
Disruption of Glioblastoma Multiforme Cell Circuits with Cinnamaldehyde Highlights Potential Targets with Implications for Novel Therapeutic Strategies
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Materials
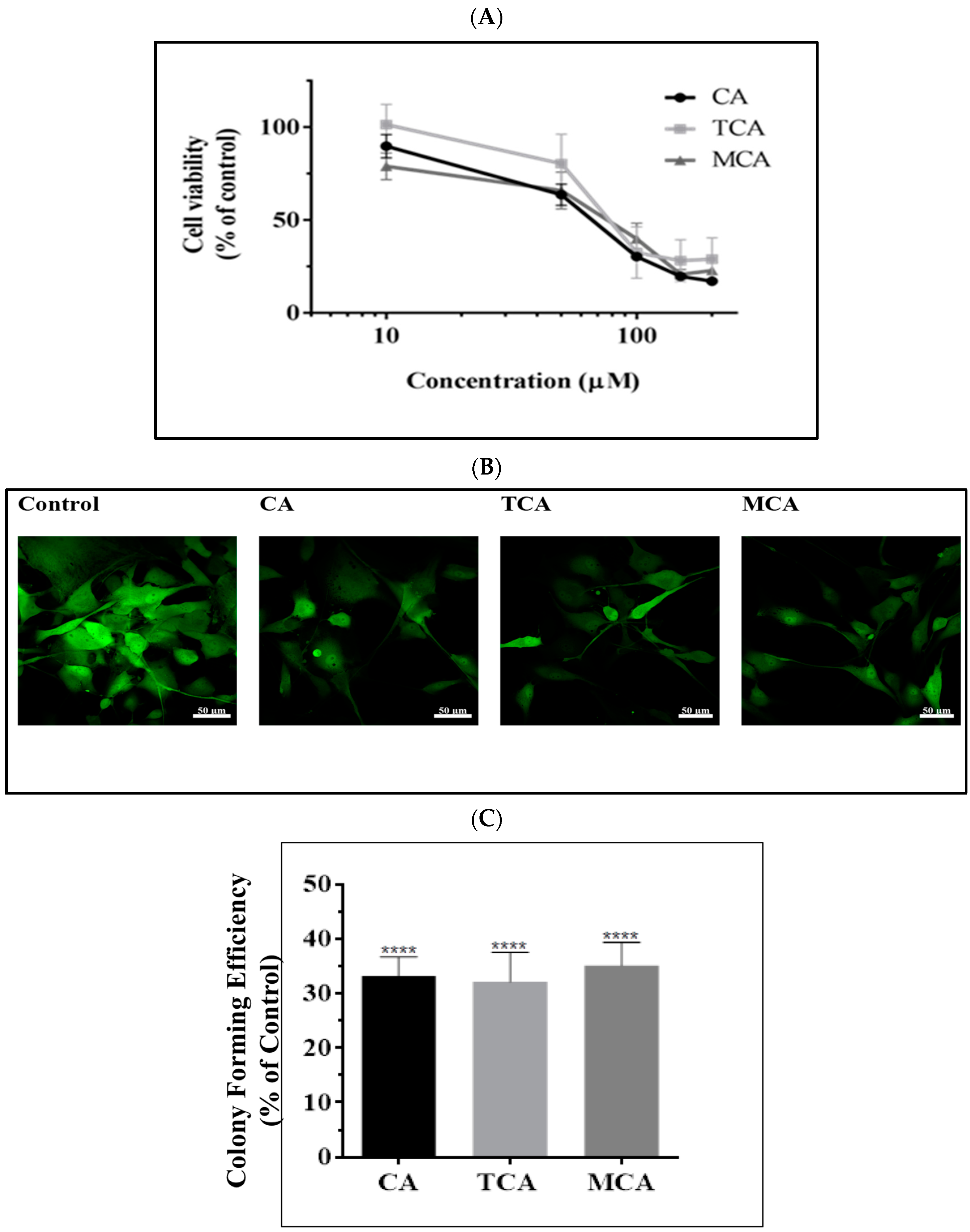
2.2. Cell Toxicity Assay
2.3. Clonogenic Assay
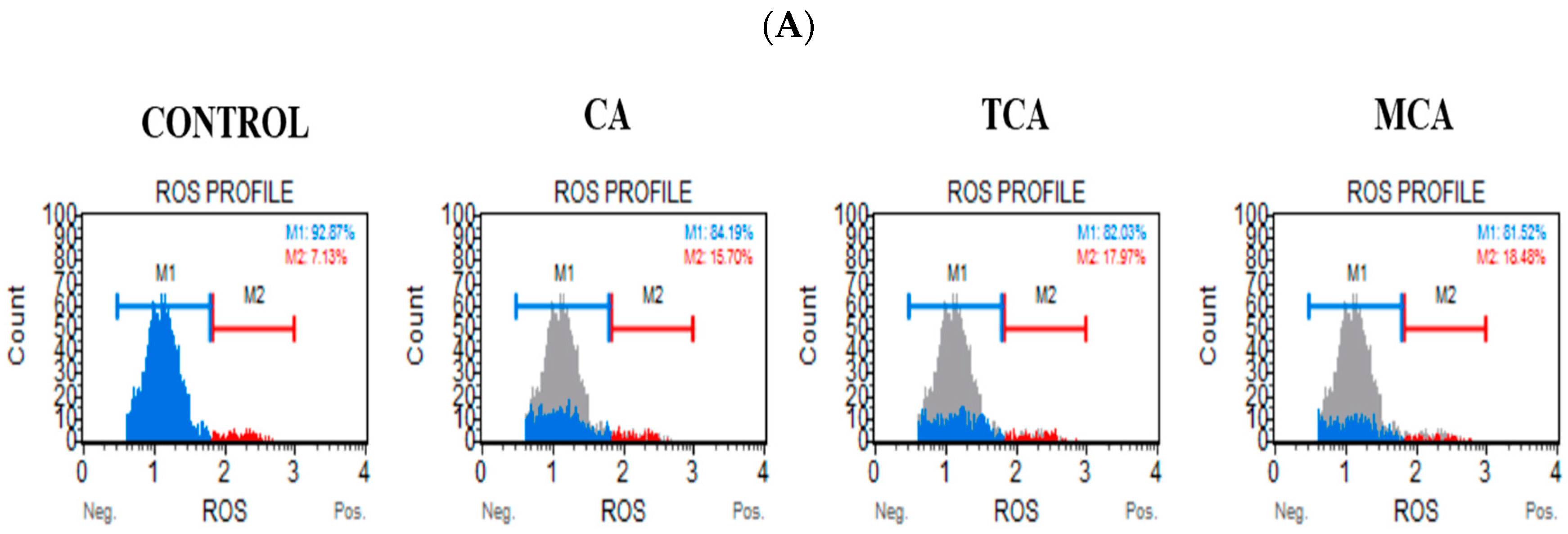
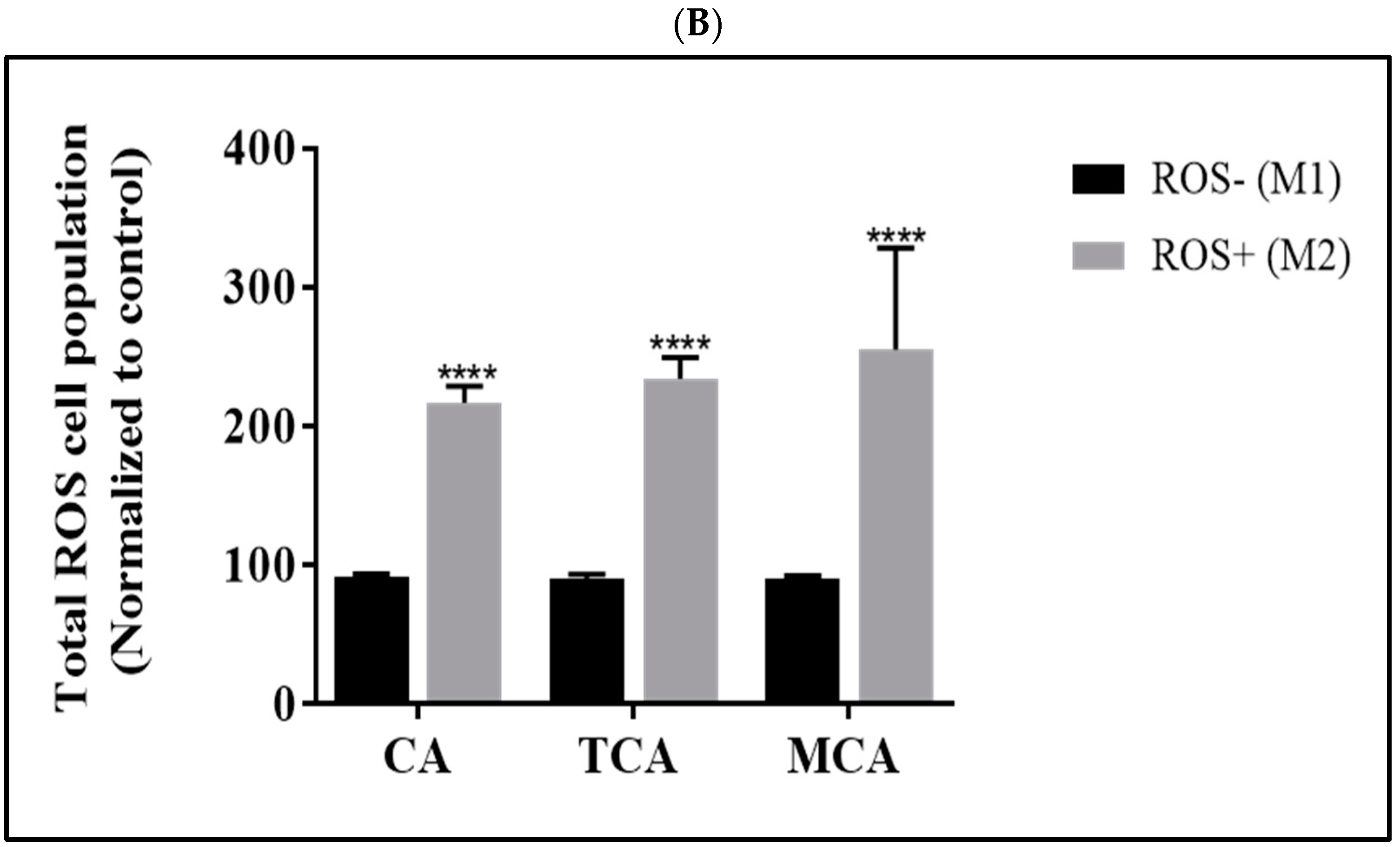
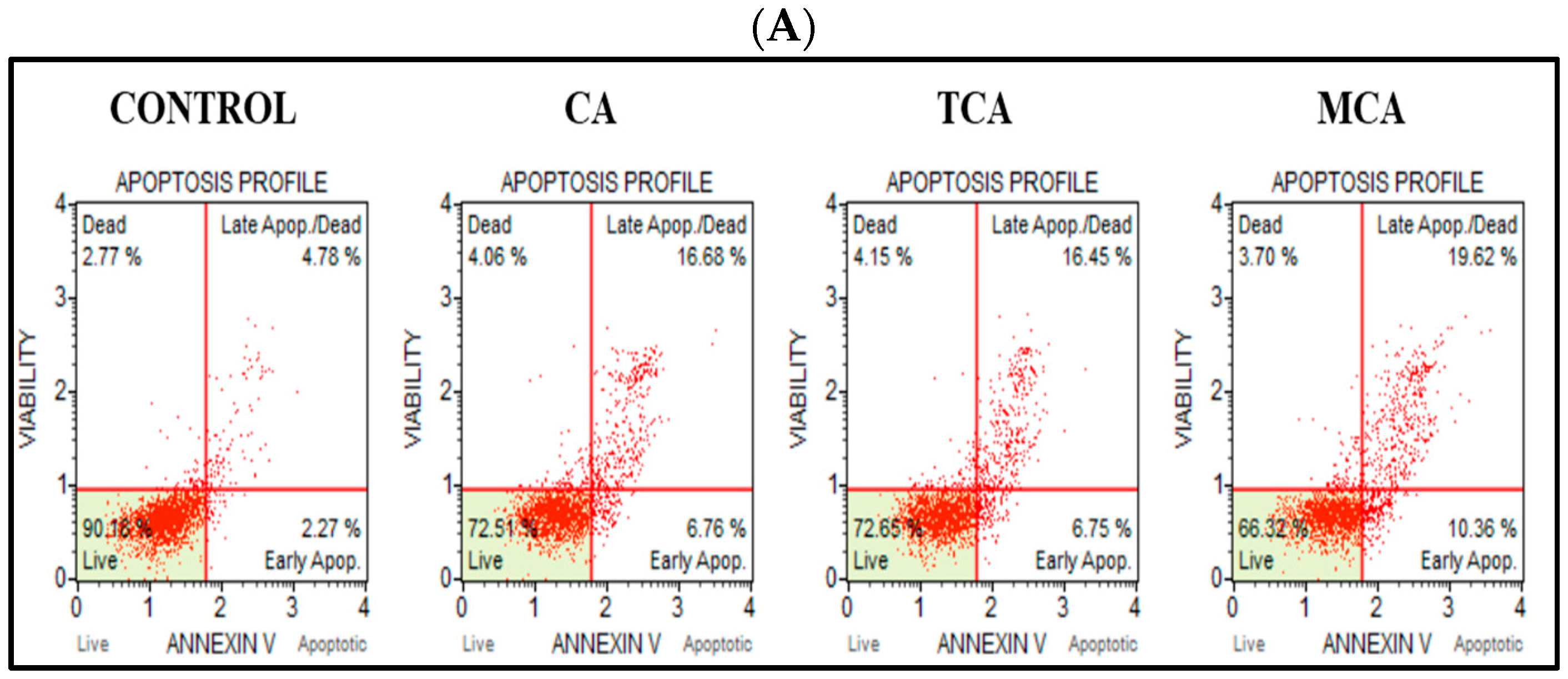
2.4. Flow Cytometry Assays
2.5. Proteomic Analysis
2.6. Statistical Analysis
3. Results
3.1. Impact of CA, TCA and MCA on U87eGFP Cells
3.2. Reactive Oxygen Species Levels Were Elevated in U87eGFP Cells after Treatment with CA, TCA and MCA
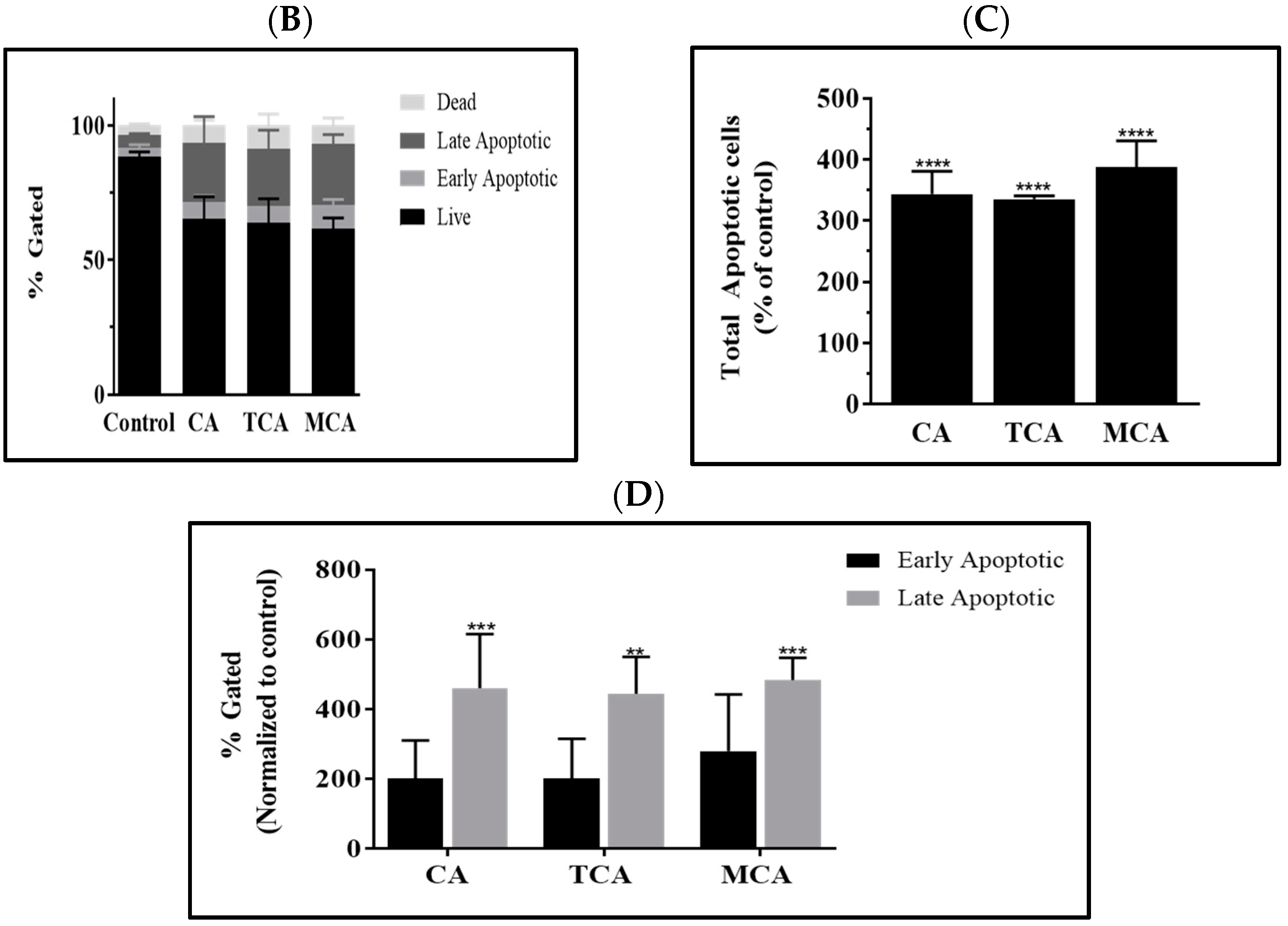
3.3. Programmed Cell-Death Pathway Was Impacted by CA, TCA and MCA in U87eGFP Cells
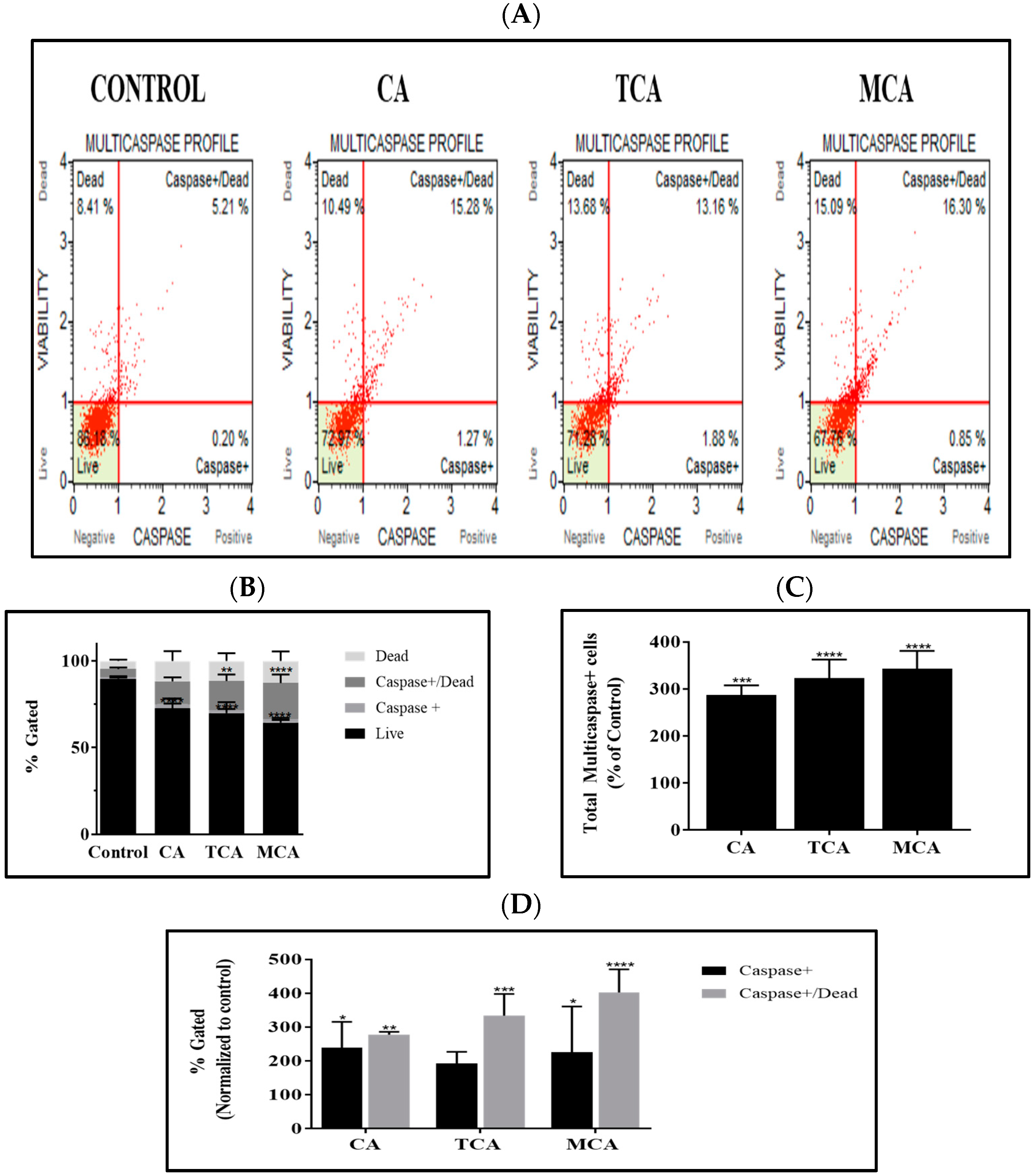
3.4. Multicaspase Was Elicited by CA, TCA and MCA in U87eGFP Cells
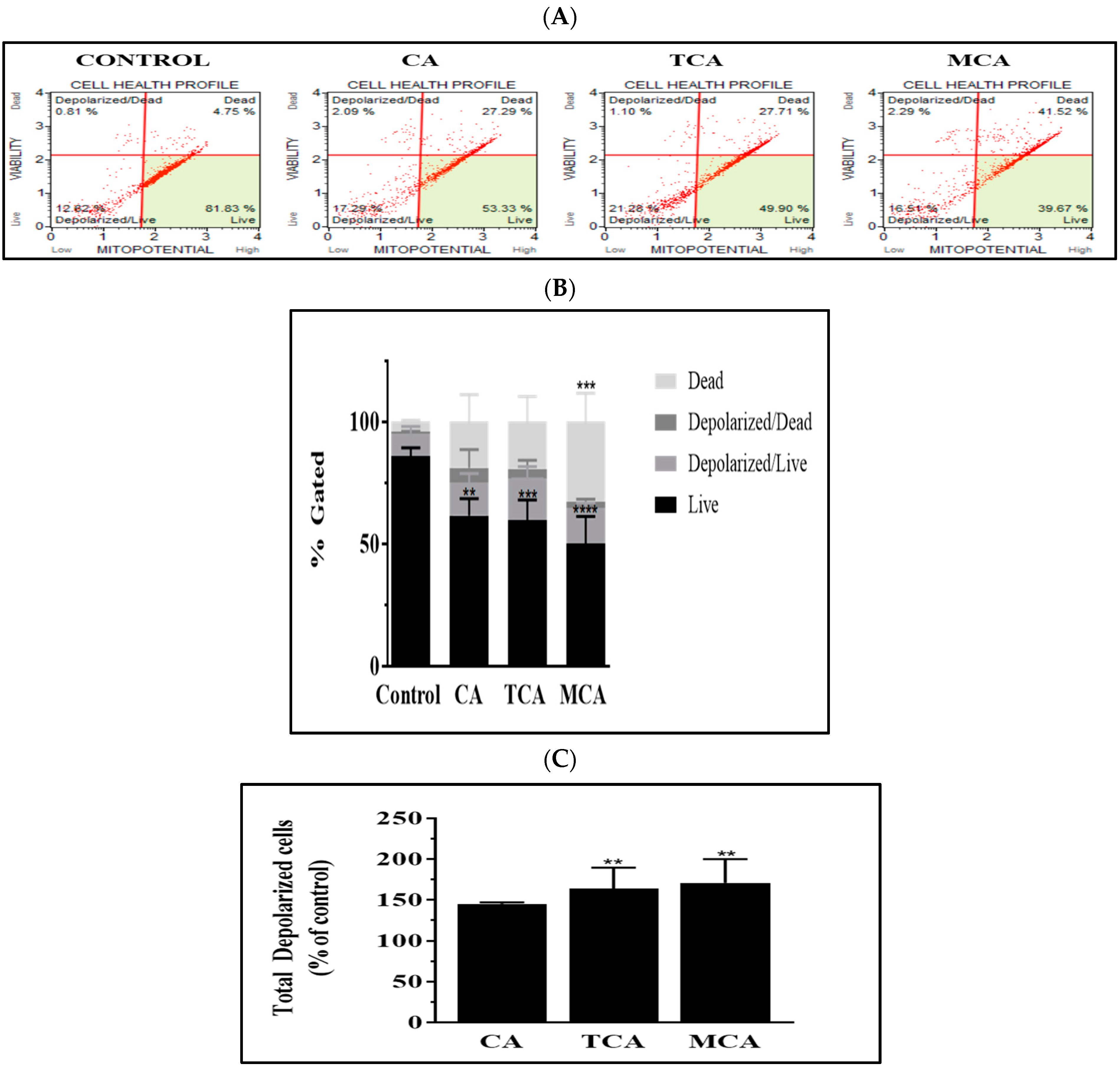
3.5. The Intrinsic Programmed Cell-Death Pathway Was Impacted by TCA and MCA in U87eGFP Cells
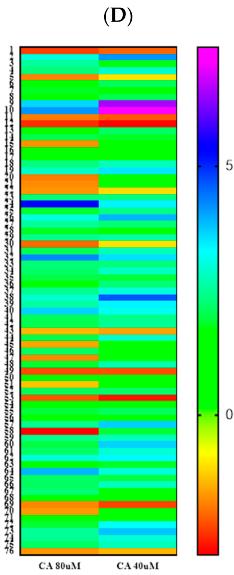
