

Transient Receptor Potential Ankyrin 1 (TRPA1) Channel as a Sensor of Oxidative Stress in Cancer Cells

Abstract
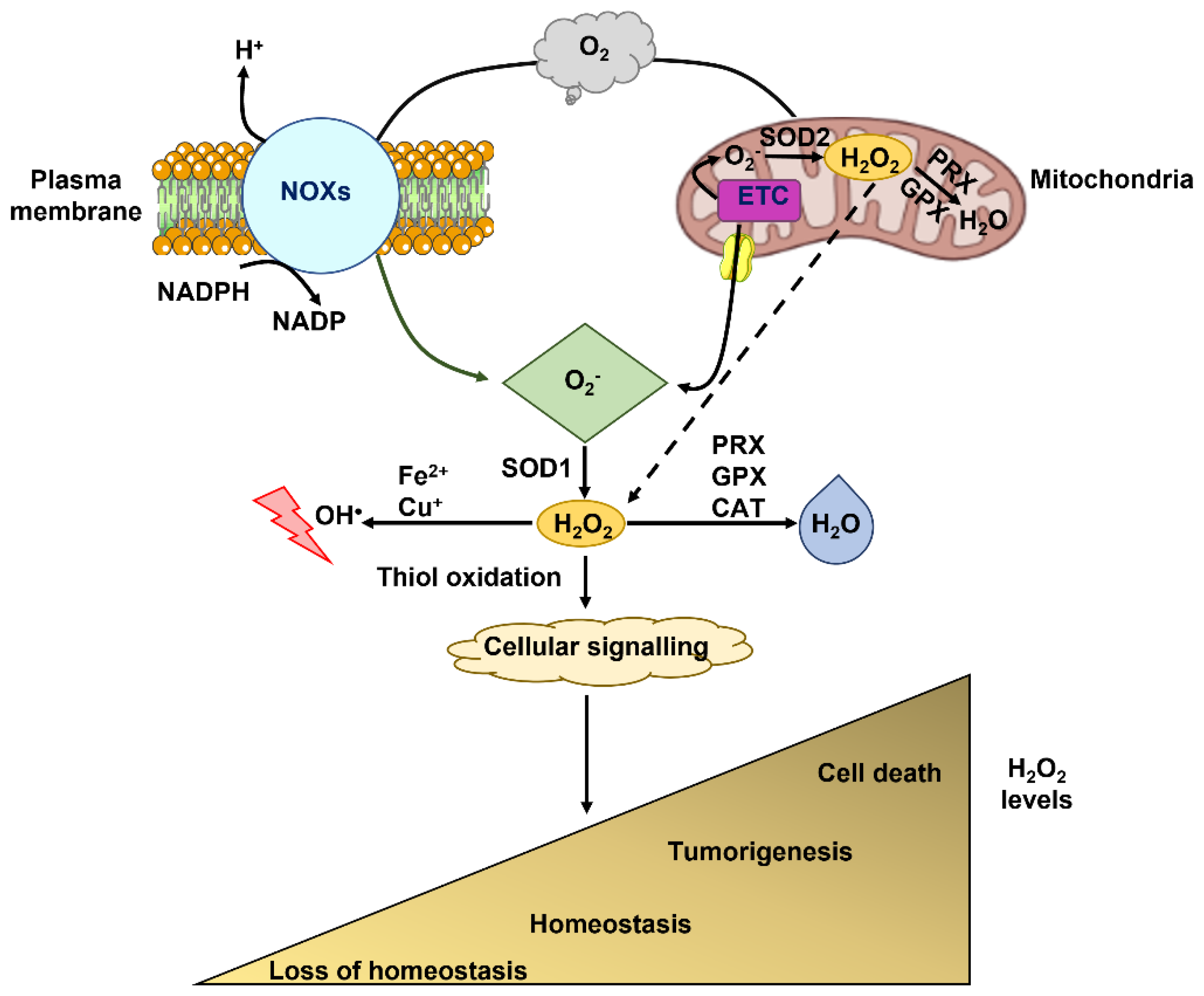
:1. Introduction
2. TRPA1: Molecular Structure, Biophysical Properties, and Pharmacological Sensitivity
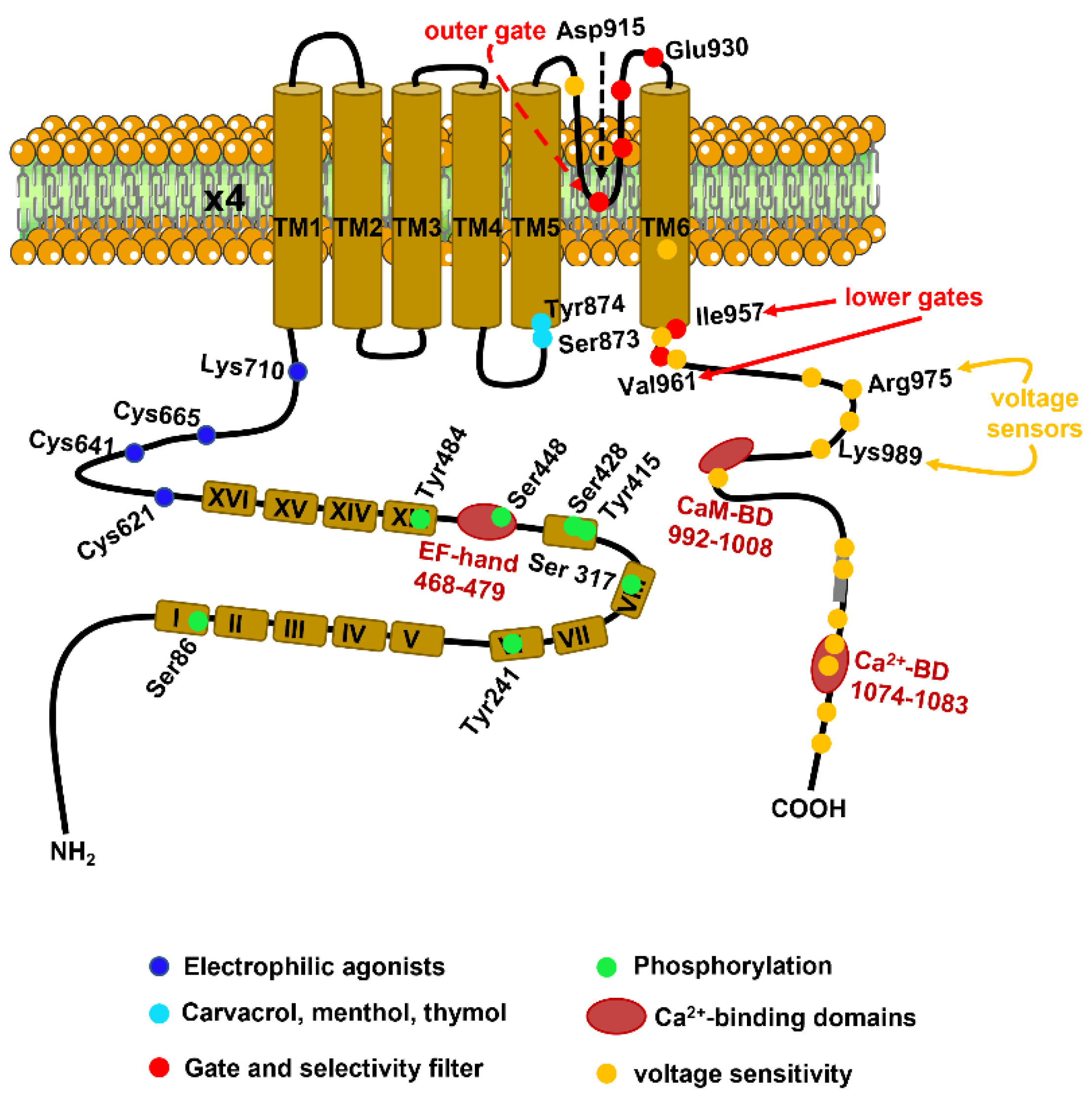
2.1. The Molecular Structure of TRPA1
2.2. Biophysical Properties and Gating Mechanisms of TRPA1
2.3. Ca2+-Dependent Regulation of TRPA1 Activity
2.4. TRPA1 Activity Is Stimulated by Physical Stimuli: Voltage, Temperature and Membrane Deformation
2.5. TRPA1 As a Sensor of Redox Signaling
3. TRPA1-Mediated Ca2+ Signals Support Tumorigenesis
4. TRPA1-Mediates ROS-Dependent Intracellular Ca2+ Signals in Cancer Cells: Survival vs. Apoptosis
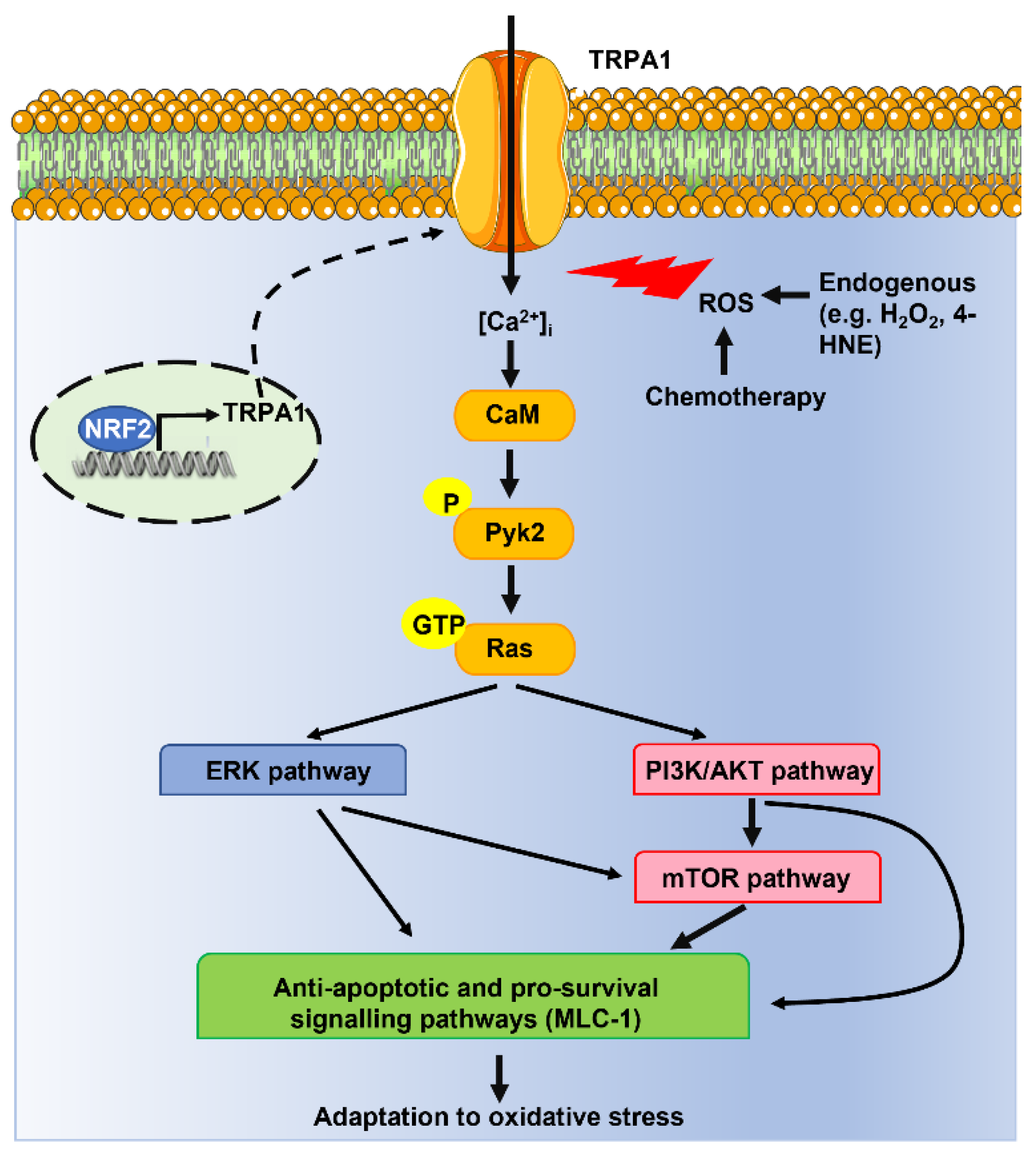
4.1. TRPA1-Mediated Ca2+ Influx Promotes Cancer Cell Survival to Oxidative Stress
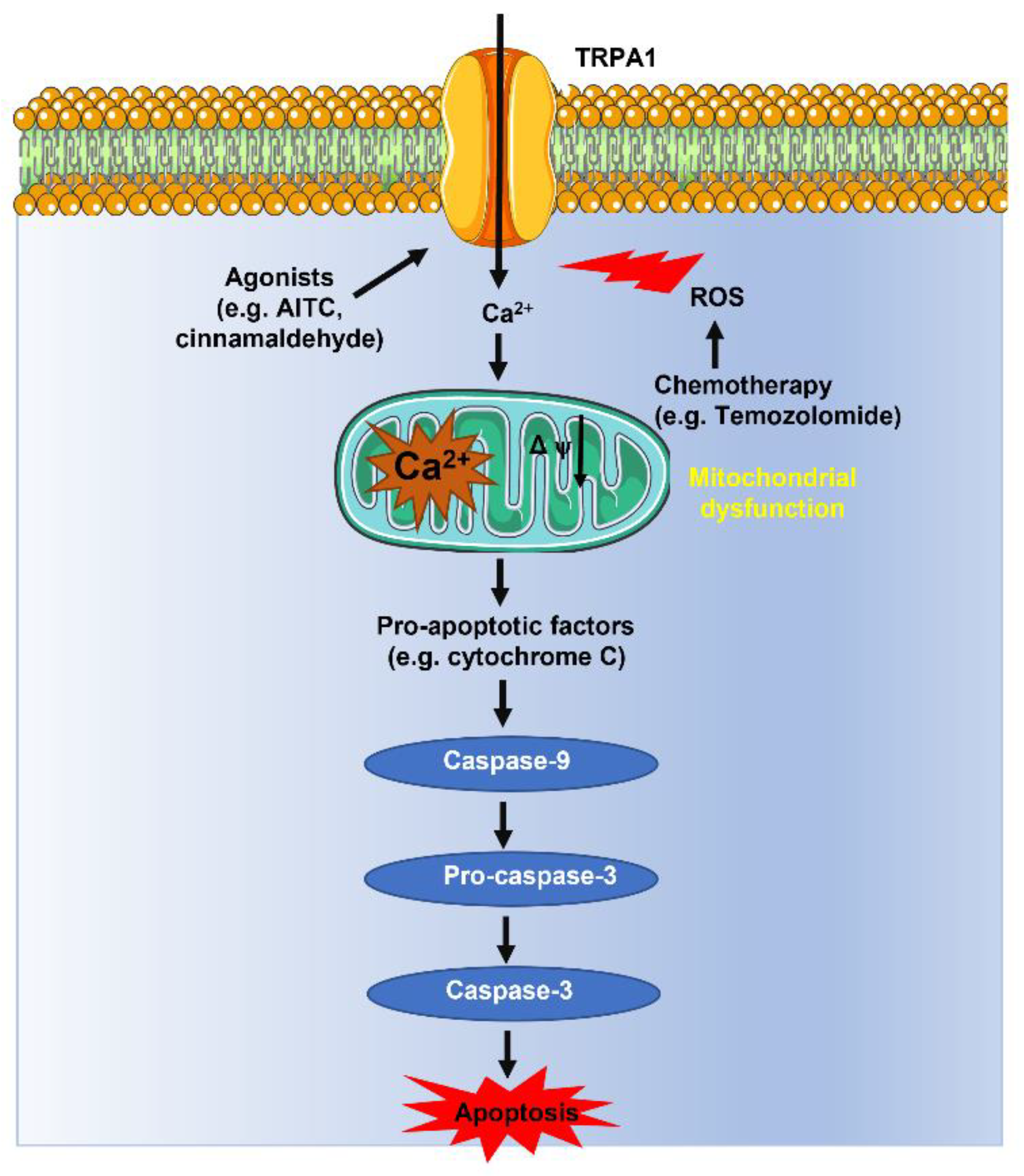
4.2. TRPA1-Mediated Ca2+ Influx Promotes ROS-Dependent Apoptosis in Cancer Cells
4.3. Why Does ROS-Sensitive TRPA1-Mediated Ca2+ Influx Exert both Anti-Cancer and Pro-Tumorigenic Effects in Cancer Cells?
5. Targeting TRPA1 to Sensitize Cancer Cells to Oxidative Stress
5.1. TRPA1 Activators
5.2. TRPA1 Blockers
5.3. Could TRPA1 Manipulation Directly Affect Oxidative Stress in the Tumor Microenvironment?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aboelella, N.S.; Brandle, C.; Kim, T.; Ding, Z.C.; Zhou, G. Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy. Cancers 2021, 13, 986. [Google Scholar] [CrossRef]
- Rezcek, C.R.; Chandel, N.S. The Two Faces of Reactive Oxygen Species in Cancer. Annu. Rev. Cancer Biol. 2017, 1, 79–89. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Remigante, A.; Morabito, R.; Spinelli, S.; Trichilo, V.; Loddo, S.; Sarikas, A.; Dossena, S.; Marino, A. d-Galactose Decreases Anion Exchange Capability through Band 3 Protein in Human Erythrocytes. Antioxidants 2020, 9, 689. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Doskey, C.M.; Buranasudja, V.; Wagner, B.A.; Wilkes, J.G.; Du, J.; Cullen, J.J.; Buettner, G.R. Tumor cells have decreased ability to metabolize H(2)O(2): Implications for pharmacological ascorbate in cancer therapy. Redox Biol. 2016, 10, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.A.; Iskandar, K.B.; Hirpara, J.L.; Clement, M.V.; Pervaiz, S. Hydrogen peroxide-mediated cytosolic acidification is a signal for mitochondrial translocation of Bax during drug-induced apoptosis of tumor cells. Cancer Res. 2004, 64, 7867–7878. