
A Reassessment of Copy Number Variations in Congenital Heart Defects: Picturing the Whole Genome

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection and Characterization of the Cohort
2.2. Molecular Karyotyping
2.3. Descriptive Statistics and Study of CNV-US
- Overlap with ClinGen Pathogenic CNV regions (see above) or ClinGen Dosage Sensitive regions (https://search.clinicalgenome.org/kb/gene-dosage, accessed on 28 February 2021) [30].
- Overlap with other CNV-US within the study cohort.
- The protein-coding gene content based on NCBI RefSeq Select genes (Updated Annotation Release 105.20190906), extracted from the UCSC Genome Browser using the Table Browser [31].
- Non-coding gene-regulatory elements contained within the CNV-US. LncRNA genes were extracted from the lncipedia high confidence set (hg19) (https://www.lncipedia.org/, accessed on 8 December 2020) [32]. Human VISTA enhancer elements were downloaded from the VISTA enhancer database (https://enhancer.lbl.gov/, accessed on 7 December 2020) [33].
- Interference with the genomic TAD structure, based on the TAD boundaries file that was provided by J. Dixon to the developers of ClinTAD (https://www.clintad.com/, accessed on 4 January 2021) and publicly available on github [34,35]. This TAD boundaries file was generated using H1 human embryonic stem cells, chromosome build GRCh37, a bin size of 40 kb, and a window size of 2 Mb. To evaluate the potential disruption of TAD-related gene-enhancer interactions, we considered the protein-coding gene content (NCBI RefSeq Select genes) and the human VISTA enhancer elements within the relevant TAD domains.
2.4. Data Browsing
2.4.1. Dosage Sensitivity of Protein-Coding Genes
2.4.2. Expression Data of Protein-Coding and lncRNA Genes
2.4.3. Protein-Coding Gene Function Annotation
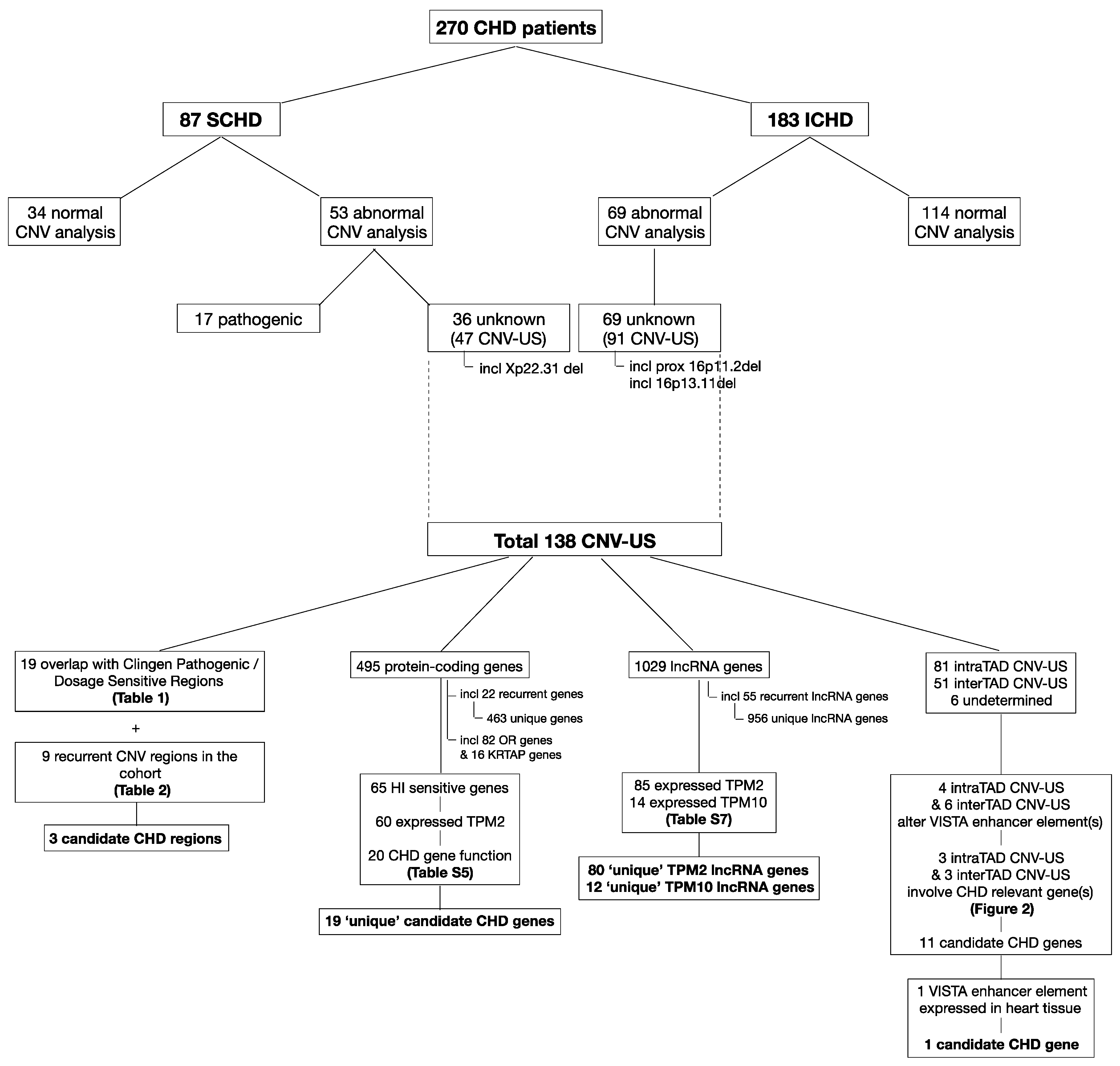
3. Results
3.1. Study Cohort and Clinical Characteristics
3.2. CNV Analyses
3.3. Comprehensive Reassessment of CNV-US
3.3.1. CNV Descriptives: CNV Type, Size and Parental Inheritance
3.3.2. Overlap with Known Dosage Sensitive Regions and Recurrence in the Study Cohort
3.3.3. Protein-Coding Gene Content
3.3.4. lncRNAs
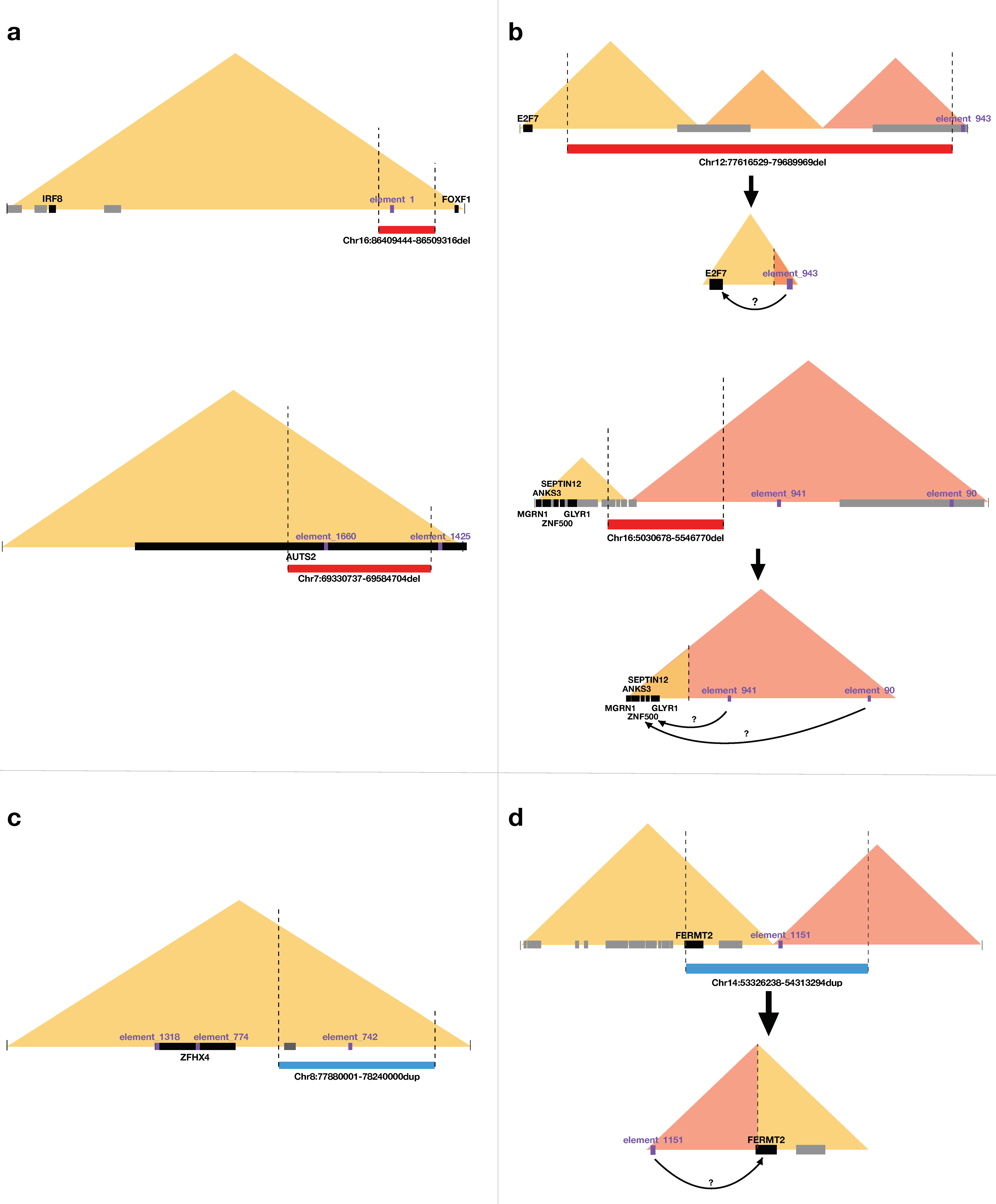
3.3.5. Enhancers and Interference with TAD-Related Gene-Enhancer Interactions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [Green Version]
