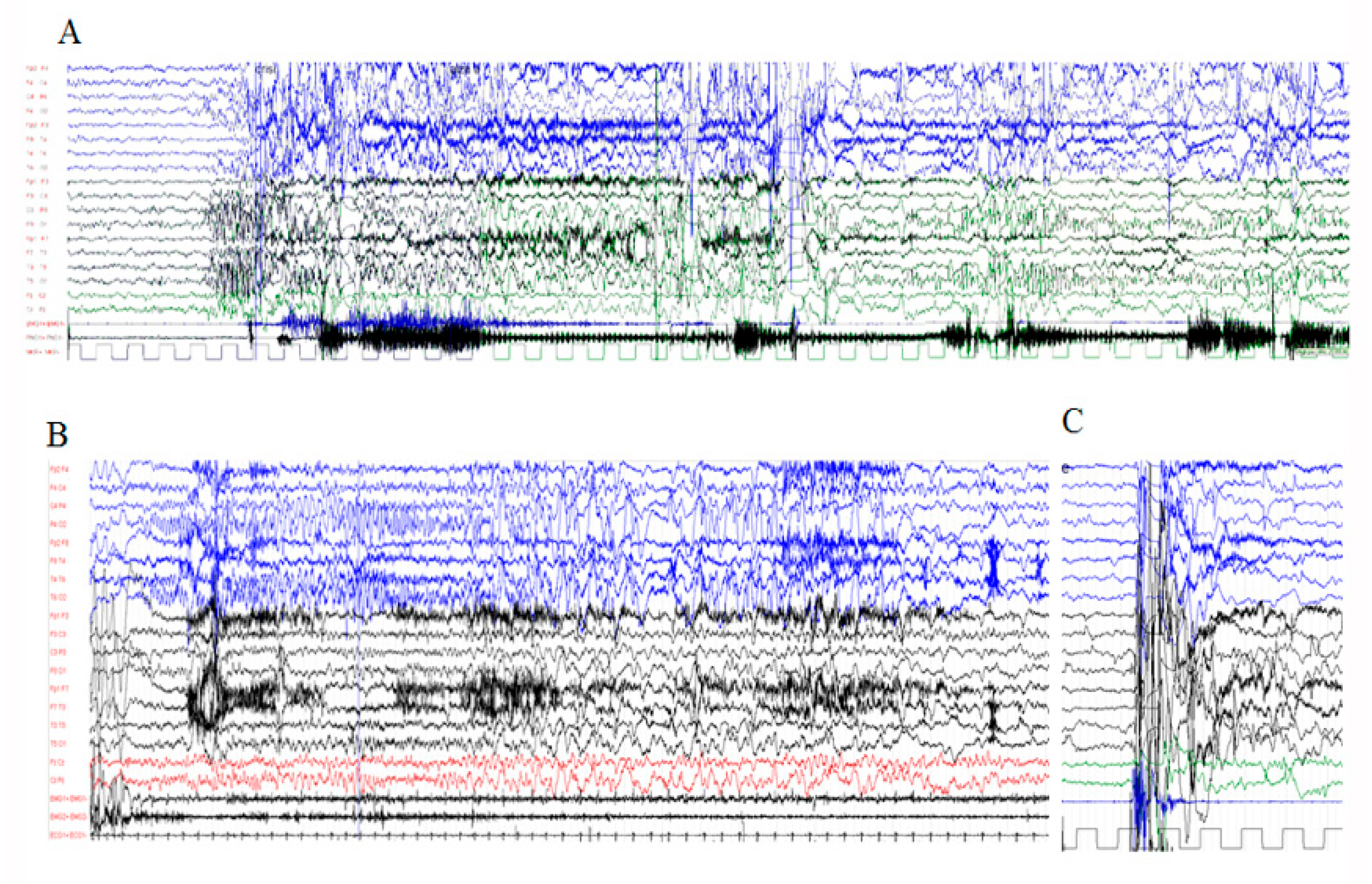
3.2.2. Electrophysiological Assessment
3.3. Other Paroxysmal Phenomena
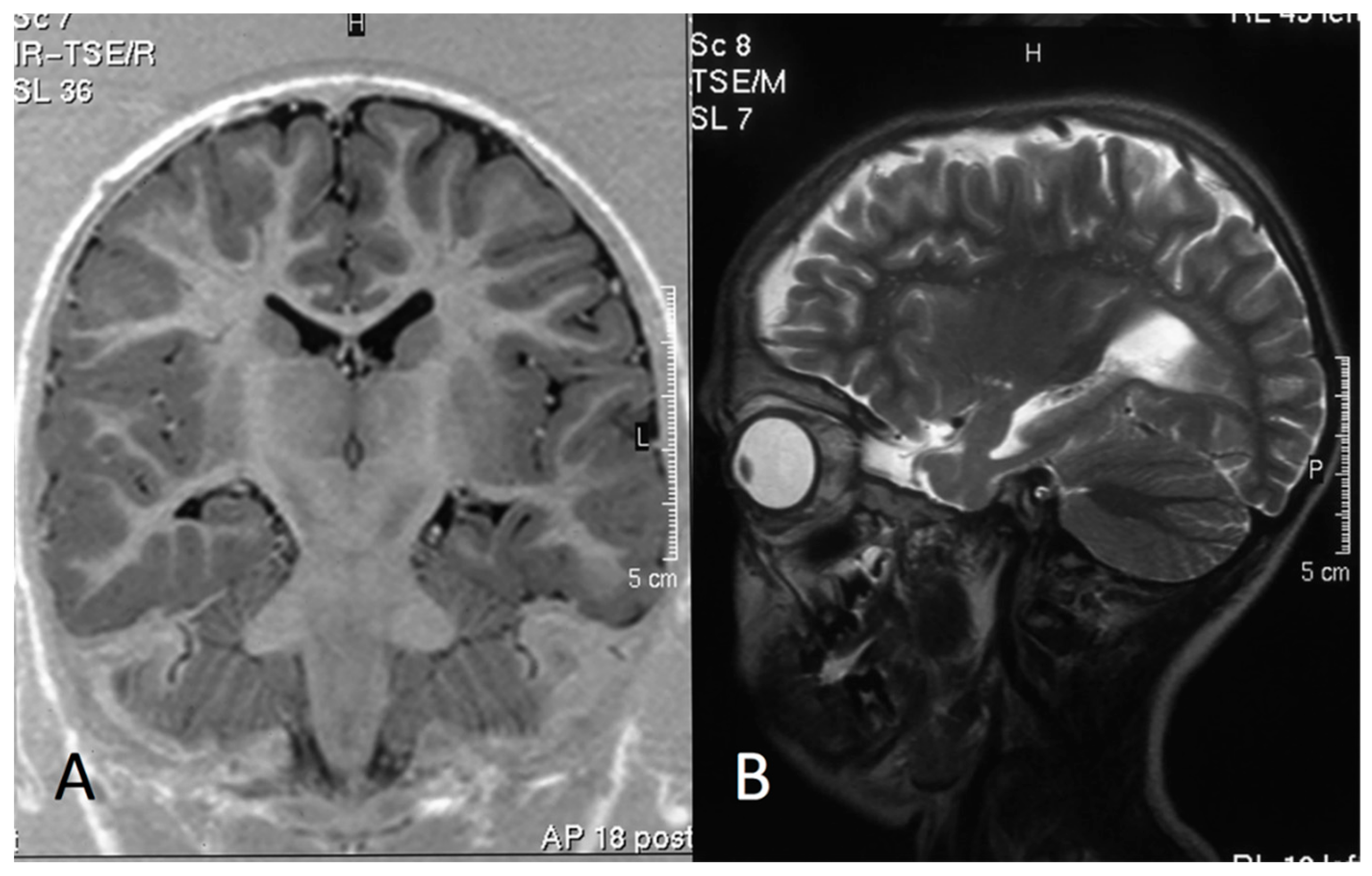
3.4. MRI Data
3.5. Structural Analyses and Genotype–Phenotype Correlations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethical Publication Statement
References
- Krens, S.F.; Spaink, H.P.; Snaar-Jagalska, B.E. Functions of the MAPK family in vertebrate-development. FEBS Lett. 2006, 580, 4984–4990. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.M.; Huganir, R.L. MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 2004, 5, 173–183. [Google Scholar] [CrossRef]
- Aoki, Y.; Niihori, T.; Inoue, S.; Matsubara, Y. Recent advances in RASopathies. J. Hum. Genet. 2016, 61, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, M.; Gelb, B.D. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: Phenotypic spectrum and molecular mechanisms. Ann. N. Y. Acad. Sci. 2010, 1214, 99–121. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.; Allanson, J.; Jadico, S.K.; Kavamura, M.I.; Noonan, J.; Opitz, J.M.; Yong, T.; Neri, J. The cardiofacious cutaneous syndrome. J. Med. Genet. 2006, 43, 833–842. [Google Scholar] [CrossRef]
- Allanson, J.E.; Annerén, G.; Aoki, Y.; Armour, C.M.; Bondeson, M.; Cave, H.; Gripp, K.W.; Kerr, B.; Nystrom, A.; Sol-Church, K.; et al. Cardio-facio-cutaneous syndrome: Does genotype predict phenotype? Am. J. Med. Genet. C Semin. Med. Genet. 2011, 157, 129–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armour, C.L.; Allanson, J.E. Further Delineation of cardio-faciocutaneous syndrome: Clinical features of 38 individuals with proven mutation. J. Med. Genet. 2008, 45, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Yoon, G.; Blaser, J.; Rauen, K.A. Neurological complications of cardio-facio-cutaneous syndrome. Dev. Med. Child Neurol. 2007, 49, 894–899. [Google Scholar] [CrossRef]