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ishigaki, Y.; Zeng, W.; Harimoto, T.; Yin, B.; Chen, Y.; Liao, S.; Liu, Y.; Sun, Y.; Zhang, X.; et al. Generation of hydroxyl radical-activatable ratiometric near-infrared bimodal probes for early monitoring of tumor response to therapy. Nat. Commun. 2021, 12, 6145. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Gamcsik, M.P.; Kasibhatla, M.S.; Teeter, S.D.; Colvin, O.M. Glutathione levels in human tumors. Biomarkers 2012, 17, 671–691. [Google Scholar] [CrossRef]
- Reczek, C.R.; Chandel, N.S. ROS Promotes Cancer Cell Survival through Calcium Signaling. Cancer Cell 2018, 33, 949–951. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca(2+) Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef]
- Luo, M.; Zhou, L.; Huang, Z.; Li, B.; Nice, E.C.; Xu, J.; Huang, C. Antioxidant Therapy in Cancer: Rationale and Progress. Antioxidants 2022, 11, 1128. [Google Scholar] [CrossRef]
- Talavera, K.; Startek, J.B.; Alvarez-Collazo, J.; Boonen, B.; Alpizar, Y.A.; Sanchez, A.; Naert, R.; Nilius, B. Mammalian Transient Receptor Potential TRPA1 Channels: From Structure to Disease. Physiol. Rev. 2020, 100, 725–803. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Hogestatt, E.D. Trpa1. Handb. Exp. Pharmacol. 2014, 222, 583–630. [Google Scholar] [CrossRef]
- Singh, R.; Adhya, P.; Sharma, S.S. Redox-sensitive TRP channels: A promising pharmacological target in chemotherapy-induced peripheral neuropathy. Expert Opin. Ther. Targets 2021, 25, 529–545. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Shimizu, S. Significance of TRP channels in oxidative stress. Eur. J. Pharmacol. 2016, 793, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Kiss, F.; Kormos, V.; Szoke, E.; Kecskes, A.; Toth, N.; Steib, A.; Szallasi, A.; Scheich, B.; Gaszner, B.; Kun, J.; et al. Functional Transient Receptor Potential Ankyrin 1 and Vanilloid 1 Ion Channels Are Overexpressed in Human Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 1921. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, F.; Selescu, T.; Domocos, D.; Marutescu, L.; Chiritoiu, G.; Chelaru, N.R.; Dima, S.; Mihailescu, D.; Babes, A.; Cucu, D. Functional expression of the transient receptor potential ankyrin type 1 channel in pancreatic adenocarcinoma cells. Sci. Rep. 2021, 11, 2018. [Google Scholar] [CrossRef]