- Vissers, L.E.; de Vries, B.B.; Osoegawa, K.; Janssen, I.M.; Feuth, T.; Choy, C.O.; Straatman, H.; van der Vliet, W.; Huys, E.H.; van Rijk, A.; et al. Array-based comparative genomic hybridization for the genomewide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003, 73, 1261–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw-Smith, C.; Redon, R.; Rickman, L.; Rio, M.; Willatt, L.; Fiegler, H.; Firth, H.; Sanlaville, D.; Winter, R.; Colleaux, L.; et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J. Med. Genet. 2004, 41, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devriendt, K.; Vermeesch, J.R. Chromosomal phenotypes and submicroscopic abnormalities. Hum. Genom. 2004, 1, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Menten, B.; Maas, N.; Thienpont, B.; Buysse, K.; Vandesompele, J.; Melotte, C.; de Ravel, T.; Van Vooren, S.; Balikova, I.; Backx, L.; et al. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: A new series of 140 patients and review of published reports. J. Med. Genet. 2006, 43, 625–633. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Southard, A.E.; Edelmann, L.J.; Gelb, B.D. Role of copy number variants in structural birth defects. Pediatrics 2012, 129, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Buysse, K.; Delle Chiaie, B.; Van Coster, R.; Loeys, B.; De Paepe, A.; Mortier, G.; Speleman, F.; Menten, B. Challenges for CNV interpretation in clinical molecular karyotyping: Lessons learned from a 1001 sample experience. Eur. J. Med. Genet. 2009, 52, 398–403. [Google Scholar] [CrossRef]
- Gijsbers, A.C.; Schoumans, J.; Ruivenkamp, C.A. Interpretation of array comparative genome hybridization data: A major challenge. Cytogenet. Genome Res. 2011, 135, 222–227. [Google Scholar] [CrossRef]
- Breckpot, J.; Thienpont, B.; Arens, Y.; Tranchevent, L.C.; Vermeesch, J.R.; Moreau, Y.; Gewillig, M.; Devriendt, K. Challenges of interpreting copy number variation in syndromic and non-syndromic congenital heart defects. Cytogenet Genome Res. 2011, 135, 251–259. [Google Scholar] [CrossRef]
- Nowakowska, B. Clinical interpretation of copy number variants in the human genome. J. Appl. Genet. 2017, 58, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.T.; Hu, J.W.; Yin, R.; Xu, L. Long noncoding RNA: An emerging paradigm of cancer research. Tumour Biol. 2013, 34, 613–620. [Google Scholar] [CrossRef]
- Valton, A.L.; Dekker, J. TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev. 2016, 36, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [Green Version]
- Spielmann, M.; Mundlos, S. Looking beyond the genes: The role of non-coding variants in human disease. Hum. Mol. Genet. 2016, 25, R157–R165. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Mertens, L.; de Ravel, T.; Eyskens, B.; Boshoff, D.; Maas, N.; Fryns, J.P.; Gewillig, M.; Vermeesch, J.R.; Devriendt, K. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur. Heart J. 2007, 28, 2778–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, F.; Larsen, L.A.; Zhang, L.; Tümer, Z.; Tommerup, N.; Chen, W.; Jacobsen, J.R.; Schubert, M.; Jurkatis, J.; Tzschach, A.; et al. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J. Med. Genet. 2008, 45, 704–709. [Google Scholar] [CrossRef]