- Niihori, T.; Aoki, Y.; Narumi, Y.; Neri, G.; Cave, H.; Verloes, A.; Okamoto, N.; Hennekam, R.C.M.; Gillessen-Kaesbach, G.; Wieczorek, D.; et al. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 2006, 38, 294–296. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Tetsu, O.; Tidyman, W.E.; Estep, A.L.; Conger, B.A.; Santa Cruz, M.; McCormick, F.; Rauen, K.A. Germline mutations in genes within the MAPK pathway cause Cardio-facio-cutaneous syndrome. Science 2006, 311, 1287–1290. [Google Scholar] [CrossRef]
- Adachi, M.; Abe, Y.; Aoki, Y.; Matsubara, Y. Epilepsy in RAS/MAPK syndrome: Two cases of cardio-facio-cutaneous syndrome with epileptic encephalopathy and a literature review. Seizures 2012, 21, 55–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuzono, S.; Fukai, R.; Noda, M.; Miyake, N.; Lee, S.; Kaku, N.; Sanefuji, M.; Akamine, S.; Kanno, S.; Ishizaki, Y.; et al. An acute encephalopathy with reduced diffusion in BRAF-associated cardio-facio-cutaneous syndrome. Brain Dev. 2019, 41, 378–381. [Google Scholar] [CrossRef]
- Wakusawa, K.; Kobayashi, S.; Abe, Y.; Tanaka, S.; Endo, W.; Inui, T.; Iwaki, M.; Watanabe, S.; Togashi, N.; Nara, T.; et al. A girl with Cardio-facio-cutaneous syndrome complicated with status epilepticus and acute encephalopathy. Brain Dev. 2014, 36, 61–63. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [PubMed]
- Fisher, R.S. An overview of the 2017 ILAE operational classification of seizure types. Epilepsy Behav. 2017, 70, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus-Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Bleck, T.P. Refractory status epilepticus. Curr. Opin. Crit. Care 2005, 11, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Kim, M.; Kim, M.J.; Kim, H.; Ock, C.; Keam, B.; Kim, T.M.; Kim, D.; Kim, J.; Heo, D.S. Clinical application of next-generation sequencing-based panel to BRAF wild-type advanced melanoma identifies key oncogenic alterations and therapeutic strategies. Mol. Cancer Ther. 2020, 19, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Ognjenović, J.; Banerjee, S.; Karandur, D.; Merk, A.; Kulhanek, K.; Wong, K.; Roose, J.P.; Subramaniam, S.; Kuriyan, J. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science 2019, 366, 109–115. [Google Scholar] [CrossRef]
