
Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum


Abstract
:1. Introduction
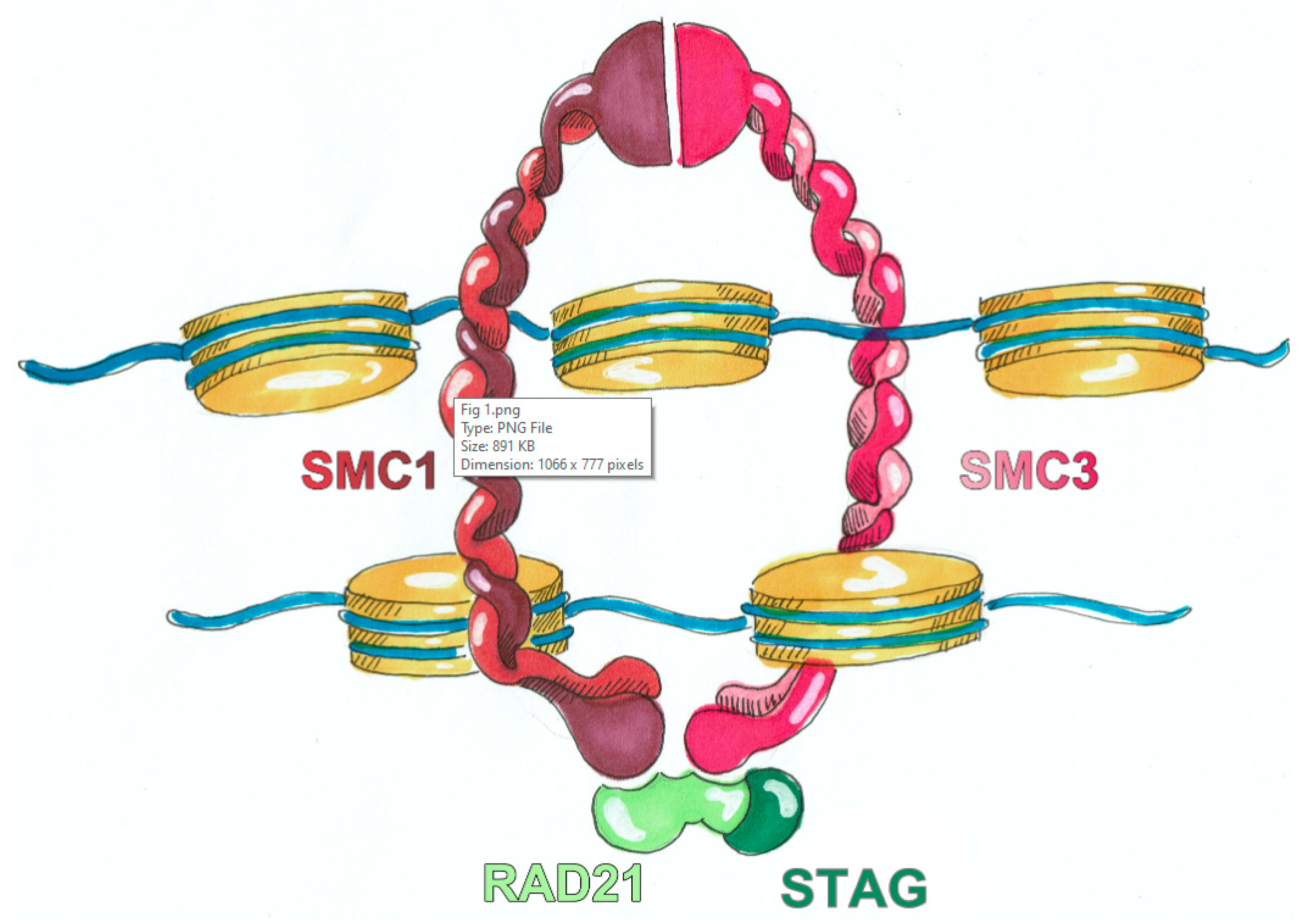
2. Biological Basis of CdLS
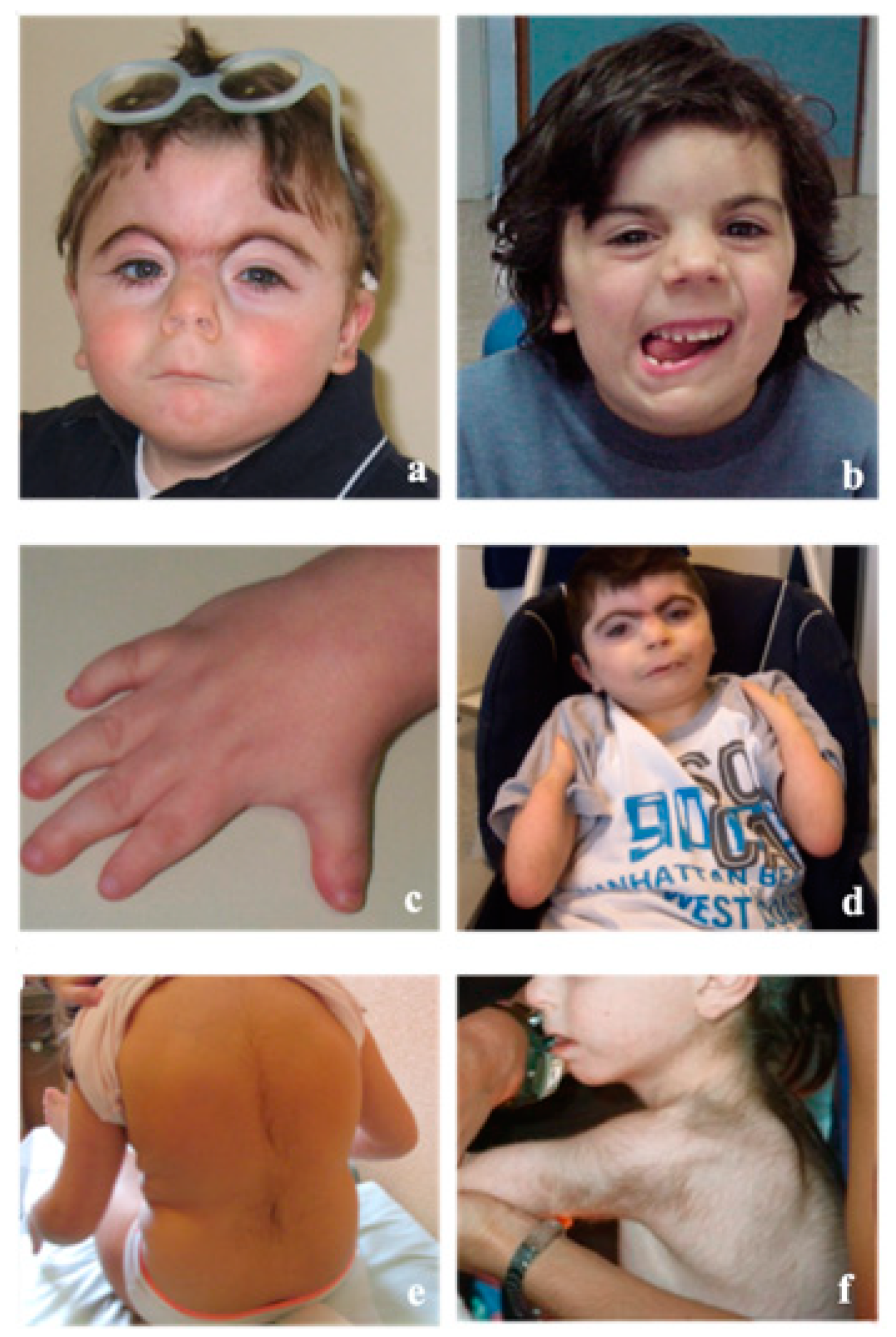
3. Clinical Features

{kind=link}
{kind=link}
| Medical Problem | Prevalence |
|---|---|
| Gastrointestinal problems | |
| -Feeding problems | frequent |
| -Enteral nutrition | 40% |
| -Gastro esophageal reflux | 60–75% |
| -Batter esophagus | 9% |
| -Eosinophilic esophagitis | 16% |
| -Constipation | 10–15% |
| -Gassiness | 48% |
| Neurology | |
| -Seizures | 45% (SMC1A gene) 15% (NIPBL gene) |
| -Autonomic nervous system disfunction | 81% (mild) 26% (severe) |
| -Sleep problems | 12–72% |
| Orthopedics | |
| -Perthes disease | 4% |
| -Leg length differences | 46% |
| -Congenital hip dislocation | 10% |
| -Scoliosis | One third of patients after 10 years |
| -Kyphosis | A quarter of patients |
| -Joint contractures | 18–25% |
| -Bunions | 75% |
| Visual problems | |
| -Palpebral ptosis | 37% (unilateral) 44% (bilateral) |
| -Blepharitis | 25% |
| -Nystagmus | 14–17% |
| -Strabismus | 16–26% |
| -Visual impairment | 44–53% |
| -Pigmented peripapillary ring | 83% |
| Hearing problems | |
| -Conductive hearing loss | 75% |
| -Neurosensorial hearing loss | 25% |
| -Otitis media with effusion | 80–85% |
| Immunological defects | 33% (only one specific report) [44] |
| Thrombocytopenia | rare |
| Cancer | No increase of risk |
4. Genotype–Phenotype Correlations
5. From Biological Basis to Future Treatment Options
5.1. Lithium
5.2. L-Leucin
5.3. Antioxidant
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Mannini, L.; Cucco, F.; Quarantotti, V.; Krantz, I.D.; Musio, A. Mutation spectrum and genotype-phenotype correlation in Cornelia de Lange syndrome. Hum. Mutat. 2013, 34, 1589–1596. [Google Scholar] [CrossRef] [Green Version]
- De Lange, C.C. Sur un type nouveau de degenerescence (typus Amstelodamensis). Arch. Med. Enfants 1933, 36, 713–719. [Google Scholar]
- Meinecke, P.; Hayek, H. Brief historical note on the Brachmann-de Lange syndrome: A patient closely resembling the case described by Brachmann in 1916. Am. J. Med. Genet. 1990, 35, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, W.R.C. Ein Fall von symmetrischer Monodaktylie durch Ulnadefekt, mit symmetrischer Flughautbildung in den Ellenbeugen, sowie anderen Abnormitäten (Zwerghaftigkeit, Halsrippen, Behaarung) (A case of symmetrical monodactyly, representing ulnar deficiency, with symmetrical antecubital webbing and other abnormalities, (dwarfism, cervical ribs, hirsutism)). Jarb. Kinder Phys. Erzie 1916, 84, 225–235. [Google Scholar]
- Vrolik, W. Tabulae ad Illustrandam Embryogenesin Hominis et Mammalium Tam Naturalem Quam Abnormem; Amstelodami: London, UK, 1849. [Google Scholar]
- Berg, J.; McCreary, B.; Ridler, M.A.; Smith, G. The De Lange Syndrome; Pergamon Press: Oxford, UK, 1970. [Google Scholar]