3.6. Proteomic Analysis Reveals Entities of Pivotal Signaling Pathways Differentially Regulated in U87eGFP Cells after Administration of CA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Davis, F.G.; Freels, S.; Grutsch, J.; Barlas, S.; Brem, S. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: An analysis based on Surveillance, Epidemiology, and End Results (SEER) data, 1973–1991. J. Neurosurg. 1998, 88, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Betancur, M.; Patel, G.D.; Valmikinathan, C.M.; Mukhatyar, V.J.; Vakharia, A.; Pai, S.B.; Brahma, B.; MacDonald, T.J.; Bellamkonda, R.V. Guiding intracortical brain tumour cells to an extracortical cytotoxic hydrogel using aligned polymeric nanofibres. Nat. Mater. 2014, 13, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Guntuku, L.; Naidu, V.G.; Yerra, V.G. Mitochondrial Dysfunction in Gliomas: Pharmacotherapeutic Potential of Natural Compounds. Curr. Neuropharmacol. 2016, 14, 567–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef]
- Osoba, D.; Brada, M.; Yung, W.K.; Prados, M. Health-related quality of life in patients treated with temozolomide versus procarbazine for recurrent glioblastoma multiforme. J. Clin. Oncol. 2000, 18, 1481–1491. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Lodise, O.; Patil, K.; Karshenboym, I.; Prombo, S.; Chukwueke, C.; Pai, S.B. Inhibition of Prostate Cancer Cells by 4,5-Dicaffeoylquinic Acid through Cell Cycle Arrest. Prostate Cancer 2019, 2019, 4520645. [Google Scholar] [CrossRef] [Green Version]