- Sarkozy, A.; Carta, C.; Moretti, S.; Zampino, G.; Digilio, M.C.; Pantaleoni, F.; Scioletti, A.P.; Esposito, G.; Cordeddu, V.; Lepri, F. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum. Hum. Mutat. 2009, 30, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Kalser, J.; Cross, J.H. The Epileptic Encephalopathy Jungle—From Dr West to the Concepts of Aetiology-Related and Developmental Encephalopathies. Curr. Opin. Neurol. 2018, 31, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Muromoto, S.; Miyabayashi, T.; Nagai, K.; Yamamura-Suzuki, S.; Anzai, M.; Takezawa, Y.; Sato, R.; Okubo, Y.; Endo, W.; Inui, T. Leucine-485 deletion variant of BRAF may exhibit the severe end of the clinical spectrum of CFC syndrome. J. Hum. Genet. 2019, 64, 499–504. [Google Scholar] [CrossRef]
- Blumke, I.; Mühlebner, A. Neuropathological work-up of focal cortical dysplasias using the new ILAE consensus classification system—practical guideline article invited by the Euro-CNS Research Committee. Clin. Neuropathol. 2011, 30, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, B.; Hitz, C.; Hölter, S.M.; Kühn, R.; Vogt Weisenhorn, D.M.; Wurst, W. Differential mRNA distribution of components of the ERK/MAPK signalling cascade in the adult mouse brain. J. Comp. Neurol. 2007, 500, 542–556. [Google Scholar] [CrossRef] [PubMed]
- English, J.D.; Sweatt, J.D. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J. Biol. Chem. 1997, 272, 19103–19106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.P.; Ohno, M.; Giese, K.P.; Kuhn, R.; Chen, R.L.; Silva, A.J. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J. Neurosci. Res. 2006, 83, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, G.; Verrotti, A.; Domizio, S.; Angeiozzi, B.; Chiarelli, F.; Neri, G. The cardio-facio-cutaneous syndrome: A long term follow-up of two patients, with special reference to the neurological features. Childs Nerv. Syst. 1997, 13, 238–241. [Google Scholar] [CrossRef]
