Principles of Genetic Engineering


Abstract
:1. Introduction
2. Principles of Genetic Engineering
2.1. Types of Genetic Modifications
2.2. Genetic Engineering with CRISPR/Cas9
2.3. Locus-Specific Genetic Engineering Vectors in Mouse and Rat Zygotes
2.4. Gene Editing in Immortalized Cell Lines
2.5. Viruses and Transposons as Genetic Engineering Vectors
2.6. Genetic Engineering Using Retroviruses
2.7. Gene Targeting Using Adeno-Associated Virus
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Graham, F.L.; van der Eb, A.J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973, 52, 456–467. [Google Scholar] [CrossRef]
- Neumann, E.; Schaefer-Ridder, M.; Wang, Y.; Hofschneider, P.H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982, 1, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, M.R. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell 1980, 22 Pt 2, 479–488. [Google Scholar] [CrossRef]
- Qasba, P.K.; Aposhian, H.V. DNA and gene therapy: Transfer of mouse DNA to human and mouse embryonic cells by polyoma pseudovirions. Proc. Natl. Acad. Sci. USA 1971, 68, 2345–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermonat, P.L.; Muzyczka, N. Use of adeno-associated virus as a mammalian DNA cloning vector: Transduction of neomycin resistance into mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA 1984, 81, 6466–6470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Kucherlapati, R.S.; Eves, E.M.; Song, K.Y.; Morse, B.S.; Smithies, O. Homologous recombination between plasmids in mammalian cells can be enhanced by treatment of input DNA. Proc. Natl. Acad. Sci. USA 1984, 81, 3153–3157. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.R.; Capecchi, M.R. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 1987, 51, 503–512. [Google Scholar] [CrossRef]
- Mansour, S.L.; Thomas, K.R.; Capecchi, M.R. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: A general strategy for targeting mutations to non-selectable genes. Nature 1988, 336, 348–352. [Google Scholar] [CrossRef]
- Auwerx, J.; Avner, P.; Baldock, R.; Ballabio, A.; Balling, R.; Barbacid, M.; Berns, A.; Bradley, A.; Brown, S.; Carmeliet, P.; et al. The European dimension for the mouse genome mutagenesis program. Nat. Genet. 2004, 36, 925–927. [Google Scholar] [PubMed] [Green Version]
- International Mouse Knockout Consortium; Collins, F.S.; Rossant, J.; Wurst, W. A mouse for all reasons. Cell 2007, 128, 9–13. [Google Scholar] [PubMed] [Green Version]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacheiro, P.; Haendel, M.A.; Smedley, D.; International Mouse Phenotyping Consortium and the Monarch Initiative. New models for human disease from the International Mouse Phenotyping Consortium. Mamm. Genome 2019, 30, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Teucher, M.; Anastassiadis, K.; Skarnes, W.; Stewart, A.F. A recombineering pipeline to make conditional targeting constructs. Methods Enzymol. 2010, 477, 125–144. [Google Scholar]
- Testa, G.; Zhang, Y.; Vintersten, K.; Benes, V.; Pijnappel, W.W.; Chambers, I.; Smith, A.J.; Smith, A.G.; Stewart, A.F. Engineering the mouse genome with bacterial artificial chromosomes to create multipurpose alleles. Nat. Biotechnol. 2003, 21, 443–447. [Google Scholar] [CrossRef]
- Valenzuela, D.M.; Murphy, A.J.; Frendewey, D.; Gale, N.W.; Economides, A.N.; Auerbach, W.; Poueymirou, W.T.; Adams, N.C.; Rojas, J.; Yasenchak, J.; et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat. Biotechnol. 2003, 21, 652–659. [Google Scholar] [CrossRef]
- Osoegawa, K.; Tateno, M.; Woon, P.Y.; Frengen, E.; Mammoser, A.G.; Catanese, J.J.; Hayashizaki, Y.; de Jong, P.J. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 2000, 10, 116–128. [Google Scholar]
- Hu, Y.; Xie, Y.; Wang, Y.; Chen, X.; Smith, D.E. Development and characterization of a novel mouse line humanized for the intestinal peptide transporter PEPT1. Mol. Pharm. 2014, 11, 3737–3746. [Google Scholar] [CrossRef] [Green Version]
- Ranatunga, D.; Hedrich, C.M.; Wang, F.; McVicar, D.W.; Nowak, N.; Joshi, T.; Feigenbaum, L.; Grant, L.R.; Stäger, S.; Bream, J.H. A human IL10 BAC transgene reveals tissue-specific control of IL-10 expression and alters disease outcome. Proc. Natl. Acad. Sci. USA 2009, 106, 17123–17128. [Google Scholar] [CrossRef] [Green Version]
