
A Case Series of Familial ARID1B Variants Illustrating Variable Expression and Suggestions to Update the ACMG Criteria

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Ascertainment
2.2. Clinical Data Collection
2.3. Transcript
2.4. DNA Methylation
2.5. Analyses of Facial Features
2.5.1. Computer Vision Algorithms
2.5.2. Face2Gene
3. Results
3.1. DNA Methylation
3.2. Facial Analyses
3.2.1. Computer Vision Algorithms
3.2.2. Face2Gene
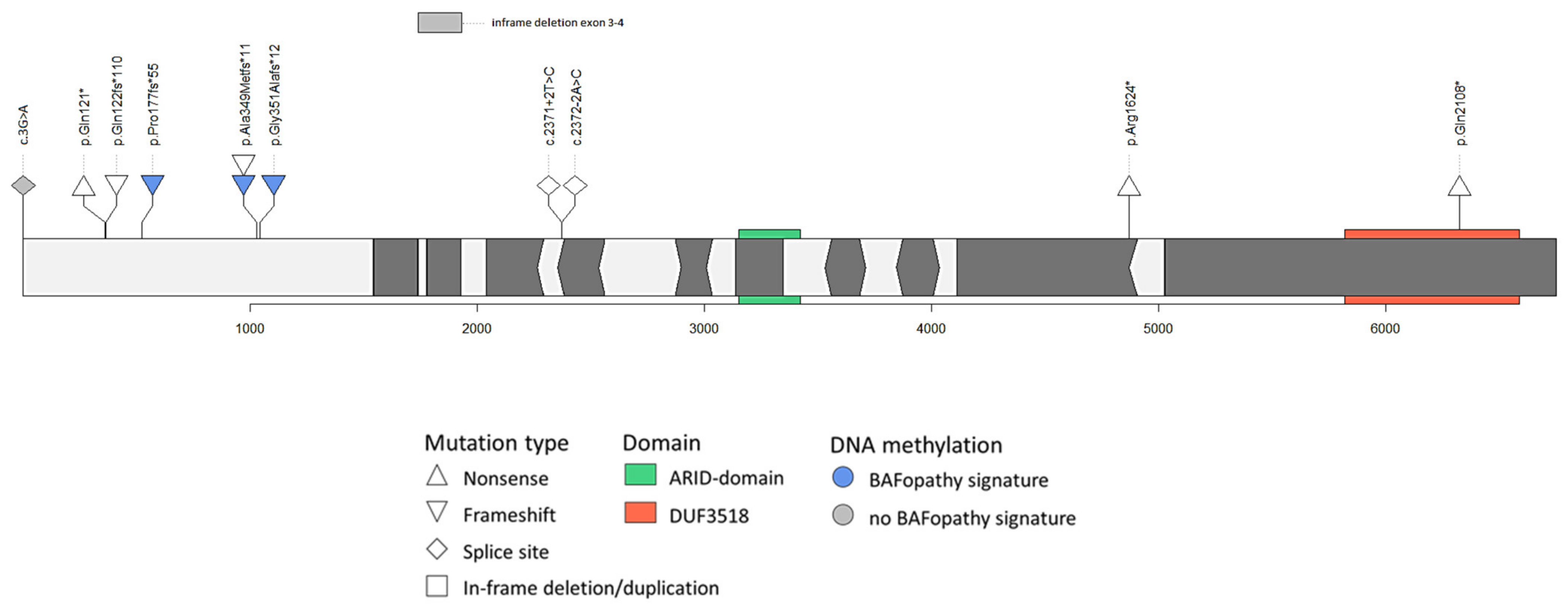
3.3. Variant Interpretation
3.3.1. Variants at the 5′ end of Exon 1 of ARID1B
Case 1
Case 2
Case 3
Case 4
Case 5
Case 6
Case 7
3.3.2. Splice Sites of in-Frame Exons or in-Frame Deletions in ARID1B
Case 8
Case 9
Case 10
3.3.3. Mosaicism in Parents
Case 11
Case 12
4. Discussion
4.1. Variable Expression
- Use caution interpreting LOF variants at the extreme 5′ and 3′ ends of a gene.
- If variants in related genes lead to the same signature, care must be taken to exclude the relevant variants in those genes.