- Preus, M.; Rex, A.P. Definition and diagnosis of the Brachmann-De Lange syndrome. Am. J. Med. Genet. 1983, 16, 301–312. [Google Scholar] [CrossRef]
- Kline, A.D.; Krantz, I.D.; Sommer, A.; Kliewer, M.; Jackson, L.G.; FitzPatrick, D.R.; Levin, A.V.; Selicorni, A. Cornelia de Lange syndrome: Clinical review, diagnostic and scoring systems, and anticipatory guidance. Am. J. Med. Genet. Part A 2007, 143, 1287–1296. [Google Scholar] [CrossRef]
- Van Allen, M.I.; Filippi, G.; Siegel-Bartelt, J.; Yong, S.-L.; McGillivray, B.; Zuker, R.M.; Smith, C.R.; Magee, J.F.; Ritchie, S.; Toi, A.; et al. Clinical variability within Brachmann-de Lange syndrome: A proposed classification system. Am. J. Med. Genet. 1993, 47, 947–958. [Google Scholar] [CrossRef]
- Selicorni, A.; Lalatta, F.; Livini, E.; Briscioli, V.; Piguzzi, T.; Bagozzi, D.C.; Mastroiacovo, P.; Zampino, G.; Gaeta, G.; Pugliese, A.; et al. Variability of the Brachmann-de Lange Syndrome. Am. J. Med. Genet. 1993, 47, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Bay, C.; Mauk, J.; Radcliffe, J.; Kaplan, P. Mild Brachmann-de Lange syndrome. Delineation of the clinical phenotype, and characteristic behaviors in a six-year-old boy. Am. J. Med. Genet. 1993, 47, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Clericuzio, C.L. Mild mental retardation with classic somatic phenotype in the Brachmann-de Lange syndrome. Am. J. Med. Genet. 1993, 47, 992–994. [Google Scholar] [CrossRef]
- Saal, H.M.; Samango-Sprouse, C.A.; Rodnan, L.A.; Rosenbaum, K.N.; Custer, D.A. Brachmann-de Lange syndrome with normal IQ. Am. J. Med. Genet. 1993, 47, 995–998. [Google Scholar] [CrossRef]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.M.; Kamphausen, S.B.; Zenker, M.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Pellicer, A.; Ascaso, Á.; Trujillano, L.; Gil-Salvador, M.; Arnedo, M.; Lucia-Campos, C.; Antoñanzas-Pérez, R.; Marcos-Alcalde, I.; Parenti, I.; Bueno-Lozano, G.; et al. Evaluating Face2Gene as a Tool to Identify Cornelia de Lange Syndrome by Facial Phenotypes. Int. J. Mol. Sci. 2020, 21, 1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panaitescu, A.M.; Duta, S.; Gica, N.; Botezatu, R.; Nedelea, F.; Peltecu, G.; Veduta, A. A broader perspective on the prenatal diagnosis of cornelia de lange syndrome: Review of the literature and case presentation. Diagnostics 2021, 11, 142. [Google Scholar] [CrossRef]
- Avagliano, L.; Parenti, I.; Grazioli, P.; Di Fede, E.; Parodi, C.; Mariani, M.; Kaiser, F.J.; Selicorni, A.; Gervasini, C.; Massa, V. Chromatinopathies: A focus on Cornelia de Lange syndrome. Clin. Genet. 2020, 97, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, F.D.; Calderon, L.; Wang, Y.F.; Georgieva, R.; Guo, Y.; Cvetesic, N.; Kaur, M.; Dharmalingam, G.; Krantz, I.D.; Lenhard, B.; et al. Neuronal genes deregulated in Cornelia de Lange Syndrome respond to removal and re-expression of cohesin. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Revenkova, E.; Focarelli, M.L.; Susani, L.; Paulis, M.; Bassi, M.T.; Mannini, L.; Frattini, A.; Delia, D.; Krantz, I.; Vezzoni, P.; et al. Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum. Mol. Genet. 2009, 18, 418–427. [Google Scholar] [CrossRef] [Green Version]
