
Two Decades after Mandibuloacral Dysplasia Discovery: Additional Cases and Comprehensive View of Disease Characteristics

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Approval
2.2. Screening of a Gene Panel by Next-Generation Sequencing
2.3. Sanger Sequencing
3. Case Reports
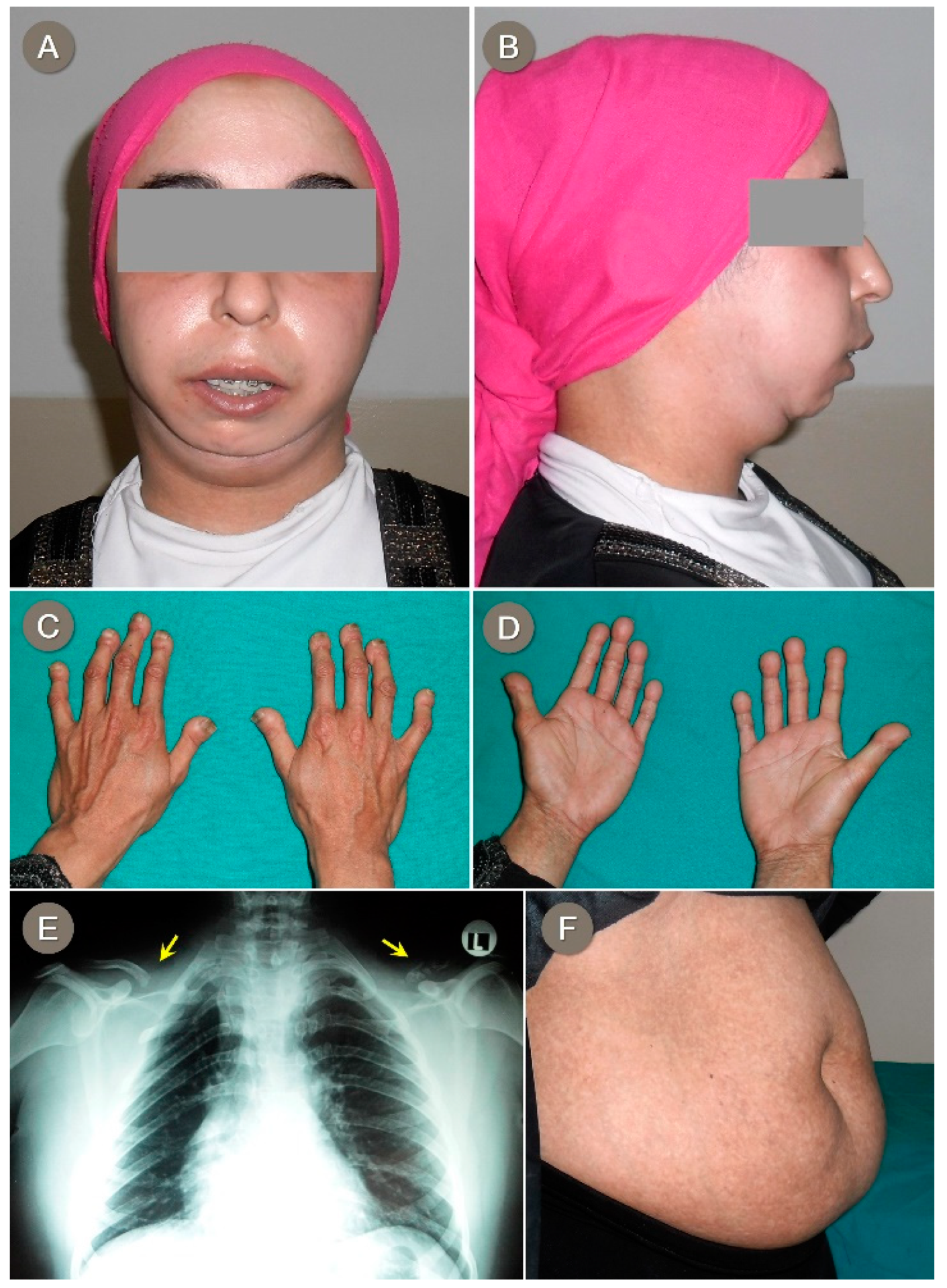
3.1. Proband 1
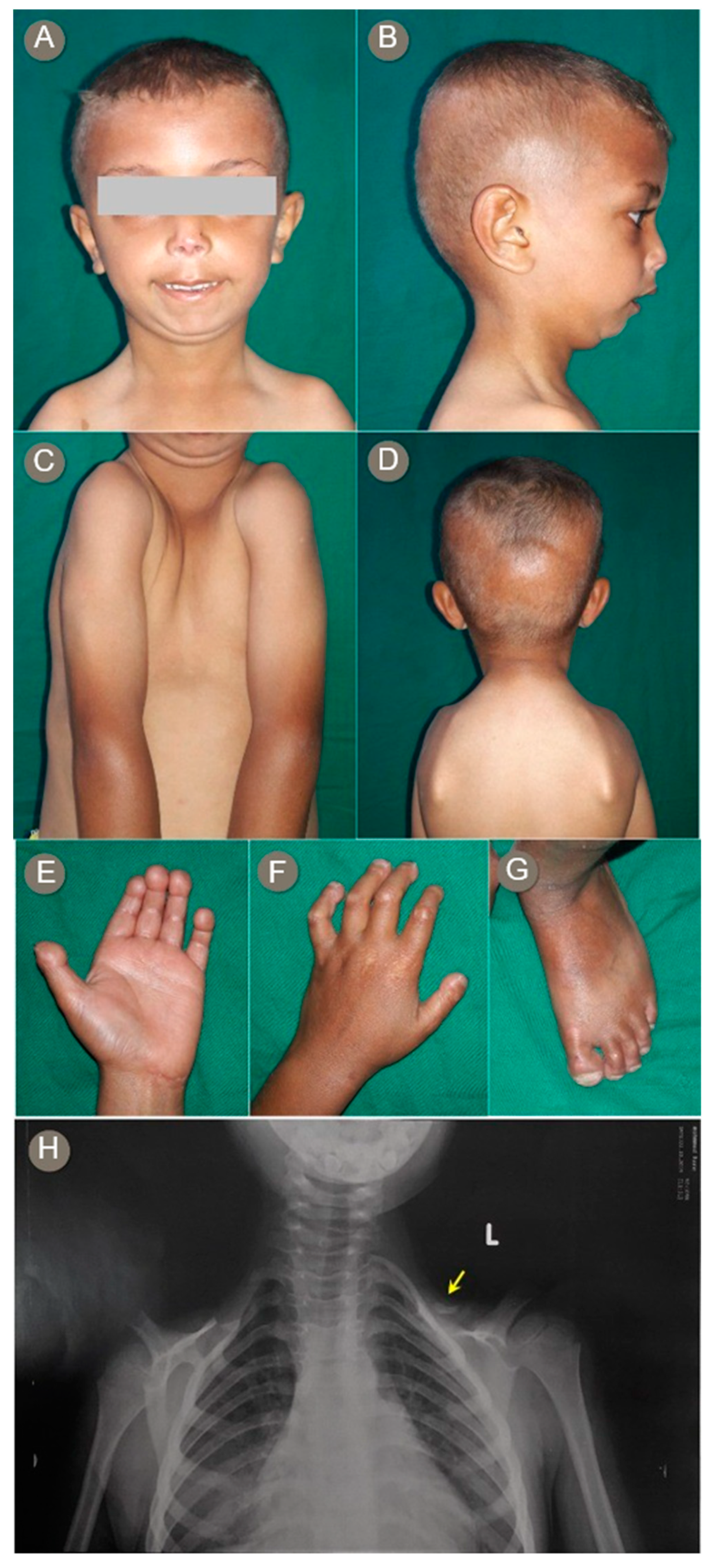
3.2. Proband 2
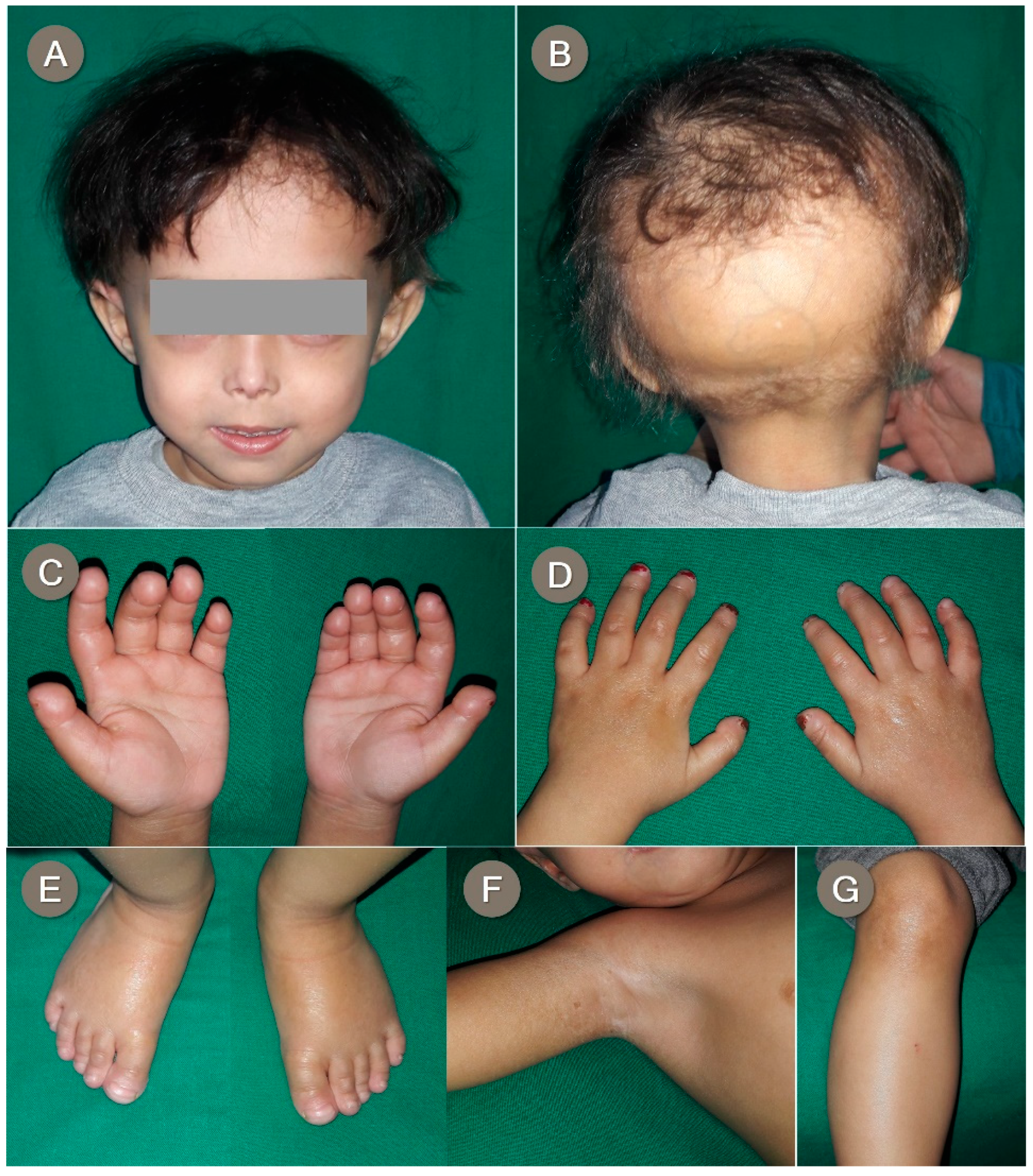
3.3. Proband 3
3.4. Comprehensive Review of the Literature
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, L.W.; Radebaugh, J.F.; Rubin, P.; Sensenbrenner, J.A.; Fiorelli, G.; McKusick, V.A. New syndrome manifested by mandibular hypoplasia, acroosteolysis, stiff joints and cutaneous atrophy (mandibuloacral dysplasia) in two unrelated boys. Birth Defects Orig. Artic. Ser. 1971, 7, 291–297. [Google Scholar]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Amer. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.K.; Fryns, J.-P.; Auchus, R.J.; Garg, A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 2003, 12, 1995–2001. [Google Scholar] [CrossRef]
- Elouej, S.; Harhouri, K.; Le Mao, M.; Baujat, G.; Nampoothiri, S.; Kayserili, H.; Menabawy, N.A.; Selim, L.; Paneque, A.L.; Kubisch, C.; et al. Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology. Nat. Commun. 2020, 11, 1–15. [Google Scholar]
- Worman, H.J.; Bonne, G. “Laminopathies”: A wide spectrum of human diseases. Exp. Cell Res. 2007, 313, 2121–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.; Cogulu, O.; Ozkinay, F.; Onay, H.; Agarwal, A.K. A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J. Clin. Endocrinol. Metab. 2005, 90, 5259–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, F.; Gullotta, F.; Columbaro, M.; Filareto, A.; D’Adamo, M.; Vielle, A.; Guglielmi, V.; Nardone, A.M.; Azzolini, V.; Grosso, E.; et al. Compound heterozygosity for mutations in LMNA in a patient with a myopathic and lipodystrophic mandibuloacral dysplasia type A phenotype. J. Clin. Endocrinol. Metab. 