- Richards, A.A.; Santos, L.J.; Nichols, H.A.; Crider, B.P.; Elder, F.F.; Hauser, N.S.; Zinn, A.R.; Garg, V. Cryptic chromosomal abnormalities identified in children with congenital heart disease. Pediatric Res. 2008, 64, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Greenway, S.C.; Pereira, A.C.; Lin, J.C.; DePalma, S.R.; Israel, S.J.; Mesquita, S.M.; Ergul, E.; Conta, J.H.; Korn, J.M.; McCarroll, S.A.; et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009, 41, 931–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breckpot, J.; Thienpont, B.; Peeters, H.; de Ravel, T.; Singer, A.; Rayyan, M.; Allegaert, K.; Vanhole, C.; Eyskens, B.; Vermeesch, J.R.; et al. Array comparative genomic hybridization as a diagnostic tool for syndromic heart defects. J. Pediatric 2010, 156, 810–817.e811–817.e814. [Google Scholar] [CrossRef] [PubMed]
- Soemedi, R.; Wilson, I.J.; Bentham, J.; Darlay, R.; Töpf, A.; Zelenika, D.; Cosgrove, C.; Setchfield, K.; Thornborough, C.; Granados-Riveron, J.; et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 2012, 91, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahed, A.C.; Gelb, B.D.; Seidman, J.G.; Seidman, C.E. Genetics of congenital heart disease: The glass half empty. Circ. Res. 2013, 112, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Andersen, T.A.; Troelsen Kde, L.; Larsen, L.A. Of mice and men: Molecular genetics of congenital heart disease. Cell. Mol. Life Sci. 2014, 71, 1327–1352. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Picker, J.; Zheng, Z.; Zhang, X.; Wang, J.; Hisama, F.; Brown, D.W.; Mullen, M.P.; Harris, D.; Stoler, J.; et al. Chromosome microarray testing for patients with congenital heart defects reveals novel disease causing loci and high diagnostic yield. BMC Genom. 2014, 15, 1127. [Google Scholar] [CrossRef] [Green Version]
- Menten, B.; Pattyn, F.; De Preter, K.; Robbrecht, P.; Michels, E.; Buysse, K.; Mortier, G.; De Paepe, A.; van Vooren, S.; Vermeesch, J.; et al. arrayCGHbase: An analysis platform for comparative genomic hybridization microarrays. BMC Bioinform. 2005, 6, 124. [Google Scholar] [CrossRef] [Green Version]
- Sante, T.; Vergult, S.; Volders, P.J.; Kloosterman, W.P.; Trooskens, G.; De Preter, K.; Dheedene, A.; Speleman, F.; De Meyer, T.; Menten, B. ViVar: A comprehensive platform for the analysis and visualization of structural genomic variation. PLoS ONE 2014, 9, e113800. [Google Scholar] [CrossRef] [Green Version]