- Castronovo, P.; Gervasini, C.; Cereda, A.; Masciadri, M.; Milani, D.; Russo, S.; Selicorni, A.; Larizza, L. Premature chromatid separation is not a useful diagnostic marker for Cornelia de Lange syndrome. Chromosom. Res. 2009, 17, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, Z.; Bando, M.; Itoh, T.; Deardorff, M.A.; Clark, D.; Kaur, M.; Tandy, S.; Kondoh, T.; Rappaport, E.; et al. Transcriptional Dysregulation in NIPBLand Cohesin Mutant Human Cells. PLoS Biol. 2009, 7, e1000119. [Google Scholar] [CrossRef] [PubMed]
- Gimigliano, A.; Mannini, L.; Bianchi, L.; Puglia, M.; Deardorff, M.A.; Menga, S.; Krantz, I.D.; Musio, A.; Bini, L. Proteomic profile identifies dysregulated pathways in Cornelia de Lange syndrome cells with distinct mutations in SMC1A and SMC3 genes. J. Proteome Res. 2012, 11, 6111–6123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Mannini, L.; Lamaze, F.C.; Cucco, F.; Amato, C.; Quarantotti, V.; Rizzo, I.M.; Krantz, I.D.; Bilodeau, S.; Musio, A. Mutant cohesin affects RNA polymerase II regulation in Cornelia de Lange syndrome. Sci. Rep. 2015, 5, 16803. [Google Scholar] [CrossRef] [Green Version]
- Cukrov, D.; Newman, T.A.C.; Leask, M.; Leeke, B.; Sarogni, P.; Patimo, A.; Kline, A.D.; Krantz, I.D.; Horsfield, J.A.; Musio, A. Antioxidant treatment ameliorates phenotypic features of SMC1A-mutated Cornelia de Lange syndrome in vitro and in vivo. Hum. Mol. Genet. 2018, 27, 3002–3011. [Google Scholar] [CrossRef] [PubMed]
- Cucco, F.; Sarogni, P.; Rossato, S.; Alpa, M.; Patimo, A.; Latorre, A.; Magnani, C.; Puisac, B.; Ramos, F.J.; Pié, J.; et al. Pathogenic variants in EP300 and ANKRD11 in patients with phenotypes overlapping Cornelia de Lange syndrome. Am. J. Med. Genet. Part A 2020, 182, 1690–1696. [Google Scholar] [CrossRef] [PubMed]
- Litwin, I.; Pilarczyk, E.; Wysocki, R. The Emerging Role of Cohesin in the DNA Damage Response. Genes 2018, 9, 581. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.P.; McKinney, S.; Gerton, J.L. Persistent DNA Damage and Senescence in the Placenta Impacts Developmental Outcomes of Embryos. Dev. Cell 2020, 54, 333.e7–347.e7. [Google Scholar] [CrossRef]
- Olley, G.; Pradeepa, M.M.; Grimes, G.R.; Piquet, S.; Polo, S.E.; FitzPatrick, D.R.; Bickmore, W.A.; Boumendil, C. Cornelia de Lange syndrome-associated mutations cause a DNA damage signalling and repair defect. Nat. Commun. 2021, 12, 3127. [Google Scholar] [CrossRef]