2007, 92, 4467–4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Hegele, R.A. LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090). J. Hum. Genet. 2003, 48, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Plasilova, M.; Chattopadhyay, C.; Pal, P.; Schaub, N.A.; Buechner, S.A.; Mueller, H.; Miny, P.; Ghosh, A.; Heinimann, K. Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome. J. Med. Genet. 2004, 41, 609–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstraeten, V.L.; Broers, J.L.; van Steensel, M.A.; Zinn-Justin, S.; Ramaekers, F.C.; Steijlen, P.M.; Kamps, M.; Kuijpers, H.J.; Merckx, D.; Smeets, H.J.; et al. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum. Mol. Genet. 2006, 15, 2509–2522. [Google Scholar] [CrossRef] [Green Version]
- Zirn, B.; Kress, W.; Grimm, T.; Berthold, L.D.; Neubauer, B.; Kuchelmeister, K.; Müller, U.; Hahn, A. Association of homozygous LMNA mutation R471C with new phenotype: Mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy. Am. J. Med.Genet. Part A 2008, 146, 1049–1054. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Kazachkova, I.; Ten, S.; Garg, A. Severe mandibuloacral dysplasia-associated lipodystrophy and progeria in a young girl with a novel homozygous Arg527Cys LMNA mutation. J. Clin. Endocrinol. Metab. 2008, 93, 4617–4623. [Google Scholar] [CrossRef] [Green Version]
- Al-Haggar, M.; Madej-Pilarczyk, A.; Kozlowski, L.; Bujnicki, J.M.; Yahia, S.; Abdel-Hadi, D.; Shams, A.; Ahmad, N.; Hamed, S.; Puzianowska-Kuznicka, M. A novel homozygous p. Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome. Eur. J. Hum. Genet. 2012, 20, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Amr, K.; Ibrahim, M.; El-Kamah, G. Mandibuloacral Dysplasia Mutation Detection in Three Egyptian Families: A Report of a Novel Mutation. Life Sci. J. 2012, 9, 940–944. [Google Scholar]
- Gruenbaum, Y.; Foisner, R. Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Ann. Rev. Biochem. 2015, 84, 131–164. [Google Scholar] [CrossRef]