- Copeland, N.G.; Jenkins, N.A.; Court, D.L. Recombineering: A powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001, 2, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Ciotta, G.; Hofemeister, H.; Maresca, M.; Fu, J.; Sarov, M.; Anastassiadis, K.; Stewart, A.F. Recombineering BAC transgenes for protein tagging. Methods 2011, 53, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Zheng, C.; Doughty, M.L.; Losos, K.; Didkovsky, N.; Schambra, U.B.; Nowak, N.J.; Joyner, A.; Leblanc, G.; Hatten, M.E.; et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 2003, 425, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Said, R.; Zheng, L.; Saunders, T.; Zeidler, M.; Papagerakis, S.; Papagerakis, P. Generation of Amelx-iCre mice supports ameloblast-specific role for Stim1. J. Dent. Res. 2019, 98, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Van Keuren, M.L.; Gavrilina, G.B.; Filipiak, W.E.; Zeidler, M.G.; Saunders, T.L. Generating transgenic mice from bacterial artificial chromosomes: Transgenesis efficiency, integration and expression outcomes. Transgenic Res. 2009, 18, 769–785. [Google Scholar] [CrossRef] [Green Version]
- Zeidler, M.G.; Saunders, T.L. Transgene recombineering in bacterial artificial chromosomes. Methods Mol. Biol. 2019, 1874, 43–69. [Google Scholar]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Epinat, J.C.; Arnould, S.; Chames, P.; Rochaix, P.; Desfontaines, D.; Puzin, C.; Patin, A.; Zanghellini, A.; Pâques, F.; Lacroix, E. A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res. 2003, 31, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Arnould, S.; Delenda, C.; Grizot, S.; Desseaux, C.; Pâques, F.; Silva, G.H.; Smith, J. The I-CreI meganuclease and its engineered derivatives: Applications from cell modification to gene therapy. Protein Eng. Des. Sel. 2011, 24, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Ménoret, S.; Fontanière, S.; Jantz, D.; Tesson, L.; Thinard, R.; Rémy, S.; Usal, C.; Ouisse, L.H.; Fraichard, A.; Anegon, I. Generation of Rag1-knockout immunodeficient rats and mice using engineered meganucleases. FASEB J. 2013, 27, 703–711. [Google Scholar] [CrossRef]
- Bogdanove, A.J.; Bohm, A.; Miller, J.C.; Morgan, R.D.; Stoddard, B.L. Engineering altered protein-DNA recognition specificity. Nucleic Acids Res. 2018, 46, 4845–4871. [Google Scholar] [CrossRef]
- Porteus, M.H.; Carroll, D. Gene targeting using zinc finger nucleases. Nat. Biotechnol. 2005, 23, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Dahlborg, E.J.; Goodwin, M.J.; Cade, L.; Zhang, F.; Cifuentes, D.; Curtin, S.J.; Blackburn, J.S.; Thibodeau-Beganny, S.; Qi, Y.; et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat. Methods 2011, 8, 67–69. [Google Scholar] [CrossRef]
- Wright, D.A.; Thibodeau-Beganny, S.; Sander, J.D.; Winfrey, R.J.; Hirsh, A.S.; Eichtinger, M.; Fu, F.; Porteus, M.H.; Dobbs, D.; Voytas, D.F.; et al. Standardized reagents and protocols for engineering zinc finger nucleases by modular assembly. Nat. Protoc. 2006, 1, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Fisher, D.A.; Kouranova, E.; McCoy, A.; Forbes, K.; Wu, Y.; Henry, R.; Ji, D.; Chambers, A.; Warren, J.; et al. Whole-rat conditional gene knockout via genome editing. Nat. Methods 2013, 10, 638–640. [Google Scholar] [CrossRef] [PubMed]
- Carbery, I.D.; Ji, D.; Harrington, A.; Brown, V.; Weinstein, E.J.; Liaw, L.; Cui, X. Targeted genome modification in mice using zinc-finger nucleases. Genetics 2010, 186, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Geurts, A.M.; Cost, G.J.; Freyvert, Y.; Zeitler, B.; Miller, J.C.; Choi, V.M.; Jenkins, S.S.; Wood, A.; Cui, X.; Meng, X.; et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 2009, 325, 433. [Google Scholar] [CrossRef] [Green Version]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Bogdanove, A.J.; Voytas, D.F. TAL effectors: Customizable proteins for DNA targeting. Science 2011, 333, 1843–1846. [Google Scholar] [CrossRef]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, C.L.; Foley, J.E.; Wright, D.A.; Müller-Lerch, F.; Rahman, S.H.; Cornu, T.I.; Winfrey, R.J.; Sander, J.D.; Fu, F.; Townsend, J.A.; et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods 2008, 5, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.; Davies, G.; Preece, C.; Puliyadi, R.; Szumska, D.; Bhattacharya, S. Site specific mutation of the Zic2 locus by microinjection of TALEN mRNA in mouse CD1, C3H and C57BL/6J oocytes. PLoS ONE 2013, 8, e60216. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Liu, M.; Chen, Z.; Shao, Y.; Pan, H.; Wei, G.; Yu, C.; Zhang, L.; Li, X.; Wang, P.; et al. High-efficiency and heritable gene targeting in mouse by transcription activator-like effector nucleases. Nucleic Acids Res. 2013, 41, e120. [Google Scholar] [CrossRef] [PubMed]
- Remy, S.; Tesson, L.; Menoret, S.; Usal, C.; De Cian, A.; Thepenier, V.; Thinard, R.; Baron, D.; Charpentier, M.; Renaud, J.B.; et al. Efficient gene targeting by homology-directed repair in rat zygotes using TALE nucleases. Genome Res. 2014, 24, 1371–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesson, L.; Usal, C.; Ménoret, S.; Leung, E.; Niles, B.J.; Remy, S.; Santiago, Y.; Vincent, A.I.; Meng, X.; Zhang, L.; et al. Knockout rats generated by embryo microinjection of TALENs. Nat. Biotechnol. 2011, 29, 695–696. [Google Scholar] [CrossRef]