- Mergler, S.; Derckx, R.; Reinach, P.S.; Garreis, F.; Bohm, A.; Schmelzer, L.; Skosyrski, S.; Ramesh, N.; Abdelmessih, S.; Polat, O.K.; et al. Calcium regulation by temperature-sensitive transient receptor potential channels in human uveal melanoma cells. Cell. Signal. 2014, 26, 56–69. [Google Scholar] [CrossRef]
- Duitama, M.; Moreno, Y.; Santander, S.P.; Casas, Z.; Sutachan, J.J.; Torres, Y.P.; Albarracin, S.L. TRP Channels as Molecular Targets to Relieve Cancer Pain. Biomolecules 2021, 12, 1. [Google Scholar] [CrossRef]
- de Almeida, A.S.; Bernardes, L.B.; Trevisan, G. TRP channels in cancer pain. Eur. J. Pharmacol. 2021, 904, 174185. [Google Scholar] [CrossRef]
- Chen, H.; Li, C.; Hu, H.; Zhang, B. Activated TRPA1 plays a therapeutic role in TMZ resistance in glioblastoma by altering mitochondrial dynamics. BMC Mol. Cell Biol. 2022, 23, 38. [Google Scholar] [CrossRef]
- Takahashi, N.; Chen, H.Y.; Harris, I.S.; Stover, D.G.; Selfors, L.M.; Bronson, R.T.; Deraedt, T.; Cichowski, K.; Welm, A.L.; Mori, Y.; et al. Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer Cell 2018, 33, 985–1003.e1007. [Google Scholar] [CrossRef]
- De Logu, F.; Souza Monteiro de Araujo, D.; Ugolini, F.; Iannone, L.F.; Vannucchi, M.; Portelli, F.; Landini, L.; Titiz, M.; De Giorgi, V.; Geppetti, P.; et al. The TRPA1 Channel Amplifies the Oxidative Stress Signal in Melanoma. Cells 2021, 10, 3131. [Google Scholar] [CrossRef]
- Kaya, M.M.; Kaya, I.; Naziroglu, M. Transient receptor potential channel stimulation induced oxidative stress and apoptosis in the colon of mice with colitis-associated colon cancer: Modulator role of Sambucus ebulus L. Mol. Biol. Rep. 2022, 50, 2207–2220. [Google Scholar] [CrossRef] [PubMed]
- Deveci, H.A.; Akyuva, Y.; Nur, G.; Naziroglu, M. Alpha lipoic acid attenuates hypoxia-induced apoptosis, inflammation and mitochondrial oxidative stress via inhibition of TRPA1 channel in human glioblastoma cell line. Biomed. Pharmacother. 2019, 111, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.A.; Stohr, S.; Meister, M.; Aigner, A.; Gudermann, T.; Buech, T.R. Stimulation of the chemosensory TRPA1 cation channel by volatile toxic substances promotes cell survival of small cell lung cancer cells. Biochem. Pharmacol. 2013, 85, 426–438. [Google Scholar] [CrossRef]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef] [PubMed]
- Himmel, N.J.; Cox, D.N. Transient receptor potential channels: Current perspectives on evolution, structure, function and nomenclature. Proc. Biol. Sci. 2020, 287, 20201309. [Google Scholar] [CrossRef]