- Filesi, I.; Gullotta, F.; Lattanzi, G.; D’Apice, M.R.; Capanni, C.; Nardone, A.M.; Columbaro, M.; Scarano, G.; Mattioli, E.; Sabatelli, P.; et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy Physiol. Genomics 2005, 23, 150–158. [Google Scholar]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S.; et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef] [Green Version]
- Camozzi, D.; D’Apice, M.R.; Schena, E.; Cenni, V.; Columbaro, M.; Capanni, C.; Maraldi, N.M.; Squarzoni, S.; Ortolani, M.; Novelli, G.; et al. Altered chromatin organization and SUN2 localization in mandibuloacral dysplasia are rescued by drug treatment. Histochem. Cell Biol. 2012, 138, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cenni, V.; D’Apice, M.R.; Garagnani, P.; Columbaro, M.; Novelli, G.; Franceschi, C.; Lattanzi, G. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res. Rev. 2018, 42, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jéru, I.; Vantyghem, M.-C.; Bismuth, E.; Cervera, P.; Barraud, S.; Auclair, M.; Vatier, C.; Lascols, O.; Savage, D.B.; Vigouroux, C.; et al. Diagnostic challenge in PLIN1-associated Familial Partial Lipodystrophy. J. Clin. Endocrinol. Metab. 2019, 104, 6025–6032. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Simha, V.; Agarwal, A.K.; Oral, E.A.; Fryns, J.P.; Garg, A. Genetic and phenotypic heterogeneity in patients with mandibuloacral dysplasia-associated lipodystrophy. J. Clin. Endocrinol. Metab. 2003, 88, 2821–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.J.; Brown, C.A.; Lupski, J.R.; Potocki, L. Mandibuloacral dysplasia caused by homozygosity for the R527H mutation in lamin A/C. J. Med. Genet. 2003, 40, 854–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garavelli, L.; D’Apice, M.; Rivieri, F.; Bertoli, M.; Wischmeijer, A.; Gelmini, C.; De Nigris, V.; Albertini, E.; Rosato, S.; Virdis, R.; et al. Mandibuloacral dysplasia type A in childhood. Am. J. Med. Genet. Part A 2009, 149, 2258–2264. [Google Scholar] [CrossRef] [PubMed]