- Wefers, B.; Meyer, M.; Ortiz, O.; Hrabé de Angelis, M.; Hansen, J.; Wurst, W.; Kühn, R. Direct production of mouse disease models by embryo microinjection of TALENs and oligodeoxynucleotides. Proc. Natl. Acad. Sci. USA 2013, 110, 3782–3787. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Cheah, S.S.; Behringer, R.R. Gene-targeting strategies. Methods Mol. Biol. 2000, 136, 455–463. [Google Scholar] [PubMed]
- Doyle, A.; McGarry, M.P.; Lee, N.A.; Lee, J.J. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Res. 2012, 21, 327–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, T.L. Gene targeting vector design for embryonic stem cell modifications. In Advanced Protocols for Animal Transgenesis: An ISTT Manual; Pease, S., Saunders, T.L., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 57–79. [Google Scholar]
- Filipiak, W.E.; Hughes, E.D.; Gavrilina, G.B.; LaForest, A.K.; Saunders, T.L. Next generation transgenic rat model production. Methods Mol. Biol. 2019, 2018, 97–114. [Google Scholar] [PubMed]
- Singh, P.; Schimenti, J.C.; Bolcun-Filas, E. A mouse geneticist’s practical guide to CRISPR applications. Genetics 2015, 199, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, E.B.; Zheng, X.; Agrawal, S.; Cartee, G.D. Whole body glucoregulation and tissue-specific glucose uptake in a novel Akt substrate of 160 kDa knockout rat model. PLoS ONE 2019, 14, e0216236. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, L.; Xie, M.; Li, Y.; Huang, P.; Saunders, T.L.; Fox, D.A.; Rosenquist, R.; Lin, F. Role of complement in a rat model of paclitaxel-induced peripheral neuropathy. J. Immunol. 2018, 200, 4094–4101. [Google Scholar] [CrossRef]
- Allan, C.M.; Heizer, P.J.; Tu, Y.; Sandoval, N.P.; Jung, R.S.; Morales, J.E.; Sajti, E.; Troutman, T.D.; Saunders, T.L.; Cusanovich, D.A.; et al. An upstream enhancer regulates Gpihbp1 expression in a tissue-specific manner. J. Lipid Res. 2019, 60, 869–879. [Google Scholar] [CrossRef]
- Min, Y.L.; Li, H.; Rodriguez-Caycedo, C.; Mireault, A.A.; Huang, J.; Shelton, J.M.; McAnally, J.R.; Amoasii, L.; Mammen, P.P.A.; Bassel-Duby, R.; et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv. 2019, 5, eaav4324. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Cawley, K.; Piemontese, M.; Fujiwara, Y.; Zhao, H.; Goellner, J.J.; O’Brien, C.A. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat. Commun. 2018, 9, 2909. [Google Scholar] [CrossRef]
- Mizuhashi, K.; Ono, W.; Matsushita, Y.; Sakagami, N.; Takahashi, A.; Saunders, T.L.; Nagasawa, T.; Kronenberg, H.M.; Ono, N. Resting zone of the growth plate houses a unique class of skeletal stem cells. Nature 2018, 563, 254–258. [Google Scholar] [CrossRef]
- van Hummel, A.; Bi, M.; Ippati, S.; van der Hoven, J.; Volkerling, A.; Lee, W.S.; Tan, D.C.; Bongers, A.; Ittner, A.; Ke, Y.D.; et al. No overt deficits in aged tau-deficient C57Bl/6.Mapttm1(EGFP)Kit GFP knockin mice. PLoS ONE 2016, 11, e0163236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, B.; Posfai, E.; Rossant, J. Efficient generation of targeted large insertions by microinjection into two-cell-stage mouse embryos. Nat. Biotechnol. 2018, 36, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Wang, M.; Ye, C.; Fang, J.; Duan, Y.; Zhang, Z.; Hua, Q.; Shi, C.; Zhang, L.; Zhang, R.; et al. One-step generation of mice carrying a conditional allele together with an HA-tag insertion for the delta opioid receptor. Sci. Rep. 2017, 7, 44476. [Google Scholar] [CrossRef]
- Lai, K.M.; Gong, G.; Atanasio, A.; Rojas, J.; Quispe, J.; Posca, J.; White, D.; Huang, M.; Fedorova, D.; Grant, C.; et al. Diverse phenotypes and specific transcription patterns in twenty mouse lines with ablated LincRNAs. PLoS ONE 2015, 10, e0125522. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, L.E.; Karow, M.; Stevens, S.; Auerbach, W.; Poueymirou, W.T.; Yasenchak, J.; Frendewey, D.; Valenzuela, D.M.; Giallourakis, C.C.; Alt, F.W.; et al. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc. Natl. Acad. Sci. USA 2014, 111, 5147–5152. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Eitzman, D.T.; Westrick, R.J.; Christie, P.D.; Xu, Z.J.; Yang, A.Y.; Purkayastha, A.A.; Yang, T.L.; Metz, A.L.; Gallagher, K.P.; et al. Spontaneous thrombosis in mice carrying the factor V Leiden mutation. Blood 2000, 96, 4222–4226. [Google Scholar] [CrossRef]
- de Ligt, J.; Veltman, J.A.; Vissers, L.E. Point mutations as a source of de novo genetic disease. Curr. Opin. Genet. Dev. 2013, 23, 257–263. [Google Scholar] [CrossRef]
- Lubeck, B.A.; Lapinski, P.E.; Bauler, T.J.; Oliver, J.A.; Hughes, E.D.; Saunders, T.L.; King, P.D. Blood vascular abnormalities in Rasa1(R780Q) knockin mice: Implications for the pathogenesis of capillary malformation-arteriovenous malformation. Am. J. Pathol. 2014, 184, 3163–3169. [Google Scholar] [CrossRef] [Green Version]