- Dhir, H.; Choudhury, M.; Patil, K.; Cheung, C.; Bodlak, A.; Pardo, D.; Adams, A.; Travaglino, S.; Rojas, J.A.; Pai, S.B. Interception of Signaling Circuits of Esophageal Adenocarcinoma Cells by Resveratrol Reveals Molecular and Immunomodulatory Signatures. Cancers 2021, 13, 5811. [Google Scholar] [CrossRef]
- Jiang, H.; Patil, K.; Vashi, A.; Wang, Y.; Strickland, E.; Pai, S.B. Cellular molecular and proteomic profiling deciphers the SIRT1 controlled cell death pathways in esophageal adenocarcinoma cells. Cancer Treat. Res. Commun. 2021, 26, 100271. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Olsen, J.R.; Yuan, X.; Cheng, P.F.; Levesque, M.P.; Brokstad, K.A.; Hoffman, P.S.; Oyan, A.M.; Zhang, W.; Kalland, K.H.; et al. Small molecule promotes beta-catenin citrullination and inhibits Wnt signaling in cancer. Nat. Chem. Biol. 2018, 14, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Tonge, R.; Shaw, J.; Middleton, B.; Rowlinson, R.; Rayner, S.; Young, J.; Pognan, F.; Hawkins, E.; Currie, I.; Davison, M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics 2001, 1, 377–396. [Google Scholar] [CrossRef]