- Kosho, T.; Takahashi, J.; Momose, T.; Nakamura, A.; Sakurai, A.; Wada, T.; Yoshida, K.; Wakui, K.; Suzuki, T.; Kasuga, K.; et al. Mandibuloacral dysplasia and a novel LMNA mutation in a woman with severe progressive skeletal changes. Am. J. Med. Genet. A 2007, 143A, 2598–2603. [Google Scholar] [CrossRef]
- Luo, D.Q.; Wang, X.Z.; Meng, Y.; He, D.Y.; Chen, Y.M.; Ke, Z.Y.; Yan, M.; Huang, Y.; Chen, D.F. Mandibuloacral dysplasia type A-associated progeria caused by homozygous LMNA mutation in a family from Southern China. BMC Pediatr. 2014, 14, 256. [Google Scholar] [CrossRef] [Green Version]
- Yassaee, V.R.; Khojaste, A.; Hashemi-Gorji, F.; Ravesh, Z.; Toosi, P. A novel homozygous LMNA mutation (p. Met540Ile) causes mandibuloacral dysplasia type A. Gene 2016, 577, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Sakka, R.; Marmouch, H.; Trabelsi, M.; Achour, A.; Golli, M.; Hannachi, I.; Kerkeni, E.; Monastiri, K.; Maazoul, F.; M’rad, R. Mandibuloacral dysplasia type A in five tunisian patients. Eur. J. Med. Genet. 2021, 64, 104138. [Google Scholar]
- Avnet, S.; Pallotta, R.; Perut, F.; Baldini, N.; Pittis, M.G.; Saponari, A.; Lucarelli, E.; Dozza, B.; Greggi, T.; Maraldi, N.M.; et al. Osteoblasts from a mandibuloacral dysplasia patient induce human blood precursors to differentiate into active osteoclasts. Biochim. Biophys. Acta 2011, 1812, 711–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhe-Paganon, S.; Werner, E.D.; Chi, Y.-I.; Shoelson, S.E. Structure of the globular tail of nuclear lamin. J. Biol. Chem. 2002, 277, 17381–17384. [Google Scholar] [CrossRef] [Green Version]
- Krimm, I.; Östlund, C.; Gilquin, B.; Couprie, J.; Hossenlopp, P.; Mornon, J.-P.; Bonne, G.; Courvalin, J.-C.; Worman, H.J.; Zinn-Justin, S. The Ig-like structure of the C-terminal domain of lamin A/C, mutated in muscular dystrophies, cardiomyopathy, and partial lipodystrophy. Structure 2002, 10, 811–823. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | Proband 1 | Proband 2 | Proband 3 |