- Chappé, C.; Padovani, L.; Scavarda, D.; Forest, F.; Nanni-Metellus, I.; Loundou, A.; Mercurio, S.; Fina, F.; Lena, G.; Colin, C. Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF (V600E) mutation and expression. Brain Pathol. 2013, 23, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; de Biase, D.; Visani, M.; Giulioni, M.; Martinoni, M.; Volpi, L.; Riguzzi, P.; Rubboli, G.; Michelucci, R.; Tallini, G. Mutant BRAF in low-grade epilepsy- associated tumors and focal cortical dysplasia. Ann. Clin. Transl. Neurol. 2014, 1, 130–134. [Google Scholar] [CrossRef]
- Saito, Y.; Sasaki, M.; Hanaoka, S.; Sugai, K.; Hashimoto, T. A case of Noonan syndrome with cortical dysplasia. Pediatr. Neurol. 1997, 17, 266–269. [Google Scholar] [CrossRef]
- Aizaki, K.; Sugai, K.; Saito, Y.; Nakagawa, E.; Sasaki, M.; Aoki, Y.; Matsubara, Y. Cardio-facio-cutaneous syndrome with spasms and delayed myelination. Brain Dev. 2011, 33, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, E.; Sifakis, S.; Sol-Church, K.; Klein-Zighelboim, E.; Stabley, D.L.; Raissaki, M.; Gripp, K.W.; Kalmanti, M. CNS imaging is a key diagnostic tool in the evaluation of patients with CFC syndrome: Two cases and literature review. Am. J. Med. Genet. A 2011, 155, 605–611. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient (Age; Sex) | Follow-Up (y) | BRAF Amino Acid Substitution | Mutation Class | Age of Seizure Onset | Epilepsy Type | Neurological Features | ID/Behavioral Disorders | PNE | SD | Brain MRI |
|---|---|---|---|---|---|---|---|---|---|---|
| Group 1 | ||||||||||
| #1 (23 y; M †) | 23 | Lys601Gnl | II | 2 m | Combined focal and generalized | Tetraparesis, nystagmus | Profound Regression | Hpx | Yes | CA, HS |
| # 2 (17 y; M) | 15 | Asp638Glu | III | 2 y | Combined focal and generalized | Tetraparesis, dystonia, nystagmus | Profound/AT | Hpx | Yes | CA, bilateral HS, gliosis |
| # 3 (15 y; M) | 15 | Asp638Glu | III | 5 m | Combined focal and generalized | Tetraparesis | Profound/AT | Hpx | Yes | CA, HS, cortical dysplasia |
| #4 (14 y; F) | 12 | Phe595Leu | II | 1.5 y | Focal | Tetraparesis | Profound/AT | Hpx | Yes | CA, gliosis |
| #5 (13 y; M) | 11 | Lys 601Gnl | II | 1.5 y | Combined focal and generalized | Tetraparesis, strabismus | Severe | Hpx | Yes | CA |