- Hong, S.H.; Ismail, I.A.; Kang, S.M.; Han, D.C.; Kwon, B.M. Cinnamaldehydes in Cancer Chemotherapy. Phytother. Res. 2016, 30, 754–767. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Jayasena, T.; Poljak, A.; Braidy, N.; Zhong, L.; Rowlands, B.; Muenchhoff, J.; Grant, R.; Smythe, G.; Teo, C.; Raftery, M.; et al. Application of Targeted Mass Spectrometry for the Quantification of Sirtuins in the Central Nervous System. Sci. Rep. 2016, 6, 35391. [Google Scholar] [CrossRef] [Green Version]
- Spencer, V.A. Actin-towards a deeper understanding of the relationship between tissue context, cellular function and tumorigenesis. Cancers 2011, 3, 4269–4280. [Google Scholar] [CrossRef] [Green Version]
- Masoumi, S.; Harisankar, A.; Gracias, A.; Bachinger, F.; Fufa, T.; Chandrasekar, G.; Gaunitz, F.; Walfridsson, J.; Kitambi, S.S. Understanding cytoskeleton regulators in glioblastoma multiforme for therapy design. Drug. Des. Dev. Ther. 2016, 10, 2881–2897. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Xiong, X.; Sun, Y. The role of ribosomal proteins in the regulation of cell proliferation, tumorigenesis, and genomic integrity. Sci. China Life Sci. 2016, 59, 656–672. [Google Scholar] [CrossRef]