|---|---|---|---|
| Age | 27 years | 5 years | 2 years |
| Sex | Female | male | male |
| Age of onset | 14 years | 2 years | 1 year |
| Height for age | <P3 | <P3 | <P3 |
| Weight for age | <P3 | <P3 | <P3 |
| Microcephaly | - | - | - |
| Prominent eyes | + | - | - |
| Full cheeks | + | - | + |
| Micrognathia | +++ | ++ | + |
| Retrognathia | +++ | ++ | + |
| Dental crowding | ++ | - | + |
| Beaked/Pinched/pointed nose | + | + | + |
| Alopecia/Sparse hair | + | ++ | +++ |
| Abdominal obesity | +++ | - | - |
| Submental obesity | ++ | + | - |
| Clavicle hypoplasia | ++ | +++ | + |
| Lipodystrophy of extremities | + | + | - |
| Acroosteolysis | ++ | ++ | + |
| Finger rounding (drumstick shaped distal phalanges) | +++ | ++ | + |
| Joint contractures | + | ++ | - |
| Mottling/Hyperpigmentation | ++ | - | + |
| References | Pathogenic Variant | Genotype | Phenotype | Phenotype | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Typical or Atypical | Number of Cases Reported | Gender | Age at Investigation | Growth Retardation/Short Stature | Prominent Eyes | Beaked/Pointed Nose | Prominent Cheeks | Dental Crowding | Mandibular Hypoplasia | Clavicular Hypoplasia/Osteolysis | Acro-osteolysis | Lipodystrophy (A/B) | Mottled Skin Pigmentation | Alopecia | |||
| Novelli et al., 2002 [2]. | c.1580 G>A-p.(Arg527His) | Homozygous | Typical MADA | 9 | 4 F 5 M | 1 case: 35 years 8 cases: N/A | 9/9 | N/A | 1/9 8/9 N/A | 1/9 8/9 N/A | N/A | 9/9 | 9/9 | 9/9 | 9/9 A | 9/9 | 5/9 |
| Cao and Hegele, 2003 [8]. | c.1580 G>T-p.(Arg471Cys)/ c.1623 C>T-p. (Arg527Leu) | Compound heterozygous | Atypical MADA with progeroid features | 1 | F | 28 years | N/A | N/A | N/A | N/A | N/A | 1/1 | 1/1 | 1/1 | 1/1 A | N/A | 1/1 |
| Simha et al., 2003 [22]. | c.1580 G>A-p.(Arg527His) | Homozygous | Typical MADA | 2 | 2 F | 20 years 16 years | 2/2 | 2/2 | 2/2 | 2/2 | 2/2 | 2/2 | 2/2 | 2/2 | 2/2 A | 2/2 | 0/2 |