- Raman, L.; Dheedene, A.; De Smet, M.; Van Dorpe, J.; Menten, B. WisecondorX: Improved copy number detection for routine shallow whole-genome sequencing. Nucleic Acids Res. 2019, 47, 1605–1614. [Google Scholar] [CrossRef]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen--the Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volders, P.J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visel, A.; Minovitsky, S.; Dubchak, I.; Pennacchio, L.A. VISTA Enhancer Browser--a database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35, D88–D92. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Spector, J.D.; Wiita, A.P. ClinTAD: A tool for copy number variant interpretation in the context of topologically associated domains. J. Hum. Genet. 2019, 64, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascenção, K.; Rummel, C.; Ovchinnikova, S.; et al. Gene expression across mammalian organ development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The Human Transcription Factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnside, R.D.; Pasion, R.; Mikhail, F.M.; Carroll, A.J.; Robin, N.H.; Youngs, E.L.; Gadi, I.K.; Keitges, E.; Jaswaney, V.L.; Papenhausen, P.R.; et al. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: A susceptibility region for neurological dysfunction including developmental and language delay. Hum. Genet. 2011, 130, 517–528. [Google Scholar] [CrossRef]
- Vanlerberghe, C.; Petit, F.; Malan, V.; Vincent-Delorme, C.; Bouquillon, S.; Boute, O.; Holder-Espinasse, M.; Delobel, B.; Duban, B.; Vallee, L.; et al. 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: A series of 52 patients. Eur. J. Med. Genet. 2015, 58, 140–147. [Google Scholar] [CrossRef]
- Hannes, F.D.; Sharp, A.J.; Mefford, H.C.; de Ravel, T.; Ruivenkamp, C.A.; Breuning, M.H.; Fryns, J.P.; Devriendt, K.; Van Buggenhout, G.; Vogels, A.; et al. Recurrent reciprocal deletions and duplications of 16p13.11: The deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J. Med. Genet. 2009, 46, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Riley, K.N.; Catalano, L.M.; Bernat, J.A.; Adams, S.D.; Martin, D.M.; Lalani, S.R.; Patel, A.; Burnside, R.D.; Innis, J.W.; Rudd, M.K. Recurrent deletions and duplications of chromosome 2q11.2 and 2q13 are associated with variable outcomes. Am. J. Med Genet. Part A 2015, 167A, 2664–2673. [Google Scholar] [CrossRef] [PubMed]
- van Bon, B.W.; Balciuniene, J.; Fruhman, G.; Nagamani, S.C.; Broome, D.L.; Cameron, E.; Martinet, D.; Roulet, E.; Jacquemont, S.; Beckmann, J.S.; et al. The phenotype of recurrent 10q22q23 deletions and duplications. Eur. J. Hum. Genet. EJHG 2011, 19, 400–408. [Google Scholar] [CrossRef]
- MacDonald, J.R.; Ziman, R.; Yuen, R.K.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, 986–992. [Google Scholar] [CrossRef] [Green Version]