- Nagy, A. Cre recombinase: The universal reagent for genome tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Sauer, B.; Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popp, M.W.; Maquat, L.E. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell 2016, 165, 1319–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, S.A.; Eppig, J.T.; Smedley, D.; Simpson, E.M.; Rosenthal, N. Beyond knockouts: Cre resources for conditional mutagenesis. Mamm. Genome 2012, 23, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, K.; Fu, J.; Patsch, C.; Hu, S.; Weidlich, S.; Duerschke, K.; Buchholz, F.; Edenhofer, F.; Stewart, A.F. Dre recombinase, like Cre, is a highly efficient site-specific recombinase in E. coli, mammalian cells and mice. Dis. Model. Mech. 2009, 2, 508–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimova, M.; Abi-Ghanem, J.; Berger, N.; Surendranath, V.; Pisabarro, M.T.; Buchholz, F. Vika/vox, a novel efficient and specific Cre/loxP-like site-specific recombination system. Nucleic Acids Res. 2013, 41, e37. [Google Scholar] [CrossRef] [Green Version]
- Karimova, M.; Baker, O.; Camgoz, A.; Naumann, R.; Buchholz, F.; Anastassiadis, K. A single reporter mouse line for Vika, Flp, Dre, and Cre-recombination. Sci. Rep. 2018, 8, 14453. [Google Scholar] [CrossRef] [Green Version]
- O’Gorman, S.; Fox, D.T.; Wahl, G.M. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 1991, 251, 1351–1355. [Google Scholar] [CrossRef] [Green Version]
- Raymond, C.S.; Soriano, P. High-efficiency FLP and PhiC31 site-specific recombination in mammalian cells. PLoS ONE 2007, 2, e162. [Google Scholar] [CrossRef]
- Sauer, B.; McDermott, J. DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucleic Acids Res. 2004, 32, 6086–6095. [Google Scholar] [CrossRef]
- Cacioppo, J.A.; Koo, Y.; Lin, P.C.; Osmulski, S.A.; Ko, C.D.; Ko, C. Generation of an estrogen receptor beta-iCre knock-in mouse. Genesis 2016, 54, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Pettibone, J.R.; Yu, J.Y.; Derman, R.C.; Faust, T.W.; Hughes, E.D.; Filipiak, W.E.; Saunders, T.L.; Ferrario, C.R.; Berke, J.D. Knock-in rat lines with Cre recombinase at the dopamine D1 and adenosine 2a receptor loci. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryda, E.C.; Men, H.; Davis, D.J.; Bock, A.S.; Shaw, M.L.; Chesney, K.L.; Hankins, M.A. A novel conditional ZsGreen-expressing transgenic reporter rat strain for validating Cre recombinase expression. Sci. Rep. 2019, 9, 13330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gasperi, R.; Rocher, A.B.; Sosa, M.A.; Wearne, S.L.; Perez, G.M.; Friedrich, V.L., Jr.; Hof, P.R.; Elder, G.A. The IRG mouse: A two-color fluorescent reporter for assessing Cre-mediated recombination and imaging complex cellular relationships in situ. Genesis 2008, 46, 308–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livet, J.; Weissman, T.A.; Kang, H.; Draft, R.W.; Lu, J.; Bennis, R.A.; Sanes, J.R.; Lichtman, J.W. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007, 450, 56–62. [Google Scholar] [CrossRef]
- Schnütgen, F.; Doerflinger, N.; Calléja, C.; Wendling, O.; Chambon, P.; Ghyselinck, N.B. A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat. Biotechnol. 2003, 21, 562–565. [Google Scholar] [CrossRef]
- Schnütgen, F.; De-Zolt, S.; Van Sloun, P.; Hollatz, M.; Floss, T.; Hansen, J.; Altschmied, J.; Seisenberger, C.; Ghyselinck, N.B.; Ruiz, P.; et al. Genomewide production of multipurpose alleles for the functional analysis of the mouse genome. Proc. Natl. Acad. Sci. USA 2005, 102, 7221–7226. [Google Scholar] [CrossRef] [Green Version]
- Economides, A.N.; Frendewey, D.; Yang, P.; Dominguez, M.G.; Dore, A.T.; Lobov, I.B.; Persaud, T.; Rojas, J.; McClain, J.; Lengyel, P.; et al. Conditionals by inversion provide a universal method for the generation of conditional alleles. Proc. Natl. Acad. Sci. USA 2013, 110, E3179–E3188. [Google Scholar] [CrossRef] [Green Version]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef]
- Mandalos, N.; Saridaki, M.; Harper, J.L.; Kotsoni, A.; Yang, P.; Economides, A.N.; Remboutsika, E. Application of a novel strategy of engineering conditional alleles to a single exon gene Sox2. PLoS ONE 2012, 7, e45768. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Jasin, M.; Haber, J.E. The democratization of gene editing: Insights from site-specific cleavage and double-strand break repair. DNA Repair 2016, 44, 6–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doench, J.G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018, 19, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Koo, T.; Jee, H.G.; Cho, H.Y.; Lee, G.; Lim, D.G.; Shin, H.S.; Kim, J.S. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Basila, M.; Kelley, M.L.; Smith, A.V.B. Minimal 2’-O-methyl phosphorothioate linkage modification pattern of synthetic guide RNAs for increased stability and efficient CRISPR-Cas9 gene editing avoiding cellular toxicity. PLoS ONE 2017, 12, e0188593. [Google Scholar] [CrossRef] [Green Version]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef]