4.2. Variant Location and the PVS1 Category
4.3. Transcript Choice
4.4. Incorporating New Developments into ACMG Guidelines for the Interpretation of Sequence Variants
4.4.1. Facial Analyses
4.4.2. DNA Methylation
4.4.3. Incorporating Suggested ACMG Updates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoyer, J.; Ekici, A.B.; Endele, S.; Popp, B.; Zweier, C.; Wiesener, A.; Wohlleber, E.; Dufke, A.; Rossier, E.; Petsch, C.; et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 2012, 90, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Santen, G.W.; Aten, E.; Vulto-van Silfhout, A.T.; Pottinger, C.; van Bon, B.W.; van Minderhout, I.J.; Snowdowne, R.; van der Lans, C.A.; Boogaard, M.; Linssen, M.M.; et al. Coffin-Siris syndrome and the BAF complex: Genotype-phenotype study in 63 patients. Hum. Mutat. 2013, 34, 1519–1528. [Google Scholar] [CrossRef]
- Wieczorek, D.; Bogershausen, N.; Beleggia, F.; Steiner-Haldenstatt, S.; Pohl, E.; Li, Y.; Milz, E.; Martin, M.; Thiele, H.; Altmuller, J.; et al. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet. 2013, 22, 5121–5135. [Google Scholar] [CrossRef]
- Tsurusaki, Y.; Okamoto, N.; Ohashi, H.; Mizuno, S.; Matsumoto, N.; Makita, Y.; Fukuda, M.; Isidor, B.; Perrier, J.; Aggarwal, S.; et al. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin. Genet. 2014, 85, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluijs, P.J.; Jansen, S.; Vergano, S.A.; Adachi-Fukuda, M.; Alanay, Y.; AlKindy, A.; Baban, A.; Bayat, A.; Beck-Wödl, S.; Berry, K.; et al. The ARID1B spectrum in 143 patients: From nonsyndromic intellectual disability to Coffin–Siris syndrome. Genet. Med. 2018. [Google Scholar] [CrossRef] [Green Version]
- Santen, G.W.; Clayton-Smith, J.; ARID1B-CSS Consortium. The ARID1B phenotype: What we have learned so far. Am. J. Med. Genet. Part C Semin. Med. Genet. 2014, 166C, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Van der Donk, R.; Jansen, S.; Schuurs-Hoeijmakers, J.H.M.; Koolen, D.A.; Goltstein, L.C.M.J.; Hoischen, A.; Brunner, H.G.; Kemmeren, P.; Nellåker, C.; Vissers, L.E.L.M.; et al. Next-generation phenotyping using computer vision algorithms in rare genomic neurodevelopmental disorders. Genet. Med. 2019, 21, 1719–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aref-Eshghi, E.; Bend, E.G.; Hood, R.L.; Schenkel, L.C.; Carere, D.A.; Chakrabarti, R.; Nagamani, S.C.S.; Cheung, S.W.; Campeau, P.M.; Prasad, C.; et al. BAFopathies’ DNA methylation epi-signatures demonstrate diagnostic utility and functional continuum of Coffin-Siris and Nicolaides-Baraitser syndromes. Nat. Commun. 2018, 9, 4885. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Kerkhof, J.; Pedro, V.P.; Groupe, D.I.F.; Barat-Houari, M.; Ruiz-Pallares, N.; Andrau, J.C.; Lacombe, D.; Van-Gils, J.; Fergelot, P.; et al. Evaluation of DNA Methylation Episignatures for Diagnosis and Phenotype Correlations in 42 Mendelian Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 356–370. [Google Scholar] [CrossRef]
- Sadikovic, B.; Levy, M.A.; Kerkhof, J.; Aref-Eshghi, E.; Schenkel, L.; Stuart, A.; McConkey, H.; Henneman, P.; Venema, A.; Schwartz, C.E.; et al. Clinical epigenomics: Genome-wide DNA methylation analysis for the diagnosis of Mendelian disorders. Genet. Med. 2021, 23, 1065–1074. [Google Scholar] [CrossRef]
- Levy, M.A.; Kerkhof, J.; Belmonte, F.R.; Kaufman, B.A.; Bhai, P.; Brady, L.; Bursztyn, L.; Tarnopolsky, M.; Rupar, T.; Sadikovic, B. Validation and clinical performance of a combined nuclear-mitochondrial next-generation sequencing and copy number variant analysis panel in a Canadian population. Am. J. Med. Genet. Part A 2021, 185, 486–499. [Google Scholar] [CrossRef]