- Beunders, G.; van de Kamp, J.; Vasudevan, P.; Morton, J.; Smets, K.; Kleefstra, T.; de Munnik, S.A.; Schuurs-Hoeijmakers, J.; Ceulemans, B.; Zollino, M.; et al. A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. J. Med. Genet. 2016, 53, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Szot, J.O.; Cuny, H.; Blue, G.M.; Humphreys, D.T.; Ip, E.; Harrison, K.; Sholler, G.F.; Giannoulatou, E.; Leo, P.; Duncan, E.L.; et al. A Screening Approach to Identify Clinically Actionable Variants Causing Congenital Heart Disease in Exome Data. Circ. Genom. Precis. Med. 2018, 11, e001978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batsukh, T.; Pieper, L.; Koszucka, A.M.; von Velsen, N.; Hoyer-Fender, S.; Elbracht, M.; Bergman, J.E.; Hoefsloot, L.H.; Pauli, S. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum. Mol. Genet. 2010, 19, 2858–2866. [Google Scholar] [CrossRef] [Green Version]
- Shanks, M.O.; Lund, L.M.; Manni, S.; Russell, M.; Mauban, J.R.; Bond, M. Chromodomain helicase binding protein 8 (Chd8) is a novel A-kinase anchoring protein expressed during rat cardiac development. PLoS ONE 2012, 7, e46316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.C.; Tsai, R.Y.; Chung, T.H. Role of catenins in the development of gap junctions in rat cardiomyocytes. J. Cell. Biochem. 2003, 88, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Toyofuku, T.; Yabuki, M.; Otsu, K.; Kuzuya, T.; Hori, M.; Tada, M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J. Biol. Chem. 1998, 273, 12725–12731. [Google Scholar] [CrossRef] [Green Version]
- Rhee, D.Y.; Zhao, X.Q.; Francis, R.J.; Huang, G.Y.; Mably, J.D.; Lo, C.W. Connexin 43 regulates epicardial cell polarity and migration in coronary vascular development. Development 2009, 136, 3185–3193. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Vranckx, R.; Khau Van Kien, P.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.M.; Brunotte, F.; et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef]
- Lu, C.C.; Liu, M.M.; Clinton, M.; Culshaw, G.; Argyle, D.J.; Corcoran, B.M. Developmental pathways and endothelial to mesenchymal transition in canine myxomatous mitral valve disease. Vet. J. 2015, 206, 377–384. [Google Scholar] [CrossRef]
- Lorenz, K.; Schmitt, J.P.; Schmitteckert, E.M.; Lohse, M.J. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat. Med. 2009, 15, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Ranish, J.A.; Hauschka, S.D. Quantitative proteomic identification of MAZ as a transcriptional regulator of muscle-specific genes in skeletal and cardiac myocytes. Mol. Cell. Biol. 2008, 28, 6521–6535. [Google Scholar] [CrossRef] [Green Version]