| Shen et al., 2003 [23]. | c.1580 G>A-p.(Arg527His) | Homozygous | Typical MADA | 1 | M | 12 years | 0/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 A | 1/1 | 0/1 |
| Plasilova et al., 2004 [9]. | c.1626 G>C-p.(Lys542Asn) | Homozygous | Typical MADA | 4 | 2 F 2 M | 4.5 years 10 years 15 years 17 years | 4/4 | 4/4 | 4/4 | 4/4 | 3/4 | 4/4 | 4/4 | 4/4 | 4/4 B | 4/4 | 4/4 |
| Verstraeten et al., 2006 [10]. | c.1583 C>T-p.(Thr528Met)/ c.1619 T>C-p.(Met540Thr) | Compound heterozygous | Atypical MADA with progeroid features | 1 | M | 18–24 months | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 A | 0/1 | 1/1 |
| Kosho et al., 2007 [25]. | c.1585 G>A-p.(Ala529Thr) | Homozygous | Typical MADA | 1 | F | mid-20s | 1/1 | 1/1 | 1/1 | 1/1 | 0/1 | 1/1 | 1/1 | 1/1 | 1/1 A | 1/1 | 1/1 |
| Lombardi et al., 2007 [7]. | c.1580 G> A-p.(Arg527His)/ c.1318 G> A-p.(Val440Met) | Compound heterozygous | Typical MADA with skeletal muscle involvement | 1 | F | 22 years | 0/1 | N/A | 1/1 | N/A | 0/1 | 0/1 | 0/1 | 1/1 | N/A | 0/1 | 0/1 |
| Agarwal et al., 2008 [12]. | c.1579 C>T-p.(Arg527Cys) | Homozygous | 1 | F | 7 years | 1/1 | N/A | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | |
| Zirn et al., 2008 [11]. | c.1623 C>T-p.(Arg471Cys) | Homozygous | Typical MADA | 1 | F | 3 years | 0/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 A | 0/1 | 0/1 |
| Garavelli et al., 2009 [24]. | c.1580 G>A-p.(Arg527His)/ c.1318 G>A-p.(Val440Met) | Homozygous | 2 | 1 F 1 M | 4 years 5 years | 0/2 | 2/2 | 2/2 | 2/2 | 1/2 | 1/2 | 2/2 | 2/2 | 2/2 | 1/2 | N/A | |
| Amr, Mostafa, and El-Kamah, 2012 [14]. | c.1580 G>T-p.(Arg527Leu)/ c.1579 C>T-p.(Arg527Cys) | Homozygous | MADA with lipodystrophy and progeroid features | 4 | 2 F 2 M | 9 years 18 years 7 years 8 years | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 | 4/4 A | N/A | N/A |