- Fellmann, C.; Gowen, B.G.; Lin, P.C.; Doudna, J.A.; Corn, J.E. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2017, 16, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.R.; Haeussler, M.; Watanabe, C.; Janakiraman, V.; Lund, J.; Modrusan, Z.; Stinson, J.; Bei, Q.; Buechler, A.; Yu, C.; et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods 2018, 15, 512–514. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef] [Green Version]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Schick, J.A.; Seisenberger, C.; Beig, J.; Bürger, A.; Iyer, V.; Maier, V.; Perera, S.; Rosen, B.; Skarnes, W.C.; Wurst, W. CRISPR-Cas9 enables conditional mutagenesis of challenging loci. Sci. Rep. 2016, 6, 32326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertsenstein, M.; Nutter, L.M.J. Engineering point mutant and epitope-tagged alleles in mice using Cas9 RNA-guided nuclease. Curr. Protoc. Mouse Biol. 2018, 8, 28–53. [Google Scholar] [CrossRef] [PubMed]
- Lanza, D.G.; Gaspero, A.; Lorenzo, I.; Liao, L.; Zheng, P.; Wang, Y.; Deng, Y.; Cheng, C.; Zhang, C.; Seavitt, J.R.; et al. Comparative analysis of single-stranded DNA donors to generate conditional null mouse alleles. BMC Biol. 2018, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Quadros, R.M.; Miura, H.; Harms, D.W.; Akatsuka, H.; Sato, T.; Aida, T.; Redder, R.; Richardson, G.P.; Inagaki, Y.; Sakai, D.; et al. Easi-CRISPR: A robust method for one-step generation of mice carrying conditional and insertion alleles using long ssDNA donors and CRISPR ribonucleoproteins. Genome Biol. 2017, 18, 92. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Ji, D.; Fisher, D.A.; Wu, Y.; Briner, D.M.; Weinstein, E.J. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat. Biotechnol. 2011, 29, 64–67. [Google Scholar] [CrossRef]
- Yao, X.; Zhang, M.; Wang, X.; Ying, W.; Hu, X.; Dai, P.; Meng, F.; Shi, L.; Sun, Y.; Yao, N.; et al. Tild-CRISPR allows for efficient and precise gene knockin in mouse and human cells. Dev. Cell 2018, 45, 526–536. [Google Scholar] [CrossRef] [Green Version]
- Miura, H.; Quadros, R.M.; Gurumurthy, C.B.; Ohtsuka, M. Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat. Protoc. 2018, 13, 195–215. [Google Scholar] [CrossRef]
- Chu, V.T.; Weber, T.; Graf, R.; Sommermann, T.; Petsch, K.; Sack, U.; Volchkov, P.; Rajewsky, K.; Kühn, R. Efficient generation of Rosa26 knock-in mice using CRISPR/Cas9 in C57BL/6 zygotes. BMC Biotechnol. 2016, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paix, A.; Folkmann, A.; Goldman, D.H.; Kulaga, H.; Grzelak, M.J.; Rasoloson, D.; Paidemarry, S.; Green, R.; Reed, R.R.; Seydoux, G. Precision genome editing using synthesis-dependent repair of Cas9-induced DNA breaks. Proc. Natl. Acad. Sci. USA 2017, 114, E10745–E10754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker-Thornburg, J. Breeding strategies for genetically modified mice. Methods Mol. Biol. 2020, 2066, 163–169. [Google Scholar]
- Codner, G.F.; Mianné, J.; Caulder, A.; Loeffler, J.; Fell, R.; King, R.; Allan, A.J.; Mackenzie, M.; Pike, F.J.; McCabe, C.V.; et al. Application of long single-stranded DNA donors in genome editing: Generation and validation of mouse mutants. BMC Biol. 2018, 16, 70. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.T.; Zhang, M.; Deng, J.M.; Usman, S.J.; Smith, C.N.; Parker-Thornburg, J.; Swinton, P.G.; Martin, J.F.; Behringer, R.R. Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev. Biol. 2014, 393, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Geng, L.; Xin, W.; Huang, D.W.; Feng, G. A universal cloning vector using vaccinia topoisomerase I. Mol. Biotechnol. 2006, 33, 23–28. [Google Scholar]
- Dubose, A.J.; Lichtenstein, S.T.; Narisu, N.; Bonnycastle, L.L.; Swift, A.J.; Chines, P.S.; Collins, F.S. Use of microarray hybrid capture and next-generation sequencing to identify the anatomy of a transgene. Nucleic Acids Res. 2013, 41, e70. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, L.O.; Splinter, E.; Davis, T.L.; Urban, R.; He, H.; Braun, R.E.; Chesler, E.J.; Kumar, V.; van Min, M.; Ndukum, J.; et al. Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res. 2019, 29, 494–505. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, P.K.; Bellott, D.W.; Cho, T.J.; Pyntikova, T.; Page, D.C. Locating and characterizing a transgene integration site by nanopore sequencing. G3 2019, 9, 1481–1486. [Google Scholar] [CrossRef] [Green Version]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-López, M.; García-Pérez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heffron, F.; McCarthy, B.J.; Ohtsubo, H.; Ohtsubo, E. DNA sequence analysis of the transposon Tn3: Three genes and three sites involved in transposition of Tn3. Cell 1979, 18, 1153–1163. [Google Scholar] [CrossRef]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Cary, L.C.; Goebel, M.; Corsaro, B.G.; Wang, H.G.; Rosen, E.; Fraser, M.J. Transposon mutagenesis of baculoviruses: Analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology 1989, 172, 156–169. [Google Scholar] [CrossRef]