- Jaber, N.; Dou, Z.; Chen, J.S.; Catanzaro, J.; Jiang, Y.P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J.; et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, L.; Schwengberg, S.; Denecke, B. Transcriptome analysis in cardiomyocyte-specific differentiation of murine embryonic stem cells reveals transcriptional regulation network. Gene Expr. Patterns 2014, 16, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Singh, A.R.; Zhao, Y.; Du, T.; Huang, Y.; Wan, X.; Mukhopadhyay, D.; Wang, Y.; Wang, N.; Zhang, P. TRIM28 regulates sprouting angiogenesis through VEGFR-DLL4-Notch signaling circuit. FASEB J. 2020, 34, 14710–14724. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.I.; Wang, Y.T.; Wang, Y.S.; Wu, C.Y.; Li, C.C. Involvement of BIG1 and BIG2 in regulating VEGF expression and angiogenesis. FASEB J. 2019, 33, 9959–9973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrington, C.B.; Dowse, B.R.; Bleyl, S.B.; Bowles, N.E. Non-synonymous variants in pre-B cell leukemia homeobox (PBX) genes are associated with congenital heart defects. Eur. J. Med. Genet. 2012, 55, 235–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sailani, M.R.; Makrythanasis, P.; Valsesia, A.; Santoni, F.A.; Deutsch, S.; Popadin, K.; Borel, C.; Migliavacca, E.; Sharp, A.J.; Duriaux Sail, G.; et al. The complex SNP and CNV genetic architecture of the increased risk of congenital heart defects in Down syndrome. Genome Res. 2013, 23, 1410–1421. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Ronemus, M.; Yamrom, B.; Lee, Y.H.; Leotta, A.; Kendall, J.; Marks, S.; Lakshmi, B.; Pai, D.; Ye, K.; et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011, 70, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburton, D.; Ronemus, M.; Kline, J.; Jobanputra, V.; Williams, I.; Anyane-Yeboa, K.; Chung, W.; Yu, L.; Wong, N.; Awad, D.; et al. The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum. Genet. 2014, 133, 11–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Lupski, J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015, 24, R102–R110. [Google Scholar] [CrossRef] [Green Version]
- D’Haene, E.; Vergult, S. Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genet. Med. 2021, 23, 34–46. [Google Scholar] [CrossRef]
- Melo, U.S.; Schöpflin, R.; Acuna-Hidalgo, R.; Mensah, M.A.; Fischer-Zirnsak, B.; Holtgrewe, M.; Klever, M.; Türkmen, S.; Heinrich, V.; Pluym, I.D.; et al. Hi-C Identifies Complex Genomic Rearrangements and TAD-Shuffling in Developmental Diseases. Am. J. Hum. Genet. 2020, 106, 872–884. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| CNV-US | Known Dosage Sensitive CNV Regions | Interpretation | ||
|---|---|---|---|---|
| Chr1:145388355-145832995dup | 1q21.1 recurrent (TAR) region (BP2-BP3, proximal) (ISCA-37428) | 99.5% overlap | TS score 1 | No definite link with CHD |