| Al-Haggar et al., 2012 [13]. | c.1580 G>T-p.(Arg527Leu) | Homozygous | Atypical MADA with progeroid features | 3 | 3 F | 2.5 years 5 years 3 years | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 | 3/3 A | 2/3 | 2/3 |
| Luo et al., 2014 [26]. | c.1579 C>T-p.(Arg527Cys) | Homozygous | Typical MADA | 3 | 2 F 1 M | 10 months 12 months 8 months | 3/3 | 2/3 | 2/3 | 2/3 | 2/3 | 3/3 | 2/3 | 3/3 | 3/3 B | 3/3 | 3/3 |
| Yassaee et al., 2016 [27]. | c.1620 G>A-p.(Met540Ile) | Homozygous | Typical MADA | 1 | M | 10 months | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 B | N/A | N/A |
| Sakka et al., 2021 [28]. | c.1580 G>A-p.(Arg527His) | Homozygous | Typical MADA with growth hormone deficiency and cardiomyopathy | 5 | 1 F 4 M | 12 years 11 years 3 years 9 years 48 years | 4/5 | 5/5 | 5/5 | 4/5 | 5/5 | 5/5 | 4/5 | 5/5 | 5/5 | 5/5 A | 1/5 |
| Current study | P1: c.1580 G>A-p.(Arg527His) P2/P3: c.1580 G>T-p.(Arg527Leu) | Homozygous | 3 | 1 F 2 M | 27 years 5 years 2 years | 3/3 | 1/3 | 3/3 | 2/3 | 2/3 | 3/3 | 3/3 | 3/3 | 3/3 | 2/3 | 3/3 | |
| Total | 43 | 23 F 20 M | 34/43 | 28/43 | 33/43 | 30/43 | 27/43 | 41/43 | 40/43 | 43/43 | 42/43 | 31/43 | 22/43 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jéru, I.; Nabil, A.; El-Makkawy, G.; Lascols, O.; Vigouroux, C.; Abdalla, E. Two Decades after Mandibuloacral Dysplasia Discovery: Additional Cases and Comprehensive View of Disease Characteristics. Genes 2021, 12, 1508. https://doi.org/10.3390/genes12101508
Jéru I, Nabil A, El-Makkawy G, Lascols O, Vigouroux C, Abdalla E. Two Decades after Mandibuloacral Dysplasia Discovery: Additional Cases and Comprehensive View of Disease Characteristics. Genes. 2021; 12(10):1508. https://doi.org/10.3390/genes12101508
Chicago/Turabian StyleJéru, Isabelle, Amira Nabil, Gehad El-Makkawy, Olivier Lascols, Corinne Vigouroux, and Ebtesam Abdalla. 2021. "Two Decades after Mandibuloacral Dysplasia Discovery: Additional Cases and Comprehensive View of Disease Characteristics" Genes 12, no. 10: 1508. https://doi.org/10.3390/genes12101508