- Cadiñanos, J.; Bradley, A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007, 35, e87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowarz, E.; Löscher, D.; Marschalek, R. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol. J. 2015, 10, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Huls, H.; Kebriaei, P.; Cooper, L.J. A new approach to gene therapy using Sleeping Beauty to genetically modify clinical-grade T cells to target CD19. Immunol. Rev. 2014, 257, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Grabundzija, I.; Irgang, M.; Mátés, L.; Belay, E.; Matrai, J.; Gogol-Döring, A.; Kawakami, K.; Chen, W.; Ruiz, P.; Chuah, M.K.; et al. Comparative analysis of transposable element vector systems in human cells. Mol. Ther. 2010, 18, 1200–1209. [Google Scholar] [CrossRef]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef]
- Yusa, K.; Zhou, L.; Li, M.A.; Bradley, A.; Craig, N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 2011, 108, 1531–1536. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, J.; Chen, T.; Wang, Y.; Xin, S.; Li, J.; Pei, G.; Kang, J. Yamanaka factors critically regulate the developmental signaling network in mouse embryonic stem cells. Cell Res. 2008, 18, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Öllinger, R.; Friedrich, M.; Ehmer, U.; Barenboim, M.; Steiger, K.; Heid, I.; Mueller, S.; Maresch, R.; Engleitner, T.; et al. CRISPR/Cas9 somatic multiplex-mutagenesis for high-throughput functional cancer genomics in mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13982–13987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, K.; Li, Y.; Wu, W.; Chen, H.; Chen, Z.; Zhang, Y.; Guo, Y.; Dong, Y. High-performance gene expression and knockout tools using sleeping beauty transposon system. Mob. DNA 2018, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Qi, X.; Du, X.; Zou, H.; Gao, F.; Feng, T.; Lu, H.; Li, S.; An, X.; Zhang, L.; et al. piggyBac mediates efficient in vivo CRISPR library screening for tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 722–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Park, J.J.; Dong, M.B.; Yang, Q.; Chow, R.D.; Peng, L.; Du, Y.; Guo, J.; Dai, X.; Wang, G.; et al. In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat. Biotechnol. 2019, 37, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Verma, I.M.; Weitzman, M.D. Gene therapy: Twenty-first century medicine. Annu. Rev. Biochem. 2005, 74, 711–738. [Google Scholar] [CrossRef] [Green Version]
- Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993, 12, 2099–2108. [Google Scholar] [CrossRef]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef]
- Cattoglio, C.; Pellin, D.; Rizzi, E.; Maruggi, G.; Corti, G.; Miselli, F.; Sartori, D.; Guffanti, A.; Di Serio, C.; Ambrosi, A.; et al. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood 2010, 116, 5507–5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Díez, I.A.; Dewey, R.A.; Böhm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Krämer, A.; Schwäble, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almarza, D.; Bussadori, G.; Navarro, M.; Mavilio, F.; Larcher, F.; Murillas, R. Risk assessment in skin gene therapy: Viral-cellular fusion transcripts generated by proviral transcriptional read-through in keratinocytes transduced with self-inactivating lentiviral vectors. Gene Ther. 2011, 18, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruggi, G.; Porcellini, S.; Facchini, G.; Perna, S.K.; Cattoglio, C.; Sartori, D.; Ambrosi, A.; Schambach, A.; Baum, C.; Bonini, C.; et al. Transcriptional enhancers induce insertional gene deregulation independently from the vector type and design. Mol. Ther. 2009, 17, 851–856. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.P.; Ciuffi, A.; Leipzig, J.; Berry, C.C.; Bushman, F.D. HIV integration site selection: Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007, 17, 1186–1194. [Google Scholar] [CrossRef] [Green Version]
- Engelman, A.; Cherepanov, P. The lentiviral integrase binding protein LEDGF/p75 and HIV-1 replication. PLoS Pathog. 2008, 4, e1000046. [Google Scholar] [CrossRef]
- Yang, Q.; Lucas, A.; Son, S.; Chang, L.J. Overlapping enhancer/promoter and transcriptional termination signals in the lentiviral long terminal repeat. Retrovirology 2007, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Keller, B.; Makalou, N.; Sutton, R.E. Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther. 2001, 12, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, N.A.; Romanowska, M.; Haritonova, N.; Foerster, J. Optimized production and concentration of lentiviral vectors containing large inserts. J. Gene Med. 2007, 9, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.I.; Linden, R.M. The cryptic life style of adeno-associated virus. Bioessays 1995, 17, 237–245. [Google Scholar] [CrossRef]