- Alvarado, M.G.; Thakore, P.; Earley, S. Transient Receptor Potential Channel Ankyrin 1: A Unique Regulator of Vascular Function. Cells 2021, 10, 1167. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Zhu, F.; Liu, B.; Chai, Z.; Wu, Q.; Hu, M.; Wang, Y.; Huang, R.; Zhang, X.; Wu, X.; et al. Intracellular TRPA1 mediates Ca2+ release from lysosomes in dorsal root ganglion neurons. J. Cell Biol. 2016, 215, 369–381. [Google Scholar] [CrossRef]
- Jaquemar, D.; Schenker, T.; Trueb, B. An ankyrin-like protein with transmembrane domains is specifically lost after oncogenic transformation of human fibroblasts. J. Biol. Chem. 1999, 274, 7325–7333. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Armache, J.P.; Gao, Y.; Cheng, Y.; Julius, D. Structure of the TRPA1 ion channel suggests regulatory mechanisms. Nature 2015, 520, 511–517. [Google Scholar] [CrossRef]
- Cordero-Morales, J.F.; Gracheva, E.O.; Julius, D. Cytoplasmic ankyrin repeats of transient receptor potential A1 (TRPA1) dictate sensitivity to thermal and chemical stimuli. Proc. Natl. Acad. Sci. USA 2011, 108, E1184–E1191. [Google Scholar] [CrossRef]
- Samad, A.; Sura, L.; Benedikt, J.; Ettrich, R.; Minofar, B.; Teisinger, J.; Vlachova, V. The C-terminal basic residues contribute to the chemical- and voltage-dependent activation of TRPA1. Biochem. J. 2011, 433, 197–204. [Google Scholar] [CrossRef]
- Meents, J.E.; Ciotu, C.I.; Fischer, M.J.M. TRPA1: A molecular view. J. Neurophysiol. 2019, 121, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. eLife 2021, 10, e63040. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Ali, S.; Earley, S. Regulation of vascular tone by transient receptor potential ankyrin 1 channels. Curr. Top. Membr. 2020, 85, 119–150. [Google Scholar] [CrossRef] [PubMed]
- Karashima, Y.; Prenen, J.; Talavera, K.; Janssens, A.; Voets, T.; Nilius, B. Agonist-induced changes in Ca(2+) permeation through the nociceptor cation channel TRPA1. Biophys. J. 2010, 98, 773–783. [Google Scholar] [CrossRef]
- Banke, T.G.; Chaplan, S.R.; Wickenden, A.D. Dynamic changes in the TRPA1 selectivity filter lead to progressive but reversible pore dilation. Am. J. Physiol. Cell Physiol. 2010, 298, C1457–C1468. [Google Scholar] [CrossRef]
- Chen, J.; Kim, D.; Bianchi, B.R.; Cavanaugh, E.J.; Faltynek, C.R.; Kym, P.R.; Reilly, R.M. Pore dilation occurs in TRPA1 but not in TRPM8 channels. Mol. Pain 2009, 5, 3. [Google Scholar] [CrossRef]