- Mills, J.A.; Herrera, P.S.; Kaur, M.; Leo, L.; McEldrew, D.; Tintos-Hernandez, J.A.; Rajagopalan, R.; Gagne, A.; Zhang, Z.; Ortiz-Gonzalez, X.R.; et al. NIPBL+/− haploinsufficiency reveals a constellation of transcriptome disruptions in the pluripotent and cardiac states. Sci. Rep. 2018, 8, 1056. [Google Scholar] [CrossRef] [Green Version]
- Pistocchi, A.; Fazio, G.; Cereda, A.; Ferrari, L.; Bettini, L.R.; Messina, G.; Cotelli, F.; Biondi, A.; Selicorni, A.; Massa, V. Cornelia de Lange Syndrome: NIPBL haploinsufficiency downregulates canonical Wnt pathway in zebrafish embryos and patients fibroblasts. Cell Death Dis. 2013, 4, e866. [Google Scholar] [CrossRef] [Green Version]
- Fazio, G.; Gaston-Massuet, C.; Bettini, L.R.; Graziola, F.; Scagliotti, V.; Cereda, A.; Ferrari, L.; Mazzola, M.; Cazzaniga, G.; Giordano, A.; et al. CyclinD1 Down-Regulation and Increased Apoptosis Are Common Features of Cohesinopathies. J. Cell. Physiol. 2016, 231, 613–622. [Google Scholar] [CrossRef]
- Avagliano, L.; Grazioli, P.; Mariani, M.; Bulfamante, G.P.; Selicorni, A.; Massa, V. Integrating molecular and structural findings: Wnt as a possible actor in shaping cognitive impairment in Cornelia de Lange syndrome. Orphanet J. Rare Dis. 2017, 12, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grazioli, P.; Parodi, C.; Mariani, M.; Bottai, D.; Di Fede, E.; Zulueta, A.; Avagliano, L.; Cereda, A.; Tenconi, R.; Wierzba, J.; et al. Lithium as a possible therapeutic strategy for Cornelia de Lange syndrome. Cell Death Discov. 2021, 7. [Google Scholar] [CrossRef]
- Miyabayashi, T.; Teo, J.-L.; Yamamoto, M.; McMillan, M.; Nguyen, C.; Kahn, M. Wnt/β-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 2007, 104, 5668–5673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, A.D.; Barr, M.; Jackson, L.G. Growth manifestations in the Brachmann-de Lange syndrome. Am. J. Med. Genet. 1993, 47, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Ajmone, P.F.; Rigamonti, C.; Dall’Ara, F.; Monti, F.; Vizziello, P.; Milani, D.; Cereda, A.; Selicorni, A.; Costantino, A. Communication, cognitive development and behavior in children with cornelia de lange syndrome (CdLS): Preliminary results. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2014, 165, 223–229. [Google Scholar] [CrossRef]
- Parma, B.; Cianci, P.; Decimi, V.; Mariani, M.; Provero, M.C.; Funari, C.; Tajè, S.; Apuril, E.; Cereda, A.; Panceri, R.; et al. Complex nutritional deficiencies in a large cohort of Italian patients with Cornelia de Lange syndrome spectrum. Am. J. Med. Genet. Part A 2020, 182, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Matute-Llorente, Á.; Ascaso, Á.; Latorre-Pellicer, A.; Puisac, B.; Trujillano, L.; Llorente, E.; Puente-Lanzarote, J.J.; Ayerza-Casas, A.; Arnedo, M.; Moreno, L.A.; et al. Targeted Gene Sequencing, Bone Health, and Body Composition in Cornelia de Lange Syndrome. Appl. Sci. 2021, 11, 710. [Google Scholar] [CrossRef]