- Kotin, R.M. Prospects for the use of adeno-associated virus as a vector for human gene therapy. Hum. Gene Ther. 1994, 5, 793–801. [Google Scholar] [CrossRef]
- Srivastava, A. Replication of the adeno-associated virus DNA termini in vitro. Intervirology 1987, 27, 138–147. [Google Scholar] [CrossRef]
- Giraud, C.; Winocour, E.; Berns, K.I. Site-specific integration by adeno-associated virus is directed by a cellular DNA sequence. Proc. Natl. Acad. Sci. USA 1994, 91, 10039–10043. [Google Scholar] [CrossRef] [Green Version]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [Green Version]
- Samulski, R.J.; Zhu, X.; Xiao, X.; Brook, J.D.; Housman, D.E.; Epstein, N.; Hunter, L.A. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J. 1991, 10, 3941–3950. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Kyöstiö, S.R.; Kotin, R.M.; Owens, R.A. Adeno-associated virus (AAV) Rep proteins mediate complex formation between AAV DNA and its integration site in human DNA. Proc. Natl. Acad. Sci. USA 1994, 91, 5808–5812. [Google Scholar] [CrossRef] [Green Version]
- McCarty, D.M.; Young, S.M., Jr.; Samulski, R.J. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu. Rev. Genet. 2004, 38, 819–845. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, K.; Lewis, S.M.; Wu, X.; Ma, C.; Munroe, D.J.; Fuess, S.; Storm, T.A.; Kay, M.A.; Nakai, H. DNA palindromes with a modest arm length of greater, similar 20 base pairs are a significant target for recombinant adeno-associated virus vector integration in the liver, muscles, and heart in mice. J. Virol. 2007, 81, 11290–11303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.G.; Trobridge, G.D.; Petek, L.M.; Jacobs, M.A.; Kaul, R.; Russell, D.W. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J. Virol. 2005, 79, 11434–11442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonelli, F.; Maguire, A.M.; Testa, F.; Pierce, E.A.; Mingozzi, F.; Bennicelli, J.L.; Rossi, S.; Marshall, K.; Banfi, S.; Surace, E.M.; et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol. Ther. 2010, 18, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Méthot, J.; Déry, S.; Brisson, D.; Essiembre, C.; Tremblay, G.; Tremblay, K.; de Wal, J.; Twisk, J.; van den Bulk, N.; et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: An open-label trial. Gene Ther. 2013, 20, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Kaeppel, C.; Beattie, S.G.; Fronza, R.; van Logtenstein, R.; Salmon, F.; Schmidt, S.; Wolf, S.; Nowrouzi, A.; Glimm, H.; von Kalle, C.; et al. A largely random AAV integration profile after LPLD gene therapy. Nat. Med. 2013, 19, 889–891. [Google Scholar] [CrossRef]
- Rosas, L.E.; Grieves, J.L.; Zaraspe, K.; La Perle, K.M.; Fu, H.; McCarty, D.M. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol. Ther. 2012, 20, 2098–2110. [Google Scholar] [CrossRef] [Green Version]
- Donsante, A.; Miller, D.G.; Li, Y.; Vogler, C.; Brunt, E.M.; Russell, D.W.; Sands, M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science 2007, 317, 477. [Google Scholar] [CrossRef] [Green Version]
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Walia, J.S.; Altaleb, N.; Bello, A.; Kruck, C.; LaFave, M.C.; Varshney, G.K.; Burgess, S.M.; Chowdhury, B.; Hurlbut, D.; Hemming, R.; et al. Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol. Ther. 2015, 23, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, X.; Danos, O.; Scherman, D.; Kichler, A. A comparison of synthetic oligodeoxynucleotides, DNA fragments and AAV-1 for targeted episomal and chromosomal gene repair. BMC Biotechnol. 2009, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Exline, C.M.; DeClercq, J.J.; Llewellyn, G.N.; Hayward, S.B.; Li, P.W.; Shivak, D.A.; Surosky, R.T.; Gregory, P.D.; Holmes, M.C.; et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015, 33, 1256–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, R.D.; Brinster, R.L. Germ-line transformation of mice. Annu. Rev. Genet. 1986, 20, 465–499. [Google Scholar] [CrossRef]
- Chiang, C.; Jacobsen, J.C.; Ernst, C.; Hanscom, C.; Heilbut, A.; Blumenthal, I.; Mills, R.E.; Kirby, A.; Lindgren, A.M.; Rudiger, S.R.; et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat. Genet. 2012, 44, 390–397. [Google Scholar] [CrossRef]
- Laboulaye, M.A.; Duan, X.; Qiao, M.; Whitney, I.E.; Sanes, J.R. Mapping transgene insertion sites reveals complex interactions between mouse transgenes and neighboring endogenous genes. Front. Mol. Neurosci. 2018, 11, 385. [Google Scholar] [CrossRef] [Green Version]
- Rossant, J.; Nutter, L.M.; Gertsenstein, M. Engineering the embryo. Proc. Natl. Acad. Sci. USA 2011, 108, 7659–7660. [Google Scholar] [CrossRef] [Green Version]