- Li, M.; Toombes, G.E.; Silberberg, S.D.; Swartz, K.J. Physical basis of apparent pore dilation of ATP-activated P2X receptor channels. Nat. Neurosci. 2015, 18, 1577–1583. [Google Scholar] [CrossRef]
- Jordt, S.E.; Bautista, D.M.; Chuang, H.H.; McKemy, D.D.; Zygmunt, P.M.; Hogestatt, E.D.; Meng, I.D.; Julius, D. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 2004, 427, 260–265. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Chang, R.B.; Waters, H.N.; McKemy, D.D.; Liman, E.R. The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J. Biol. Chem. 2008, 283, 32691–32703. [Google Scholar] [CrossRef]
- Doerner, J.F.; Gisselmann, G.; Hatt, H.; Wetzel, C.H. Transient receptor potential channel A1 is directly gated by calcium ions. J. Biol. Chem. 2007, 282, 13180–13189. [Google Scholar] [CrossRef]
- Sura, L.; Zima, V.; Marsakova, L.; Hynkova, A.; Barvik, I.; Vlachova, V. C-terminal acidic cluster is involved in Ca2+-induced regulation of human transient receptor potential ankyrin 1 channel. J. Biol. Chem. 2012, 287, 18067–18077. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Leeson-Payne, A.T.; Jaggar, J.H.; Zhang, X. Calmodulin is responsible for Ca(2+)-dependent regulation of TRPA1 Channels. Sci. Rep. 2017, 7, 45098. [Google Scholar] [CrossRef]
- Moparthi, L.; Kichko, T.I.; Eberhardt, M.; Hogestatt, E.D.; Kjellbom, P.; Johanson, U.; Reeh, P.W.; Leffler, A.; Filipovic, M.R.; Zygmunt, P.M. Human TRPA1 is a heat sensor displaying intrinsic U-shaped thermosensitivity. Sci. Rep. 2016, 6, 28763. [Google Scholar] [CrossRef]
- Moparthi, L.; Sinica, V.; Moparthi, V.K.; Kreir, M.; Vignane, T.; Filipovic, M.R.; Vlachova, V.; Zygmunt, P.M. The human TRPA1 intrinsic cold and heat sensitivity involves separate channel structures beyond the N-ARD domain. Nat. Commun. 2022, 13, 6113. [Google Scholar] [CrossRef]
- Sinica, V.; Vlachova, V. Transient receptor potential ankyrin 1 channel: An evolutionarily tuned thermosensor. Physiol. Res. 2021, 70, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Moparthi, L.; Survery, S.; Kreir, M.; Simonsen, C.; Kjellbom, P.; Hogestatt, E.D.; Johanson, U.; Zygmunt, P.M. Human TRPA1 is intrinsically cold- and chemosensitive with and without its N-terminal ankyrin repeat domain. Proc. Natl. Acad. Sci. USA 2014, 111, 16901–16906. [Google Scholar] [CrossRef] [PubMed]
- Reeh, P.W.; Fischer, M.J.M. Nobel somatosensations and pain. Pflug. Arch. 2022, 474, 405–420. [Google Scholar] [CrossRef]
