
The Phenotypic Spectrum of 15q13.3 Region Duplications: Report of 5 Patients

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gonzalez-Mantilla, A.J.; Moreno-De-Luca, A.; Ledbetter, D.H.; Martin, C.L. A Cross-Disorder Method to Identify Novel Candidate Genes for Developmental Brain Disorders. JAMA Psychiatry 2016, 73, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takumi, T.; Tamada, K. CNV biology in neurodevelopmental disorders. Curr. Opin. Neurobiol. 2018, 48, 183–192. [Google Scholar] [CrossRef]
- Makoff, A.J.; Flomen, R.H. Detailed analysis of 15q11–q14 sequence corrects errors and gaps in the public access sequence to fully reveal large segmental duplications at breakpoints for Prader-Willi, Angelman, and inv dup(15) syndromes. Genome Biol. 2007, 8, R114. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.J.; Mefford, H.C.; Li, K.; Baker, C.; Skinner, C.; Stevenson, R.E.; Schroer, R.J.; Novara, F.; De Gregori, M.; Ciccone, R.; et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008, 40, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Shen, Y.; Weiss, L.A.; Korn, J.; Anselm, I.; Bridgemohan, C.; Cox, G.F.; Dickinson, H.; Gentile, J.; Harris, D.J.; et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J. Med. Genet. 2009, 46, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Gillentine, M.A.; Berry, L.N.; Goin-Kochel, R.P.; Ali, M.A.; Ge, J.; Guffey, D.; Rosenfeld, J.A.; Hannig, V.; Bader, P.; Proud, M.; et al. The Cognitive and Behavioral Phenotypes of Individuals with CHRNA7 Duplications. J. Autism Dev. Disord. 2017, 47, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillentine, M.A.; Schaaf, C.P. The human clinical phenotypes of altered CHRNA7 copy number. Biochem. Pharmacol. 2015, 97, 352–362. [Google Scholar] [CrossRef] [Green Version]
- Online Mendelian Inherintance in Man. Available online: https://omim.org/ (accessed on 4 April 2021).
- van Bon, B.W.; Mefford, H.C.; Menten, B.; Koolen, D.A.; Sharp, A.J.; Nillesen, W.M.; Innis, J.W.; de Ravel, T.J.; Mercer, C.L.; Fichera, M.; et al. Further delineation of the 15q13 microdeletion and duplication syndromes: A clinical spectrum varying from non-pathogenic to a severe outcome. J. Med. Genet. 2009, 46, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Lowther, C.; Costain, G.; Stavropoulos, D.J.; Melvin, R.; Silversides, C.K.; Andrade, D.M.; So, J.; Faghfoury, H.; Lionel, A.C.; Marshall, C.R.; et al. Delineating the 15q13.3 microdeletion phenotype: A case series and comprehensive review of the literature. Genet. Med. 2015, 17, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Endris, V.; Hackmann, K.; Neuhann, T.M.; Grasshoff, U.; Bonin, M.; Haug, U.; Hahn, G.; Schallner, J.C.; Schröck, E.; Tinschert, S.; et al. Homozygous loss of CHRNA7 on chromosome 15q13.3 causes severe encephalopathy with seizures and hypotonia. Am. J. Med. Genet. Part A 2010, 152, 2908–2911. [Google Scholar] [CrossRef]