- Selicorni, A.; Russo, S.; Gervasini, C.; Castronovo, P.; Milani, D.; Cavalleri, F.; Bentivegna, A.; Masciadri, M.; Domi, A.; Divizia, M.T.; et al. Clinical score of 62 Italian patients with Cornelia de Lange syndrome and correlations with the presence and type of NIPBL mutation. Clin. Genet. 2007, 72, 98–108. [Google Scholar] [CrossRef]
- Bhuiyan, Z.A.; Klein, M.; Hammond, P.; Van Haeringen, A.; Mannens, M.M.A.M.; Van Berckelaer-Onnes, I.; Hennekam, R.C.M. Genotype-phenotype correlations of 39 patients with Cornelia de Lange syndrome: The Dutch experience. J. Med. Genet. 2006, 43, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cereda, A.; Mariani, M.; Rebora, P.; Sajeva, A.; Ajmone, P.F.; Gervasini, C.; Russo, S.; Kullmann, G.; Valsecchi, G.; Selicorni, A. A new prognostic index of severity of intellectual disabilities in Cornelia de Lange syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2016, 172, 179–189. [Google Scholar] [CrossRef]
- Jyonouchi, S.; Orange, J.; Sullivan, K.E.; Krantz, I.; Deardorff, M. Immunologic Features of Cornelia de Lange Syndrome. Pediatrics 2013, 132, e484–e489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, L.A.; McCallum, J.; Kaur, M.; DeScipio, C.; Yaeger, D.; Mariani, A.; Kline, A.D.; Li, H.H.; Devoto, M.; Jackson, L.G.; et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am. J. Hum. Genet. 2004, 75, 610–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, I.; Diab, F.; Gil, S.R.; Mulugeta, E.; Casa, V.; Berutti, R.; Brouwer, R.W.W.; Dupé, V.; Eckhold, J.; Graf, E.; et al. MAU2 and NIPBL Variants Impair the Heterodimerization of the Cohesin Loader Subunits and Cause Cornelia de Lange Syndrome. Cell Rep. 2020, 31, 107647. [Google Scholar] [CrossRef]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. Part A 2017, 173, 2108–2125. [Google Scholar] [CrossRef] [Green Version]
- Borck, G.; Zarhrate, M.; Bonnefont, J.P.; Munnich, A.; Cormier-Daire, V.; Colleaux, L. Incidence and clinical features of X-linked Cornelia de Lange syndrome due to SMC1L1 mutations. Hum. Mutat. 2007, 28, 205–206. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Joss, S.; Metcalfe, K.A.; Somarathi, S.; Cruden, J.; Devlin, A.M.; Donaldson, A.; DiDonato, N.; Fitzpatrick, D.; Kaiser, F.J.; et al. Heterozygous truncation mutations of the SMC1A gene cause a severe early onset epilepsy with cluster seizures in females: Detailed phenotyping of 10 new cases. Epilepsia 2017, 58, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Jansen, S.; Kleefstra, T.; Willemsen, M.H.; de Vries, P.; Pfundt, R.; Hehir-Kwa, J.Y.; Gilissen, C.; Veltman, J.A.; de Vries, B.B.A.; Vissers, L.E.L.M. De novo loss-of-function mutations in X-linked SMC1A cause severe ID and therapy-resistant epilepsy in females: Expanding the phenotypic spectrum. Clin. Genet. 2016, 90, 413–419. [Google Scholar] [CrossRef]