- Moparthi, L.; Zygmunt, P.M. Human TRPA1 is an inherently mechanosensitive bilayer-gated ion channel. Cell Calcium 2020, 91, 102255. [Google Scholar] [CrossRef]
- Sakaguchi, R.; Mori, Y. Transient receptor potential (TRP) channels: Biosensors for redox environmental stimuli and cellular status. Free Radic. Biol. Med. 2020, 146, 36–44. [Google Scholar] [CrossRef]
- Ferrera, L.; Barbieri, R.; Picco, C.; Zuccolini, P.; Remigante, A.; Bertelli, S.; Fumagalli, M.R.; Zifarelli, G.; La Porta, C.A.M.; Gavazzo, P.; et al. TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase. Int. J. Mol. Sci. 2021, 22, 8359. [Google Scholar] [CrossRef]
- Remigante, A.; Spinelli, S.; Marino, A.; Pusch, M.; Morabito, R.; Dossena, S. Oxidative Stress and Immune Response in Melanoma: Ion Channels as Targets of Therapy. Int. J. Mol. Sci. 2023, 24, 887. [Google Scholar] [CrossRef]
- Ogawa, N.; Kurokawa, T.; Fujiwara, K.; Polat, O.K.; Badr, H.; Takahashi, N.; Mori, Y. Functional and Structural Divergence in Human TRPV1 Channel Subunits by Oxidative Cysteine Modification. J. Biol. Chem. 2016, 291, 4197–4210. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Tullii, G.; Vismara, M.; Pellegata, A.F.; Lodola, F.; Guidetti, G.; Rosti, V.; Antognazza, M.R.; Moccia, F. Conjugated polymers mediate intracellular Ca(2+) signals in circulating endothelial colony forming cells through the reactive oxygen species-dependent activation of Transient Receptor Potential Vanilloid 1 (TRPV1). Cell Calcium 2022, 101, 102502. [Google Scholar] [CrossRef]
- Andersson, D.A.; Gentry, C.; Moss, S.; Bevan, S. Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J. Neurosci. 2008, 28, 2485–2494. [Google Scholar] [CrossRef]
- Bessac, B.F.; Sivula, M.; von Hehn, C.A.; Escalera, J.; Cohn, L.; Jordt, S.E. TRPA1 is a major oxidant sensor in murine airway sensory neurons. J. Clin. Investig. 2008, 118, 1899–1910. [Google Scholar] [CrossRef]
- Miyamoto, T.; Dubin, A.E.; Petrus, M.J.; Patapoutian, A. TRPV1 and TRPA1 mediate peripheral nitric oxide-induced nociception in mice. PLoS ONE 2009, 4, e7596. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.A.; Filipovic, M.R.; Gentry, C.; Eberhardt, M.; Vastani, N.; Leffler, A.; Reeh, P.; Bevan, S. Streptozotocin Stimulates the Ion Channel TRPA1 Directly: Involvement of peroxynitrite. J. Biol. Chem. 2015, 290, 15185–15196. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Morabito, R.; Remigante, A.; Gugliandolo, E.; Britti, D.; Cuzzocrea, S.; Marino, A. Susceptibility of erythrocytes from different sources to xenobiotics-induced lysis. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2019, 221, 68–72. [Google Scholar] [CrossRef]