- Lepichon, J.B.; Bittel, D.C.; Graf, W.D.; Yu, S.A. 15q13.3 homozygous microdeletion associated with a severe neurodevelopmental disorder suggests putative functions of the TRPM1, CHRNA7, and other homozygously deleted genes. Am. J. Med. Genet. Part A 2010, 152, 1300–1304. [Google Scholar] [CrossRef]
- Masurel-Paulet, A.; Andrieux, J.; Callier, P.; Cuisset, J.M.; Le Caignec, C.; Holder, M.; Thauvin-Robinet, C.; Doray, B.; Flori, E.; Alex-Cordier, M.P.; et al. Delineation of 15q13.3 microdeletions. Clin. Genet. 2010, 78, 149–161. [Google Scholar] [CrossRef]
- Szafranski, P.; Schaaf, C.P.; Person, R.E.; Gibson, I.B.; Xia, Z.; Mahadevan, S.; Wiszniewska, J.; Bacino, C.A.; Lalani, S.; Potocki, L.; et al. Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: Benign or pathological? Hum. Mutat. 2010, 31, 840–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, N.M.; Franke, B.; Mick, E.; Anney, R.J.; Freitag, C.M.; Gill, M.; Thapar, A.; O’Donovan, M.C.; Owen, M.J.; Holmans, P.; et al. Genome-wide analysis of copy number variants in attention deficit hyperactivity disorder: The role of rare variants and duplications at 15q13.3. Am. J. Psychiatry 2012, 169, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Melchior, L.; Bertelsen, B.; Debes, N.M.; Groth, C.; Skov, L.; Mikkelsen, J.D.; Brøndum-Nielsen, K.; Tümer, Z. Microduplication of 15q13.3 and Xq21.31 in a family with Tourette syndrome and comorbidities. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Szatkiewicz, J.P.; O’Dushlaine, C.; Chen, G.; Chambert, K.; Moran, J.L.; Neale, B.M.; Fromer, M.; Ruderfer, D.; Akterin, S.; Bergen, S.E.; et al. Copy number variation in schizophrenia in Sweden. Mol. Psychiatry 2014, 19, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beal, J.C. Case report: Neuronal migration disorder associated with chromosome 15q13.3 duplication in a boy with autism and seizures. J. Child Neurol. 2014, 29, NP186–NP188. [Google Scholar] [CrossRef] [PubMed]
- Bacchelli, E.; Battaglia, A.; Cameli, C.; Lomartire, S.; Tancredi, R.; Thomson, S.; Sutcliffe, J.S.; Maestrini, E. Analysis of CHRNA7 rare variants in autism spectrum disorder susceptibility. Am. J. Med. Genet. Part A 2015, 167, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Pettigrew, K.A.; Reeves, E.; Leavett, R.; Hayiou-Thomas, M.E.; Sharma, A.; Simpson, N.H.; Simpson, N.H.; Martinelli, A.; Thompson, P.; Hulme, C.; et al. Copy Number Variation Screen Identifies a Rare De Novo Deletion at Chromosome 15q13.1-13.3 in a Child with Language Impairment. PLoS ONE 2015, 10, e0134997. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Gochman, P.; Broadnax, D.D.; Rapoport, J.L.; Ahn, K. 15q13.3 Duplication in Two Patients with Childhood-Onset Schizophrenia. Am. J. Med. Genet. Part B 2016, 171, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Soler-Alfonso, C.; Carvalho, C.M.; Ge, J.; Roney, E.K.; Bader, P.I.; Kolodziejska, K.E.; Miller, R.M.; Lupski, J.R.; Stankiewicz, P.; Cheung, S.W.; et al. CHRNA7 triplication associated with cognitive impairment and neuropsychiatric phenotypes in a three-generation pedigree. Eur. J. Hum. Genet. 2014, 22, 1071–1076. [Google Scholar] [CrossRef] [Green Version]