- Smolock, E.M.; Korshunov, V.A.; Glazko, G.; Qiu, X.; Gerloff, J.; Berk, B.C. Ribosomal protein L17, RpL17, is an inhibitor of vascular smooth muscle growth and carotid intima formation. Circulation 2012, 126, 2418–2427. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, P.E.; Miralem, T.; Maines, M.D. Biliverdin reductase: A target for cancer therapy? Front. Pharmacol. 2015, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Le Francois, B.G.; Maroun, J.A.; Birnboim, H.C. Expression of thymidylate synthase in human cells is an early G(1) event regulated by CDK4 and p16INK4A but not E2F. Br. J. Cancer 2007, 97, 1242–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riganti, C.; Massaia, M. Inhibition of the mevalonate pathway to override chemoresistance and promote the immunogenic demise of cancer cells: Killing two birds with one stone. Oncoimmunology 2013, 2, e25770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Xiao, Y.; Xia, F.; Wu, P.; Zhao, T.; Xie, S.; Wang, R.; Wen, Q.; Zhou, W.; Xu, H.; et al. The mevalonate coordinates energy input and cell proliferation. Cell Death Dis. 2019, 10, 327. [Google Scholar] [CrossRef] [Green Version]
- Gruenbacher, G.; Thurnher, M. Mevalonate metabolism governs cancer immune surveillance. Oncoimmunology 2017, 6, e1342917. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadasu, E.; Kang, R.; Usmani, A.; Borlongan, C.V. Effects of Lovastatin on Brain Cancer Cells. Cell Transplant. 2022, 31. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Guminska, M.; Ignacak, J.; Kedryna, T.; Stachurska, M.B. Tumor-specific pyruvate kinase isoenzyme M2 involved in biochemical strategy of energy generation in neoplastic cells. Acta Biochim. Pol. 1997, 44, 711–724. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, J.S.; Oh, Y.; Lee, J.S.; Park, H.E.; Lee, H.; Park, Y.S.; Kyung, S.Y.; Kim, H.S.; Yoon, S. PKM2 Is Overexpressed in Glioma Tissues, and Its Inhibition Highly Increases Late Apoptosis in U87MG Cells With Low-density Specificity. In Vivo 2022, 36, 694–703. [Google Scholar] [CrossRef]
- Kefas, B.; Comeau, L.; Erdle, N.; Montgomery, E.; Amos, S.; Purow, B. Pyruvate kinase M2 is a target of the tumor-suppressive microRNA-326 and regulates the survival of glioma cells. Neuro-Oncology 2010, 12, 1102–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.; Mukherjee, J.; Ito, M.; Chaumeil, M.M.; Jalbert, L.E.; Gaensler, K.; Ronen, S.M.; Nelson, S.J.; Pieper, R.O. Changes