- Gil-Rodríguez, M.C.; Deardorff, M.A.; Ansari, M.; Tan, C.A.; Parenti, I.; Baquero-Montoya, C.; Ousager, L.B.; Puisac, B.; Hernández-Marcos, M.; Teresa-Rodrigo, M.E.; et al. De novo heterozygous mutations in SMC3 cause a range of cornelia de lange syndrome-overlapping phenotypes. Hum. Mutat. 2015, 36, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Boyle, M.I.; Jespersgaard, C.; Nazaryan, L.; Bisgaard, A.-M.; Tümer, Z. A novel RAD21 variant associated with intrafamilial phenotypic variation in Cornelia de Lange syndrome—Review of the literature. Clin. Genet. 2017, 91, 647–649. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Wilde, J.J.; Albrecht, M.; Dickinson, E.; Tennstedt, S.; Braunholz, D.; Mönnich, M.; Yan, Y.; Xu, W.; Gil-Rodríguez, M.C.; et al. RAD21 mutations cause a human cohesinopathy. Am. J. Hum. Genet. 2012, 90, 1014–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krab, L.C.; Marcos-Alcalde, I.; Assaf, M.; Balasubramanian, M.; Andersen, J.B.; Bisgaard, A.-M.; Fitzpatrick, D.R.; Gudmundsson, S.; Huisman, S.A.; Kalayci, T.; et al. Delineation of phenotypes and genotypes related to cohesin structural protein RAD21. Hum. Genet. 2020, 139, 575–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olley, G.; Ansari, M.; Bengani, H.; Grimes, G.R.; Rhodes, J.; Von Kriegsheim, A.; Blatnik, A.; Stewart, F.J.; Wakeling, E.; Carroll, N.; et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome. Nat. Genet. 2018, 50, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Harakalova, M.; van den Boogaard, M.J.; Sinke, R.; van Lieshout, S.; van Tuil, M.C.; Duran, K.; Renkens, I.; Terhal, P.A.; de Kovel, C.; Nijman, I.J.; et al. X-exome sequencing identifies a HDAC8 variant in a large pedigree with X-linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J. Med. Genet. 2012, 49, 539–543. [Google Scholar] [CrossRef]
- Parenti, I.; Gervasini, C.; Pozojevic, J.; Wendt, K.S.; Watrin, E.; Azzollini, J.; Braunholz, D.; Buiting, K.; Cereda, A.; Engels, H.; et al. Expanding the clinical spectrum of the “HDAC8-phenotype”—Implications for molecular diagnostics, counseling and risk prediction. Clin. Genet. 2016, 89, 564–573. [Google Scholar] [CrossRef]
- Parenti, I.; Gervasini, C.; Pozojevic, J.; Graul-Neumann, L.; Azzollini, J.; Braunholz, D.; Watrin, E.; Wendt, K.S.; Cereda, A.; Cittaro, D.; et al. Broadening of cohesinopathies: Exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype. Clin. Genet. 2016, 89, 74–81. [Google Scholar] [CrossRef]
- Parenti, I.; Mallozzi, M.B.; Hüning, I.; Gervasini, C.; Kuechler, A.; Agolini, E.; Albrecht, B.; Baquero-Montoya, C.; Bohring, A.; Bramswig, N.C.; et al. ANKRD11 variants: KBG syndrome and beyond. Clin. Genet. 2021, cge.13977. [Google Scholar] [CrossRef]
- Castronovo, P.; Delahaye-Duriez, A.; Gervasini, C.; Azzollini, J.; Minier, F.; Russo, S.; Masciadri, M.; Selicorni, A.; Verloes, A.; Larizza, L. Somatic mosaicism in Cornelia de Lange syndrome: A further contributor to the wide clinical expressivity? Clin. Genet. 2010, 78, 560–564. [Google Scholar] [CrossRef]