- Taylor-Clark, T.E.; Ghatta, S.; Bettner, W.; Undem, B.J. Nitrooleic acid, an endogenous product of nitrative stress, activates nociceptive sensory nerves via the direct activation of TRPA1. Mol. Pharmacol. 2009, 75, 820–829. [Google Scholar] [CrossRef]
- Bahia, P.K.; Parks, T.A.; Stanford, K.R.; Mitchell, D.A.; Varma, S.; Stevens, S.M., Jr.; Taylor-Clark, T.E. The exceptionally high reactivity of Cys 621 is critical for electrophilic activation of the sensory nerve ion channel TRPA1. J. Gen. Physiol. 2016, 147, 451–465. [Google Scholar] [CrossRef]
- Zayats, V.; Samad, A.; Minofar, B.; Roelofs, K.E.; Stockner, T.; Ettrich, R. Regulation of the transient receptor potential channel TRPA1 by its N-terminal ankyrin repeat domain. J. Mol. Model. 2013, 19, 4689–4700. [Google Scholar] [CrossRef] [PubMed]
- Hinman, A.; Chuang, H.H.; Bautista, D.M.; Julius, D. TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. USA 2006, 103, 19564–19568. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Kuwaki, T.; Kiyonaka, S.; Numata, T.; Kozai, D.; Mizuno, Y.; Yamamoto, S.; Naito, S.; Knevels, E.; Carmeliet, P.; et al. TRPA1 underlies a sensing mechanism for O2. Nat. Chem. Biol. 2011, 7, 701–711. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Duran, R.V. Prolyl hydroxylase domain enzymes and their role in cell signaling and cancer metabolism. Int. J. Biochem. Cell Biol. 2016, 80, 71–80. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Mori, Y.; Takahashi, N.; Kurokawa, T.; Kiyonaka, S. TRP channels in oxygen physiology: Distinctive functional properties and roles of TRPA1 in O(2) sensing. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 464–482. [Google Scholar] [CrossRef] [PubMed]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef]
- Armani, G.; Pozzi, E.; Pagani, A.; Porta, C.; Rizzo, M.; Cicognini, D.; Rovati, B.; Moccia, F.; Pedrazzoli, P.; Ferraris, E. The heterogeneity of cancer endothelium: The relevance of angiogenesis and endothelial progenitor cells in cancer microenvironment. Microvasc. Res. 2021, 138, 104189. [Google Scholar] [CrossRef]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Scarpellino, G.; Genova, T.; Avanzato, D.; Bernardini, M.; Bianco, S.; Petrillo, S.; Tolosano, E.; de Almeida Vieira, J.R.; Bussolati, B.; Fiorio Pla, A.; et al. Purinergic Calcium Signals in Tumor-Derived Endothelium. Cancers 2019, 11, 766. [Google Scholar] [CrossRef]