| #6 (12 y; F) | 7 | Asp638Glu | III | 1.5 y | Combined focal and generalized | Tetraparesis, strabismus | Severe Regression | Hpx | Yes | CA, HS |
| #7 (7 y; F †) | 6 | Phe595Leu | II | 7 m | Combined focal and generalized | Tetraparesis, strabismus | Profound Regression | Hpx | Yes | CA, HS, hippocampal hypoplasia |
| #8 (6 y; F) | 6 | Val487Gly | II | 8 day | Focal | Tetraparesis | Profound/AT | Hpx | Yes | CA, hippocampal hypoplasia |
| #9 (5 y; M †) | NA | Asp565Glu | I | 4 y | Combined focal and generalized | Tetraparesis, strabismus | Profound | Hpx | NA | CA, thin CC |
| #10 (3 y; M) | 3 | Pro468Ser | II | 1 m | Focal | Tetraparesis | Profound/AT | NA | Yes | CA + bilateral HS |
| Group 2 | ||||||||||
| #11 (27 y; F) | 25 | Trp531Cys | II | 2 y | Focal | Ataxia, strabismus | Severe | NA | NA | Normal |
| #12 (23 y; M) | 10 | Thr241Pro | I | 17 y | Focal | Hypotonia | Mild | NA | NA | Normal |
| #13 (22 y; F) | 20 | Lys499Asn | II | 1.6 y | Focal | Ataxia | Moderate | Hpx | Yes | Removal results of cerebellar astrocytoma |
| #14 (20 y; M) | 5 | Lys483Asn | II | 18 y | Focal | Hypotonia | Moderate | No | No | Normal |
| #15 (19 y; F) | 15 | Leu485Phe | II | 14 y | Focal | Tetraparesis, strabismus | Severe/AT | Blinking | Yes | CA |
| #16 (15 y; F) | 10 | Trp531Cys | II | 9 y | Combined focal and generalized | Tetraparesis, strabismus | Severe | Hpx | Yes | Thin CC |
| #17 (15 y; F) | 8 | Leu525Pro | II | 11 y | Focal | Tetraparesis, strabismus | Moderate | Hpx | Yes | CA, gliosis |
| #18 (12 y; F) | 2 | Gln257Arg | I | 10 y | Focal | Ataxia | Severe/AT | No | Yes | Gliosis |
| #19 (12 y; F) | 4 | Gln257Arg | I | 10 y | Focal | Ataxia | Severe | Blinking | Yes | CA, HS |
| # 20 (8 y; F) | 7 | Gln257Arg | I | 6 y | Combined focal and generalized | Tetraparesis, strabismus | Severe/AT | No | No | Normal |
| #21 (8 y; F) | 5 | Thr599Arg | II | 6 y | Combined focal and generalized | Tetraparesis, strabismus | Severe/AT | Hpx | Yes | Gliosis |
| #22 (4 y; F) | 3 | Gln257Arg | I | 4 y | Generalized | Ataxia, strabismus | Severe | No | Yes | Ventriculomegaly |