- Dingemans, A.J.M.; Stremmelaar, D.E.; van der Donk, R.; Vissers, L.; Koolen, D.A.; Rump, P.; Hehir-Kwa, J.Y.; de Vries, B.B.A. Quantitative facial phenotyping for Koolen-de Vries and 22q11.2 deletion syndrome. Eur. J. Hum. Genet. 2021. [Google Scholar] [CrossRef]
- Lord, J.; Gallone, G.; Short, P.J.; McRae, J.F.; Ironfield, H.; Wynn, E.H.; Gerety, S.S.; He, L.; Kerr, B.; Johnson, D.S.; et al. Pathogenicity and selective constraint on variation near splice sites. Genome Res. 2019, 29, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Ruzzo, E.K.; Perez-Cano, L.; Jung, J.Y.; Wang, L.K.; Kashef-Haghighi, D.; Hartl, C.; Singh, C.; Xu, J.; Hoekstra, J.N.; Leventhal, O.; et al. Inherited and De novo Genetic Risk for Autism Impacts Shared Networks. Cell 2019, 178, 850–866.e826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, K.E.; Giordano, J.; Thorsten, V.; Buchovecky, C.; Thomas, A.; Ganapathi, M.; Liao, J.; Dharmadhikari, A.V.; Revah-Politi, A.; Ernst, M.; et al. Causal Genetic Variants in Stillbirth. N. Engl. J. Med. 2020, 383, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Al-Shamsi, A.; Hertecant, J.L.; Souid, A.K.; Al-Jasmi, F.A. Whole exome sequencing diagnosis of inborn errors of metabolism and other disorders in United Arab Emirates. Orphanet J. Rare Dis. 2016, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef]
- Sekiguchi, F.; Tsurusaki, Y.; Okamoto, N.; Teik, K.W.; Mizuno, S.; Suzumura, H.; Isidor, B.; Ong, W.P.; Haniffa, M.; White, S.M.; et al. Genetic abnormalities in a large cohort of Coffin-Siris syndrome patients. J. Hum. Genet. 2019. [Google Scholar] [CrossRef]
- Smith, J.A.; Holden, K.R.; Friez, M.J.; Jones, J.R.; Lyons, M.J. A novel familial autosomal dominant mutation in ARID1B causing neurodevelopmental delays, short stature, and dysmorphic features. Am. J. Med. Genet. Part A 2016, 170, 3313–3318. [Google Scholar] [CrossRef]
- Mignot, C.; Moutard, M.L.; Rastetter, A.; Boutaud, L.; Heide, S.; Billette, T.; Doummar, D.; Garel, C.; Afenjar, A.; Jacquette, A.; et al. ARID1B mutations are the major genetic cause of corpus callosum anomalies in patients with intellectual disability. Brain 2016, 139, e64. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Yao, R.; Wang, L.; Fan, Y.; Huang, X.; Hirschhorn, J.; Dauber, A.; Shen, Y. De novo mutations in ARID1B associated with both syndromic and non-syndromic short stature. BMC Genom. 2015, 16, 701. [Google Scholar] [CrossRef] [Green Version]
- Ben-Salem, S.; Sobreira, N.; Akawi, N.A.; Al-Shamsi, A.M.; John, A.; Pramathan, T.; Valle, D.; Ali, B.R.; Al-Gazali, L. Gonadal mosaicism in ARID1B gene causes intellectual disability and dysmorphic features in three siblings. Am. J. Med. Genet. Part A 2016, 170A, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherot, E.; Keren, B.; Dubourg, C.; Carre, W.; Fradin, M.; Lavillaureix, A.; Afenjar, A.; Burglen, L.; Whalen, S.; Charles, P.; et al. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: Experience of 2 clinical units and 216 patients. Clin. Genet. 2018, 93, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.S.W.; Luk, H.M.; Mok, M.T.; Leung, S.S.; Lo, I.F.M. Genotype and phenotype in 18 Chinese patients with Coffin-Siris syndrome. Am. J. Med. Genet. Part A 2021. [Google Scholar] [CrossRef] [PubMed]