- Wiśniowiecka-Kowalnik, B.; Kastory-Bronowska, M.; Bartnik, M.; Derwińska, K.; Dymczak-Domini, W.; Szumbarska, D.; Ziemka, E.; Szczałuba, K.; Sykulski, M.; Gambin, T.; et al. Application of custom-designed oligonucleotide array CGH in 145 patients with autistic spectrum disorders. Eur. J. Hum. Genet. 2013, 21, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Coe, B.P.; Witherspoon, K.; Rosenfeld, J.A.; van Bon, B.W.; Vulto-van Silfhout, A.T.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.E.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.L.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Roza, E.; Streață, I.; Șoșoi, S.; Burada, F.; Puiu, M.; Ioana, M.; Teleanu, R.I. A 14q31.1–q32.11 deletion case: Genotype—neurological phenotype correlations in 14q interstitial deletion syndrome. Rom. Biotechnol. Lett. 2020, 25, 1677–1682. [Google Scholar] [CrossRef]
- Budisteanu, M.; Tutulan-Cunita, A.; Focsa, I.O.; Papuc, S.M.; Arghir, A. First-Tier Array CGH in Clinically Variable Entity Diagnosis: 22q13.3 Deletion Syndrome in Chromosomal Abnormalities; Çelik, T.A., Dey, S., Eds.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Sinkus, M.L.; Graw, S.; Freedman, R.; Ross, R.G.; Lester, H.A.; Leonard, S. The human CHRNA7 and CHRFAM7A genes: A review of the genetics, regulation and function. Neuropharmacology 2015, 96, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbig, I.; Mefford, H.C.; Sharp, A.J.; Guipponi, M.; Fichera, M.; Franke, A.; Muhle, H.; Kovel, C.D.; Baker, C.; Spiczak, S.; et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009, 41, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Smajlagić, D.; Lavrichenko, K.; Berland, S.; Helgeland, Ø.; Knudsen, G.P.; Vaudel, M.; Haavik, J.; Knappskog, P.M.; Njølstad, P.R.; Houge, G.; et al. Population prevalence and inheritance pattern of recurrent CNVs associated with neurodevelopmental disorders in 12,252 newborns and their parents. Eur. J. Hum. Genet. 2021, 29, 205–215. [Google Scholar] [CrossRef]
- Hoyle, E.; Genn, R.F.; Fernandes, C.; Stolerman, I.P. Impaired performance of alpha7 nicotinic receptor knockout mice in the five-choice serial reaction time task. Psychopharmacology 2006, 189, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Young, J.W.; Crawford, N.; Kelly, J.S.; Kerr, L.E.; Marston, H.M.; Spratt, C.; Finlayson, K.; Sharkey, J. Impaired attention is central to the cognitive deficits observed in alpha 7 deficient mice. Eur. Neuropsychopharmacol. 2007, 17, 145–155. [Google Scholar] [CrossRef]
- Young, J.W.; Meves, J.M.; Tarantino, I.S.; Caldwell, S.; Geyer, M.A. Delayed procedural learning in α7-nicotinic acetylcholine receptor knockout mice. Genes Brain Behav. 2011, 10, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Chen, W.; Yang, H.; Xue, M.; Schaaf, C.P. Chrna7 deficient mice manifest no consistent neuropsychiatric and behavioral phenotypes. Sci. Rep. 2017, 7, 39941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillentine, M.A.; Yin, J.; Bajic, A.; Zhang, P.; Cummock, S.; Kim, J.J.; Schaaf, C.P. Functional Consequences of CHRNA7 Copy-Number Alterations in Induced Pluripotent Stem Cells and Neural Progenitor Cells. Am. J. Hum. Genet. 