- Huisman, S.A.; Redeker, E.J.W.; Maas, S.M.; Mannens, M.M.; Hennekam, R.C.M. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J. Med. Genet. 2013, 50, 339–344. [Google Scholar] [CrossRef]
- Volkmann, C.; Bschor, T.; Köhler, S. Lithium Treatment Over the Lifespan in Bipolar Disorders. Front. Psychiatry 2020, 11, 377. [Google Scholar] [CrossRef]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics Wingless signalling in intact cells. Curr. Biol. 1996, 6, 1664–1669. [Google Scholar] [CrossRef] [Green Version]
- Poels, E.M.P.; Bijma, H.H.; Galbally, M.; Bergink, V. Lithium during pregnancy and after delivery: A review. Int. J. Bipolar Disord. 2018, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munk-Olsen, T.; Liu, X.; Viktorin, A.; Brown, H.K.; Di Florio, A.; D’Onofrio, B.M.; Gomes, T.; Howard, L.M.; Khalifeh, H.; Krohn, H.; et al. Maternal and infant outcomes associated with lithium use in pregnancy: An international collaborative meta-analysis of six cohort studies. Lancet Psychiatry 2018, 5, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Poels, E.M.P.; Schrijver, L.; Kamperman, A.M.; Hillegers, M.H.J.; Hoogendijk, W.J.G.; Kushner, S.A.; Roza, S.J. Long-term neurodevelopmental consequences of intrauterine exposure to lithium and antipsychotics: A systematic review and meta-analysis. Eur. Child. Adolesc. Psychiatry 2018, 27, 1209–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyck, L.I.; Morrow, E.M. Genetic control of postnatal human brain growth. Curr. Opin. Neurol. 2017, 30, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Smith, C.B. Lithium: A promising treatment for fragile X syndrome. ACS Chem. Neurosci. 2014, 5, 477–483. [Google Scholar] [CrossRef]
- McCamphill, P.K.; Stoppel, L.J.; Senter, R.K.; Lewis, M.C.; Heynen, A.J.; Stoppel, D.C.; Sridhar, V.; Collins, K.A.; Shi, X.; Pan, J.Q.; et al. Selective inhibition of glycogen synthase kinase 3α corrects pathophysiology in a mouse model of fragile X syndrome. Sci. Transl. Med. 2020, 12, eaam8572. [Google Scholar] [CrossRef]
- Xu, B.; Sowa, N.; Cardenas, M.E.; Gerton, J.L. L-leucine partially rescues translational and developmental defects associated with zebrafish models of Cornelia de Lange syndrome. Hum. Mol. Genet. 2015, 24, 1540–1555. [Google Scholar] [CrossRef]
- Sarogni, P.; Pallotta, M.M.; Musio, A. Cornelia de Lange syndrome: From molecular diagnosis to therapeutic approach. J. Med. Genet. 2020, 57, 289–295. [Google Scholar] [CrossRef]
- Reliene, R.; Fischer, E.; Schiestl, R.H. Effect of N -Acetyl Cysteine on Oxidative DNA Damage and the Frequency of DNA Deletions in Atm -Deficient Mice. Cancer Res. 2004, 64, 5148–5153. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, M.; Ushimaru, N.; Osada, N.; Oda, T.; Ishige, K.; Ito, Y. N-acetylcysteine selectively protects cerebellar granule cells from 4-hydroxynonenal-induced cell death. Neurosci. Res. 2006, 55, 255–263. [Google Scholar] [CrossRef] [PubMed]