- Min, Z.; Qian, C.; Ying, D. Novel ARID1B variant inherited from somatogonadal mosaic mother in siblings with Coffin-Siris syndrome 1. Exp. Ther. Med. 2021, 21, 614. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working, G. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.G.; Wilusz, J.E. Non-AUG translation: A new start for protein synthesis in eukaryotes. Genes Dev. 2017, 31, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Cummings, B.B.; Karczewski, K.J.; Kosmicki, J.A.; Seaby, E.G.; Watts, N.A.; Singer-Berk, M.; Mudge, J.M.; Karjalainen, J.; Satterstrom, F.K.; O’Donnell-Luria, A.H.; et al. Transcript expression-aware annotation improves rare variant interpretation. Nature 2020, 581, 452–458. [Google Scholar] [CrossRef]
- Johnston, J.J.; Lewis, K.L.; Ng, D.; Singh, L.N.; Wynter, J.; Brewer, C.; Brooks, B.P.; Brownell, I.; Candotti, F.; Gonsalves, S.G.; et al. Individualized iterative phenotyping for genome-wide analysis of loss-of-function mutations. Am. J. Hum. Genet. 2015, 96, 913–925. [Google Scholar] [CrossRef] [Green Version]
- Yuen, R.K.C.; Merico, D.; Bookman, M.; Howe, J.L.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Deflaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. [Google Scholar] [CrossRef]
- Pascolini, G.; Valiante, M.; Bottillo, I.; Laino, L.; Fleischer, N.; Ferraris, A.; Grammatico, P. Striking phenotypic overlap between Nicolaides-Baraitser and Coffin-Siris syndromes in monozygotic twins with ARID1B intragenic deletion. Eur. J. Med. Genet. 2019, 103739. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluijs, P.J.; Santen, G.W.E. Letter regarding the article: Striking phenotypic overlap between Nicolaides-Baraitser and Coffin-Siris syndromes in monozygotic twins with ARID1B intragenic deletion. Eur. J. Med. Genet. 2020, 63, 103813. [Google Scholar] [CrossRef]
- Fujita, T.; Ihara, Y.; Hayashi, H.; Ishii, A.; Ideguchi, H.; Inoue, T.; Imaizumi, T.; Yamamoto, T.; Hirose, S. Coffin-Siris syndrome with bilateral macular dysplasia caused by a novel exonic deletion in ARID1B. Congenit. Anom. 2020, 60, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Cappuccio, G.; Sayou, C.; Tanno, P.L.; Tisserant, E.; Bruel, A.L.; Kennani, S.E.; Sa, J.; Low, K.J.; Dias, C.; Havlovicova, M.; et al. De novo SMARCA2 variants clustered outside the helicase domain cause a new recognizable syndrome with intellectual disability and blepharophimosis distinct from Nicolaides-Baraitser syndrome. Genet. Med. 2020, 22, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Bend, E.G.; Aref-Eshghi, E.; Everman, D.B.; Rogers, R.C.; Cathey, S.S.; Prijoles, E.J.; Lyons, M.J.; Davis, H.; Clarkson, K.; Gripp, K.W.; et al. Gene domain-specific DNA methylation episignatures highlight distinct molecular entities of ADNP syndrome. Clin. Epigenetics 2019, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Baple, E.L.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; Deans, Z.C.; et al. ACGS Best Practice Guidelines for Variant Classification 2019: Association for Clinical Genetics Science (ACGS). Available online: https://www.acgs.uk.com/news/acgs-best-practice-guidelines-for-variant-classification-2019/ (accessed on 25 May 2021).
- Ellard, S.; Baple, E.L.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; Deans, Z.C.; et al. ACGS Best Practice Guidelines for Variant Classification 2020: Association for Clinical Genetics Science (ACGS). Available online: https://www.acgs.uk.com/quality/best-practice-guidelines/#VariantGuidelines (accessed on 25 May 2021).