- Mohsen, Z.; Sim, H.; Garcia-Galiano, D.; Han, X.; Bellefontaine, N.; Saunders, T.L.; Elias, C.F. Sexually dimorphic distribution of Prokr2 neurons revealed by the Prokr2-Cre mouse model. Brain Struct. Funct. 2017, 222, 4111–4129. [Google Scholar] [CrossRef]
- Cheng, T.L.; Li, S.; Yuan, B.; Wang, X.; Zhou, W.; Qiu, Z. Expanding C-T base editing toolkit with diversified cytidine deaminases. Nat. Commun. 2019, 10, 3612. [Google Scholar] [CrossRef] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 55, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, E.; Sun, Y.; Wei, W.; Yuan, T.; Ying, W.; Sun, H.; Yuan, L.; Steinmetz, L.M.; Li, Y.; Yang, H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 2019, 364, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, Y.; Uno, Y.; Yoshimi, K.; Kunihiro, Y.; Yoshimura, T.; Tanaka, T.; Ishikubo, H.; Hiraoka, Y.; Takemoto, N.; Tanaka, T.; et al. CLICK: One-step generation of conditional knockout mice. BMC Genom. 2018, 19, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, N.; Mizutani, E.; Sato, H.; Kasai, M.; Ogawa, A.; Suchy, F.; Yamaguchi, T.; Nakauchi, H. Intra-embryo gene cassette knockin by CRISPR/Cas9-mediated genome editing with adeno-associated viral vector. iScience 2018, 9, 286–297. [Google Scholar] [CrossRef] [Green Version]
- Fielder, T.J.; Montoliu, L. Transgenic production benchmarks. In Advanced Protocols for Animal Transgenesis: An ISTT Manual; Pease, S., Saunders, T.L., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 81–97. [Google Scholar]
- Riesenberg, S.; Chintalapati, M.; Macak, D.; Kanis, P.; Maricic, T.; Pääbo, S. Simultaneous precise editing of multiple genes in human cells. Nucleic Acids Res. 2019, 47, e116. [Google Scholar] [CrossRef]
- Song, J.; Yang, D.; Xu, J.; Zhu, T.; Chen, Y.E.; Zhang, J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016, 7, 10548. [Google Scholar] [CrossRef] [Green Version]
- Wilde, J.J.; Aida, T.; Wienisch, M.; Zhang, Q.; Qi, P.; Feng, G. Efficient zygotic genome editing via RAD51-enhanced interhomolog repair. bioRxiv 2018, 263699. [Google Scholar] [CrossRef]


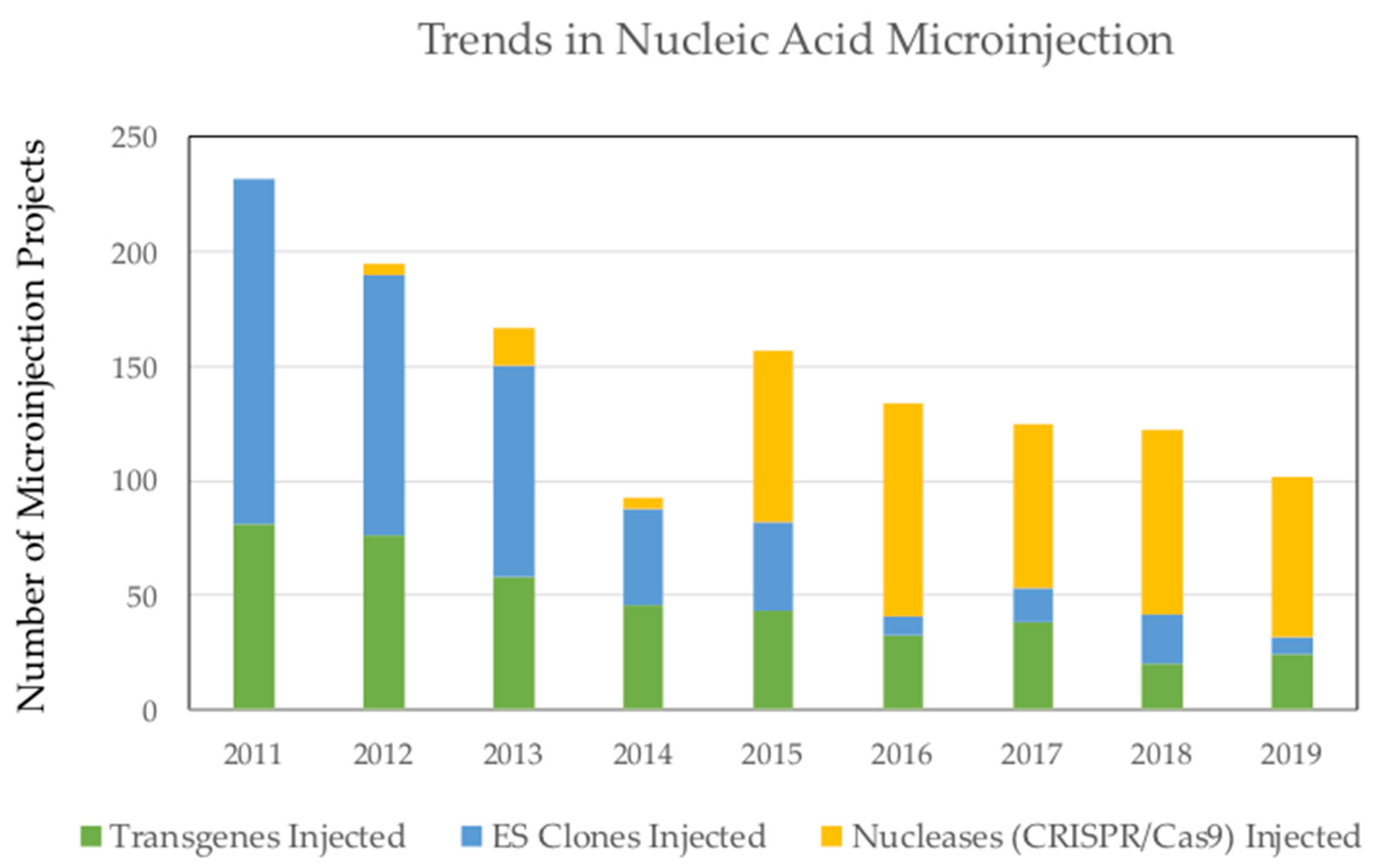{kind=link}
| Targeted Gene | Purpose 1 | Cas9 Format 2 | DNA Donor Format 3 | Efficiency 4 | Reference |
|---|---|---|---|---|---|
| Pitx1 | Conditional | RNP | ssDNA | 5.3 | [108] |
| Ambra1 | Conditional | RNP | ssDNA | 9.5 | [108] |
| Col12a1 | Conditional | RNP | ssDNA | 3.8 | [108] |
| Ubr5 | Conditional | RNP | ssDNA | 12.5 | [108] |
| Syt1 | Conditional | RNP | ssDNA | 2.2 | [108] |
| Syt9 | Conditional | RNP | ssDNA | 2.4 | [108] |
| PPP2r2a | Conditional | RNP | ssDNA | 9.1 | [108] |
| Fgf8 | Reporter | RNP | ssDNA | 7.7 | [108] |
| Slc26a5 | Reporter | RNP | ssDNA | 4.5 | [108] |
| Mafb | Reporter | RNP | ssDNA | 3.8 | [108] |
| Otoa | Reporter | RNP | ssDNA | 5.6 | [108] |
| Mmp9 | Reporter | RNP | ssDNA | 16.0 | [108] |
| Mmp13 | Reporter | RNP | ssDNA | 7.7 | [108] |
| Sox2 | Reporter | Cas9 mRNA | dsDNA | 2.0 | [63] |
| Nanog | Reporter | Cas9-mSA | BioPCR | 2.7 | [63] |
| Gata6 | Reporter | Cas9 mRNA | dsDNA | 2.0 | [63] |
| Gata6 | Reporter | Cas9-mSA | BioPCR | 5.0 | [63] |
| Cdk9 | Reporter | Cas9 mRNA | dsDNA | 4.0 | [63] |
| ROSA26 | Reporter | Cas9 mRNA | dsDNA | 1.3 | [63] |
| Cdx2 | Reporter | Cas9 mRNA | HMEJ | 5.9 | [110] |
| Cdx2 | Reporter | Cas9 mRNA | Tild | 1.9 | [110] |
| Dbh | Reporter | Cas9 mRNA | Tild | 3.6 | [110] |
| Sp8 | Reporter | Cas9 mRNA | HMEJ | 3.2 | [110] |
| Sp8 | Reporter | Cas9 mRNA | Tild | 2.0 | [110] |
| Tdtomato | Reporter | Cas9 mRNA | Tild | 3.5 | [110] |
| Nr3c2 | Conditional | Cas9 mRNA | Tild | 4.8 | [110] |
| Lhx6 | Conditional | Cas9 mRNA | Tild | 6.3 | [110] |
| Serpina3 | Conditional | Cas9 mRNA | ssDNA | 3.5 | [186] |
| Tyr | Conditional | Cas9 mRNA | ssDNA | 2.0 | [186] |
| mKIAA1322 | Conditional | Cas9 mRNA | ssDNA | 3.0 | [186] |
| Serpina3n | Conditional | Cas9 mRNA | ssDNA | 1.3 | [186] |
| Mct4 | Conditional | Cas9 mRNA | ssDNA | 1.5 | [186] |
| Rat Vapb | Conditional | Cas9 mRNA | ssDNA | 3.9 | [186] |
| ROSA26 | Reporter | RNP | AAV | 1.2 | [187] |
| ROSA26 | Reporter | RNP | AAV | 4.8 | [187] |
| Rat ROSA26 | Reporter | RNP | AAV | 4.2 | [187] |
| Rat ROSA26 | Reporter | RNP | AAV | 5.4 | [187] |
| ROSA26 | Reporter | Cas9 mRNA | dsDNA | 3.4 | [112] |
| ROSA26 | Reporter | Cas9 mRNA | dsDNA | 2.1 | [112] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lanigan, T.M.; Kopera, H.C.; Saunders, T.L. Principles of Genetic Engineering. Genes 2020, 11, 291. https://doi.org/10.3390/genes11030291
Lanigan TM, Kopera HC, Saunders TL. Principles of Genetic Engineering. Genes. 2020; 11(3):291. https://doi.org/10.3390/genes11030291
Chicago/Turabian StyleLanigan, Thomas M., Huira C. Kopera, and Thomas L. Saunders. 2020. "Principles of Genetic Engineering" Genes 11, no. 3: 291. https://doi.org/10.3390/genes11030291