| Cardinal Features | Score |
|---|---|
| Synophrys (HP:0000664) and/or thick eyebrows (HP:0000574) | 2 |
| Short nose (HP:0003196), concave nasal ridge (HP:0011120) and/or upturned nasal tip (HP:0000463) | 2 |
| Long (HP:0000343) and/or smooth philtrum (HP:0000319) | 2 |
| Thin upper lip vermilion (HP:0000219) and/or downturned corners of mouth (HP:0002714) | 2 |
| Hand oligodactyly (HP:0001180) and/or adactyly (HP:0009776) | 2 |
| Congenital diaphragmatic hernia (HP:0000776) | 2 |
| Suggestive Features | |
| Global developmental delay (HP:0001263) and/or intellectual disability (HP:0001249) | 1 |
| Prenatal growth retardation (<2 SD) (HP:0001511) | 1 |
| Postnatal growth retardation (<2 SD) (HP:0008897) | 1 |
| Microcephaly (prenatally and/or postnatally) (HP:0000252) | 1 |
| Small hands (HP:0200055) and/or feet (HP:0001773) | 1 |
| Short fifth finger (HP:0009237) | 1 |
| Hirsutism (HP:0001007) | 1 |
| Interpretation of the Score | |
| Score ≥11 points, of which at least 3 are cardinal features | classic CdLS |
| Score between 9 or 10 points, of which at least 2 are cardinal features | non-classic CdLS |
| Score between 4–8 points, of which at least 1 is cardinal feature | molecular testing for CdLS indicated |
| Score <4 points | insufficient to indicate molecular testing for CdLS |
| Malformation | Prevalence |
|---|---|
| Heart malformations (no specific defect) | 25% |
| Palate | 20% |
| Eyes Unilateral or bilateral nasolacrimal duct obstruction | 60–80% |
| Central Nervous System | 47% (in the wider cohort reported) [35] |
| Limb defects | About one third |
| Urinary tract | 10% |
| Genitalia | |
| Cryptorchidism | 80% |
| Micropenis | 37% |
| Hypospadias | 9% |
| Bicornuate uterus | 19% |
| Gastrointestinal system | |
| Intestinal malrotation | 5–10% |
| Pyloric stenosis | 7% |
| Diaphragmatic Hernia | rare |
| Gene | Overlapping Syndrome |
|---|---|
| ANKRD11 | KBG syndrome |
| AFF4 | CHOP syndrome |
| EP300 | Rubinstein-Taybi syndrome |
| KMT2A | Wiedemann-Steiner syndrome |
| TAF6 | Alazami-Yuan syndrome |
| SETD5 | Mental retardation, autosomal dominant 23 |
| ARID1B | Coffin-Siris syndrome |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selicorni, A.; Mariani, M.; Lettieri, A.; Massa, V. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes 2021, 12, 1075. https://doi.org/10.3390/genes12071075
Selicorni A, Mariani M, Lettieri A, Massa V. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes. 2021; 12(7):1075. https://doi.org/10.3390/genes12071075
Chicago/Turabian StyleSelicorni, Angelo, Milena Mariani, Antonella Lettieri, and Valentina Massa. 2021. "Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum" Genes 12, no. 7: 1075. https://doi.org/10.3390/genes12071075