{kind=link}
| Case | Exon | cDNA | Protein Change | Inheritance | Variant | GnomAD (1 May 2021) | DNA Methylation Pattern | Phenotype Suggestive for an ARID1B Related Disorder | Photograph Clusters with ARID1B Patients | CSS in Face2Gene (Rank/Similarity) | ACMG Criteria | Interpretation ACMG | Updated ACMG Criteria | Interpretation Updated ACMG | Expert Opinion |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | c.3G > A | p.(Met1?) | Not maternal | Start codon | No | no BAFopathy | + | + | 2/0.32 | PVS1, PM2 | LP | PM2, PM7, PP4 | VUS | VUS |
| 2 | 1 | c.361C > T | p.(Gln121*) | Paternal | Nonsense | No | n.a. | +/− | n.a. | n.a. | PVS1, PM2 | LP | PM2, PM7 | VUS | VUS |
| 3 | 1 | c.363_364insG | p.(Gln122fs*110) | Unknown | Frameshift | 7x | n.a. | − | n.a. | n.a. | PVS1, BS2 | VUS | PM4, BS2 | VUS | LB |
| 4 | 1 | c.521dup | p.(Pro177fs*55) | Maternal | Frameshift | No | BAFopathy | + | +(parent−) | 1/0.13 (−/0.13) | PVS1, PM2 | LP | PS5, PM2, PM7 | LP | P |
| 5 | 1 | c.1029_1056del | p.(Ala349Metfs*11) | Maternal | Frameshift | No | BAFopathy | + | + | 19/0.08 | PVS1, PM2 | LP | PVS1, PS5, PM2, PM7 | P | P |
| 6 | 1 | c.1044_1071del | p.(Ala349Metfs*11) | Maternal | Frameshift | No | n.a. | + | + | 1/0.38 | PVS1, PM2 | LP | PVS1, PM2, PM7 | P | P |
| 7 | 1 | c.1044_1062del | p.(Gly351Alafs*12) | Paternal | Frameshift | No | BAFopathy | + | +(parent−) | 7/0.31 16/0.07 | PVS1, PM2 | LP | PVS1, PS5, PM2, PM7 | P | P |
| 8 | 3–4 | exon 3–4 deletion | p.? | Paternal | In-frame deletion | - | no BAFopathy | - | n.a. | n.a. | PM2, PM4 | VUS | PM2, PM4 | VUS | LB |
| 9 | 7 | c.2371+2T > C | r.spl? | Unknown | Splice site | No | n.a. | + | n.a. | n.a. | PM2, PM4, PP4 | VUS | PM2, PM4, PP4 | VUS | VUS |
| 10 | 8 | c.2372-2A > C | r.spl? | Maternal | Splice site | 1x | n.a. | + | n.a. | n.a. | PP4 | VUS | PP4 | VUS | VUS |
| 11 | 18 | c.4870C > T | p.(Arg1624*) | Father is inconclusively mosaic. Mother is negative. Siblings. | Nonsense | No | n.a. | + | n.a. | n.a. | PVS1, PM2, PP4 | P | PVS1, PM2, PP4 | P | P |
| 12 | 20 | c.6322C > T | p.(Gln2108*) | Paternal, mosaic father | Nonsense | No | n.a. | + | +(parent+) | 1/0.78 1/0.34 | PVS1, PM2, PP4 | P | PVS1, PM2, PM7, PP1, PP4 | P | P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van der Sluijs, P.J.; Alders, M.; Dingemans, A.J.M.; Parbhoo, K.; van Bon, B.W.; Dempsey, J.C.; Doherty, D.; den Dunnen, J.T.; Gerkes, E.H.; Milller, I.M.; et al. A Case Series of Familial ARID1B Variants Illustrating Variable Expression and Suggestions to Update the ACMG Criteria. Genes 2021, 12, 1275. https://doi.org/10.3390/genes12081275
van der Sluijs PJ, Alders M, Dingemans AJM, Parbhoo K, van Bon BW, Dempsey JC, Doherty D, den Dunnen JT, Gerkes EH, Milller IM, et al. A Case Series of Familial ARID1B Variants Illustrating Variable Expression and Suggestions to Update the ACMG Criteria. Genes. 2021; 12(8):1275. https://doi.org/10.3390/genes12081275
Chicago/Turabian Stylevan der Sluijs, Pleuntje J., Mariëlle Alders, Alexander J. M. Dingemans, Kareesma Parbhoo, Bregje W. van Bon, Jennifer C. Dempsey, Dan Doherty, Johan T. den Dunnen, Erica H. Gerkes, Ilana M. Milller, and et al. 2021. "A Case Series of Familial ARID1B Variants Illustrating Variable Expression and Suggestions to Update the ACMG Criteria" Genes 12, no. 8: 1275. https://doi.org/10.3390/genes12081275