| Group 3 | ||||||||||
| #23 (53 y; F) | 7 | Ala246Pro | I | - | - | Normal | NA | No | NA | NA |
| #24 (43 y; F) | 10 | Thr241Met | I | - | - | Normal | NA | No | NA | NA |
| #25 (27 y; F) | 6 | Thr241Arg | I | - | - | Normal | Mild | NA | NA | NA |
| #26 (24 y; F) | 17 | Gln709Arg | I | - | - | Normal | Mild | No | Yes | Normal |
| #27 (19 y; M) | 14 | Glu501Lys | II | - | - | Normal | Severe | NA | NA | NA |
| #28 (15 y; M) | 6 | Leu245Phe | I | - | - | Normal | Mild | NA | NA | NA |
| #29 (14 y; F) | 8 | Gly469Glu | II | - | - | Normal | Moderate | No | NA | Thin CC |
| #30 (12 y; M) | 2 | Thr470Pro | II | - | - | Normal | Moderate | No | No | Gliosis |
| #31 (11 y; F) | 7 | Gln257Arg | I | - | - | Normal | Moderate | NA | NA | Thin CC |
| #32 (7 y; F) | 5 | Gln257Arg | I | - | - | Normal | Moderate | No | Yes | Thin CC, cerebellar malrotation |
| #33 (7 y; F) | 5 | Trp531Arg | II | - | - | Normal | Moderate | No | Yes | Normal |
| #34 (3 y; F) | 2 | Asn581Asp | II | - | - | Normal | Mild | No | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Battaglia, D.I.; Gambardella, M.L.; Veltri, S.; Contaldo, I.; Chillemi, G.; Veredice, C.; Quintiliani, M.; Leoni, C.; Onesimo, R.; Verdolotti, T.; et al. Epilepsy and BRAF Mutations: Phenotypes, Natural History and Genotype-Phenotype Correlations. Genes 2021, 12, 1316. https://doi.org/10.3390/genes12091316
Battaglia DI, Gambardella ML, Veltri S, Contaldo I, Chillemi G, Veredice C, Quintiliani M, Leoni C, Onesimo R, Verdolotti T, et al. Epilepsy and BRAF Mutations: Phenotypes, Natural History and Genotype-Phenotype Correlations. Genes. 2021; 12(9):1316. https://doi.org/10.3390/genes12091316
Chicago/Turabian StyleBattaglia, Domenica I., Maria Luigia Gambardella, Stefania Veltri, Ilaria Contaldo, Giovanni Chillemi, Chiara Veredice, Michela Quintiliani, Chiara Leoni, Roberta Onesimo, Tommaso Verdolotti, and et al. 2021. "Epilepsy and BRAF Mutations: Phenotypes, Natural History and Genotype-Phenotype Correlations" Genes 12, no. 9: 1316. https://doi.org/10.3390/genes12091316