in pyruvate metabolism detected by magnetic resonance imaging are linked to DNA damage and serve as a sensor of temozolomide response in glioblastoma cells. Cancer Res. 2014, 74, 7115–7124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinat, C.; Patel, C.B.; Haywood, T.; Murty, S.; Naya, L.; Castillo, J.B.; Reyes, S.T.; Phillips, M.; Buccino, P.; Shen, B.; et al. A Clinical PET Imaging Tracer ([(18)F]DASA-23) to Monitor Pyruvate Kinase M2-Induced Glycolytic Reprogramming in Glioblastoma. Clin. Cancer Res. 2021, 27, 6467–6478. [Google Scholar] [CrossRef] [PubMed]









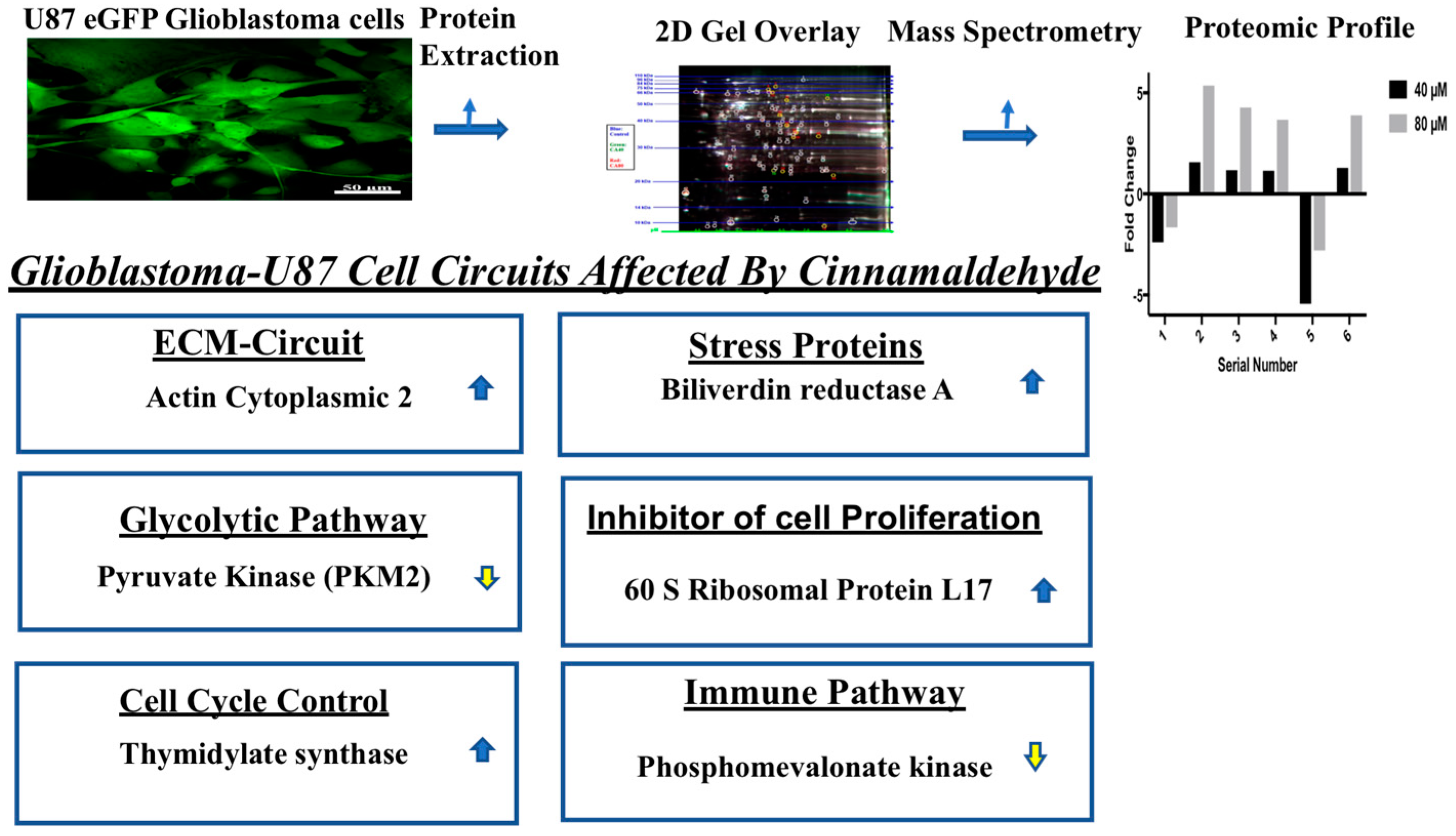
 ) and downregulated (
) and downregulated ( ) entities in glioblastoma cells after treatment with cinnamaldehyde.
) and downregulated () entities in glioblastoma cells after treatment with cinnamaldehyde.
) entities in glioblastoma cells after treatment with cinnamaldehyde.
) and downregulated () entities in glioblastoma cells after treatment with cinnamaldehyde.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fold Change | Protein ID | |
|---|---|---|
| 40 µM | 80 µM | |
| −2.4 | −1.66 | Pyruvate Kinase 2 (PKM2) |
| +1.56 | +5.36 | Actin cytoplasmic 2 |