| Chr2:60998688-61093639dup | 2p15p16.1 region (ISCA-37408) | 2.8% overlap | TS score 1 | No link with CHD |
| Chr2:112650001-112740000del | 2q13 recurrent region (ISCA-37496) | 5.3% overlap | HI score 2 | Linked to CHD; excl CHD candidate gene BCL2L11 |
| Chr5:1005001-1290000dup | 5p15 terminal (Cri du chat syndrome) region (ISCA-37390) | 2.5% overlap | TS score 2 | No clear link with CHD |
| Chr10:84054763-84073574del | 10q22.3q23.2 recurrent region (LCR-3/4-flanked) (ISCA-37424) | 0.3% overlap | HI score 3 | Linked to CHD; excl CHD candidate gene BMPR1A |
| Chr10:88004601-88065186del | 10q22.3q23.2 recurrent region (LCR-3/4-flanked) (ISCA-37424) | 0.9% overlap | HI score 3 | Linked to CHD; excl CHD candidate gene BMPR1A |
| Chr15:22755001-23085000del | 15q11.2 recurrent region (BP1-BP2) (ISCA-37448) | 97.7% overlap | HI score 2 | Link with CHD under debate |
| Chr15:22765628-23167699del | 15q11.2 recurrent region (BP1-BP2) (ISCA-37448) | 100% overlap | HI score 2 | Link with CHD under debate |
| Chr15:22765628-23208842dup | 15q11.2 recurrent region (BP1-BP2) (ISCA-37448) | 100% overlap | TS score 40 | - |
| Chr15:22765628-23208842dup | 15q11.2 recurrent region (BP1-BP2) (ISCA-37448) | 100% overlap | TS score 40 | - |
| Chr15:24005491-24470088dup | 15q11q13 recurrent (PWS/AS) region (BP2-BP3, Class 1) (ISCA-37404) | 10.0% overlap | TS score 3 | No clear link with CHD |
| Chr16:14968855-16292181del | 16p13.11 recurrent region (BP2-BP3) (ISCA-37415) | 100% overlap | HI score 3 | Link with CHD under debate |
| Chr16:29656684-30197290del | 16p11.2 recurrent region (proximal, BP4-BP5) (ISCA-37400) | 98.3% overlap | HI score 3 | Link with CHD under debate |
| Chr17:15257416-15482813dup | 17p12 recurrent (HNPP/CMT1A) region (ISCA-37436) | 12.5% overlap | TS score 3 | No link with CHD |
| Chr17:18148172-18662098dup | 17p11.2 recurrent (SMS/PLS) region (ISCA-37418) | 15.1% overlap | TS score 3 | Linked to CHD; excl CHD candidate gene RAI1 |
| Chr17:58372095-58588996dup | 17q23.1q23.2 recurrent region (ISCA-37501) | 10.0% overlap | TS score 2 | Link with CHD unclear |
| ChrX:6467006-8131751del * | Xp22.31 recurrent region (ISCA-37417) | 99.3% overlap | HI score 3 | No link with CHD |
| ChrX:6467006-8131751dup * | Xp22.31 recurrent region (ISCA-37417) | 99.3% overlap | TS score 40 | - |
| ChrX:7515001-8130000dup | Xp22.31 recurrent region (ISCA-37417) | 36.5% overlap | TS score 40 | - |
| CNV-US | Smallest Region of Overlap (SRO) (Protein-Coding Genes) | Interpretation |
|---|---|---|
| Chr4:135455435-137460949dup Chr4:135700662-135829279dup | Chr4:135700662-135829279 (no protein-coding genes) | One nearly identical duplication in Decipher, classified ‘likely benign’. Two partially overlapping duplications in the same boundaries in DGV. |