2017, 101, 874–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meganathan, K.; Prakasam, R.; Baldridge, D.; Gontarz, P.; Zhang, B.; Urano, F.; Bonni, A.; Constantino, J.N.; Kroll, K.L. Alterations in neuronal physiology, development, and function associated with a common duplication of chromosome 15 involving CHRNA7. BioRxiy 2020. [Google Scholar] [CrossRef] [Green Version]
- Moreno-De-Luca, D.; Sanders, S.J.; Willsey, A.J.; Mulle, J.G.; Lowe, J.K.; Geschwind, D.H.; State, M.W.; Martin, C.L.; Ledbetter, D.H. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry 2013, 18, 1090–1095. [Google Scholar] [CrossRef] [Green Version]
- Boronat, S.; Mehan, W.A.; Shaaya, E.A.; Thibert, R.L.; Caruso, P. Hippocampal abnormalities in magnetic resonance imaging (MRI) of 15q duplication syndromes. J. Child Neurol. 2015, 30, 333–338. [Google Scholar] [CrossRef]


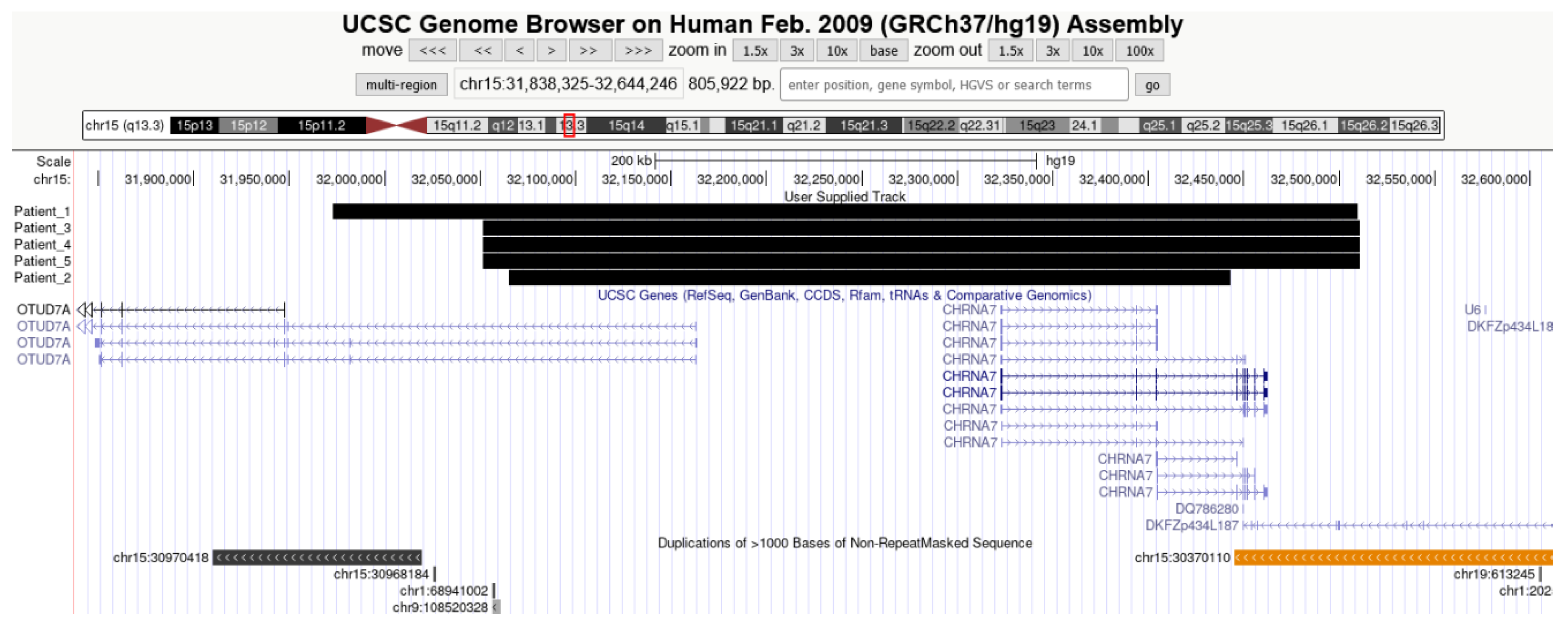{kind=link}
| Pt No. | Gender | Age at Presentation | Dysmorphic Features | ID/DD (IQ/DQ) | Speech Delay | ASD | Feeding Difficulties | Hypotonia | Epilepsy | EEG Anomalies | Brain IRM Anomalies |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | M | 26 months → 9 years | − | + (51 → 45) | + | + | − | + | − | + | + |
| 2. | M | 15 years | − | + (40) | + | + | − | + | + | + | + |
| 3. | M | 2 yrs 4 months | − | + (39) | + | + | − | + | − | NA | − |
| 4. | F | 4 years | − | − (73) | + | + | − | + | − | NA | NA |
| 5. | M | 10 years | − | − (90) | + | + | − | + | − | + | − |
| Patient No. | Size | ISCN Formula | OMIM Genes |
|---|---|---|---|
| 1. | 537,280 | arr[GRCh37] 15q13.3(31864691x2,31972646_32509926x3,32631629x2) | OTUD7A, CHRNA7 |
| 2. | 378,087 | arr[GRCh37] 15q13.3(31972706x2,32065000_32443078x3,32445199x2) | OTUD7A, CHRNA7 |
| 3. | 459,630 | arr[GRCh37] 15q13.3(32021793x2,32051233_32510863x3,32914080x2) | OTUD7A, CHRNA7 |
| 4. | 459,630 | arr[GRCh37] 15q13.3(32021793x2,32051233_32510863x3,32914080x2) | OTUD7A, CHRNA7 |
| 5. | 459,630 | arr[GRCh37] 15q13.3(32021793x2,32051233_32510863x3,32914080x2) | OTUD7A, CHRNA7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budisteanu, M.; Papuc, S.M.; Streata, I.; Cucu, M.; Pirvu, A.; Serban-Sosoi, S.; Erbescu, A.; Andrei, E.; Iliescu, C.; Ioana, D.; et al. The Phenotypic Spectrum of 15q13.3 Region Duplications: Report of 5 Patients. Genes 2021, 12, 1025. https://doi.org/10.3390/genes12071025
Budisteanu M, Papuc SM, Streata I, Cucu M, Pirvu A, Serban-Sosoi S, Erbescu A, Andrei E, Iliescu C, Ioana D, et al. The Phenotypic Spectrum of 15q13.3 Region Duplications: Report of 5 Patients. Genes. 2021; 12(7):1025. https://doi.org/10.3390/genes12071025
Chicago/Turabian StyleBudisteanu, Magdalena, Sorina Mihaela Papuc, Ioana Streata, Mihai Cucu, Andrei Pirvu, Simona Serban-Sosoi, Alina Erbescu, Emanuela Andrei, Catrinel Iliescu, Doina Ioana, and et al. 2021. "The Phenotypic Spectrum of 15q13.3 Region Duplications: Report of 5 Patients" Genes 12, no. 7: 1025. https://doi.org/10.3390/genes12071025