| +1.18 | +4.27 | Biliverdin reductase A |
| +1.15 | +3.67 | Thymidylate synthase |
| −5.44 | −2.81 | Phosphomevalonate kinase |
| +1.29 | +3.89 | 60 S ribosomal protein L17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, S.; Patil, K.; Thompson, E.W.; Nakhai, S.A.; Kim, Y.N.; Haynes, C.; Bryant, C.; Pai, S.B. Disruption of Glioblastoma Multiforme Cell Circuits with Cinnamaldehyde Highlights Potential Targets with Implications for Novel Therapeutic Strategies. Cells 2023, 12, 1277. https://doi.org/10.3390/cells12091277
Srivastava S, Patil K, Thompson EW, Nakhai SA, Kim YN, Haynes C, Bryant C, Pai SB. Disruption of Glioblastoma Multiforme Cell Circuits with Cinnamaldehyde Highlights Potential Targets with Implications for Novel Therapeutic Strategies. Cells. 2023; 12(9):1277. https://doi.org/10.3390/cells12091277
Chicago/Turabian StyleSrivastava, Shraddha, Ketki Patil, Elizabeth W. Thompson, Shadi A. Nakhai, Yoo Na Kim, Casey Haynes, Crystal Bryant, and S. Balakrishna Pai. 2023. "Disruption of Glioblastoma Multiforme Cell Circuits with Cinnamaldehyde Highlights Potential Targets with Implications for Novel Therapeutic Strategies" Cells 12, no. 9: 1277. https://doi.org/10.3390/cells12091277