| Chr7:11221210-12462629dup Chr7:12300173-12462629del | Chr7:12300173-12462629 (VWDE) | Two comparable deletions in Decipher, both classified ‘CNV-US’. Multiple comparable CNVs in DGV. |
| Chr9:195001-405000dup Chr9:210001-540000dup | Chr9:210001-405000 (DOCK8) | Multiple comparable CNVs in Decipher, most classified ‘CNV-US’. Multiple comparable CNVs in DGV. |
| Chr9:107409506-107729796dup Chr9:107409509-107769094dup | Chr9:107409509-107729769 (OR13D1, NIPSNAP3A, NIPSNAP3B, ABCA1) | Four comparable duplications in Decipher, three classified ‘CNV-US’, one classified ‘likely pathogenic’. No comparable CNVs in DGV. |
| Chr15:22755001-23085000del Chr15:22765628-23167699del Chr15:22765628-23208842dup Chr15:22765628-23208842dup | Chr15:22765628-23085000 (CYFIP1, NIPA1,NIPA2, TUBGCP5) | Known ClinGen Pathogenic and Dosage Sensitive CNV region (ISCA-37448). |
| Chr21:43014314-48090258del Chr21:47591379-47671404dup | Chr21:47591379-47671404 (SPATC1L, LSS, MCM3AP) | No comparable CNVs in Decipher. No comparable CNVs in DGV. |
| ChrX:61091-437220del * ChrY:61091-819199del | ChrX/Y:61091-437220 (PLCXD1, GTPBP6, PPP2R3B) | No comparable CNVs in Decipher. No comparable CNVs in DGV. |
| ChrX:6467006-8131751dup * ChrX:6467006-8131751del * ChrX:7515001-8130000dup | ChrX:7515001-8130000 (VCX, PNPLA4) | Multiple comparable CNVs in Decipher, all but one classified ‘CNV-US’. Few comparable CNVs in DGV. |
| ChrX:130610000-130950000dup ChrX:130631863-130960558dup * | ChrX:130631863-130950000 (OR13H1) | Multiple comparable CNVs in Decipher, most classified ‘CNV-US’ or ‘benign’. No comparable CNVs in DGV. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meerschaut, I.; Vergult, S.; Dheedene, A.; Menten, B.; De Groote, K.; De Wilde, H.; Muiño Mosquera, L.; Panzer, J.; Vandekerckhove, K.; Coucke, P.J.; et al. A Reassessment of Copy Number Variations in Congenital Heart Defects: Picturing the Whole Genome. Genes 2021, 12, 1048. https://doi.org/10.3390/genes12071048
Meerschaut I, Vergult S, Dheedene A, Menten B, De Groote K, De Wilde H, Muiño Mosquera L, Panzer J, Vandekerckhove K, Coucke PJ, et al. A Reassessment of Copy Number Variations in Congenital Heart Defects: Picturing the Whole Genome. Genes. 2021; 12(7):1048. https://doi.org/10.3390/genes12071048
Chicago/Turabian StyleMeerschaut, Ilse, Sarah Vergult, Annelies Dheedene, Björn Menten, Katya De Groote, Hans De Wilde, Laura Muiño Mosquera, Joseph Panzer, Kristof Vandekerckhove, Paul J. Coucke, and et al. 2021. "A Reassessment of Copy Number Variations in Congenital Heart Defects: Picturing the Whole Genome" Genes 12, no. 7: 1048. https://doi.org/10.3390/genes12071048