- Scarpellino, G.; Munaron, L.; Cantelmo, A.R.; Fiorio Pla, A. Calcium-Permeable Channels in Tumor Vascularization: Peculiar, Sensors of Microenvironmental Chemical and Physical Cues. Rev. Physiol. Biochem. Pharmacol. 2020, 182, 111–137. [Google Scholar] [CrossRef]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca(2+) Signalling and Cancer Hallmarks: Two-Pore Channels on the Move, TRPML1 Lags Behind! Cancers 2018, 11, 27. [Google Scholar] [CrossRef]
- Zhong, T.; Zhang, W.; Guo, H.; Pan, X.; Chen, X.; He, Q.; Yang, B.; Ding, L. The regulatory and modulatory roles of TRP family channels in malignant tumors and relevant therapeutic strategies. Acta Pharm. Sin. B 2022, 12, 1761–1780. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Laforenza, U.; Cattaneo, F.; Ruffinatti, F.A.; Poletto, V.; Massa, M.; Tancredi, R.; Zuccolo, E.; Khdar, A.D.; Riccardi, A.; et al. VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 2017, 8, 95223–95246. [Google Scholar] [CrossRef] [PubMed]
- Fliniaux, I.; Germain, E.; Farfariello, V.; Prevarskaya, N. TRPs and Ca(2+) in cell death and survival. Cell Calcium 2018, 69, 4–18. [Google Scholar] [CrossRef]
- Perna, A.; Sellitto, C.; Komici, K.; Hay, E.; Rocca, A.; De Blasiis, P.; Lucariello, A.; Moccia, F.; Guerra, G. Transient Receptor Potential (TRP) Channels in Tumor Vascularization. Int. J. Mol. Sci. 2022, 23, 4253. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Ohta, T. Membrane translocation of transient receptor potential ankyrin 1 induced by inflammatory cytokines in lung cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 587–593. [Google Scholar] [CrossRef]
- Derouiche, S.; Mariot, P.; Warnier, M.; Vancauwenberghe, E.; Bidaux, G.; Gosset, P.; Mauroy, B.; Bonnal, J.L.; Slomianny, C.; Delcourt, P.; et al. Activation of TRPA1 Channel by Antibacterial Agent Triclosan Induces VEGF Secretion in Human Prostate Cancer Stromal Cells. Cancer Prev. Res. (Phila.) 2017, 10, 177–187. [Google Scholar] [CrossRef]
- Moccia, F.; Poletto, V. May the remodeling of the Ca(2)(+) toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochim. Biophys. Acta 2015, 1853, 1958–1973. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient Receptor Potential Channel Expression Signatures in Tumor-Derived Endothelial Cells: Functional Roles in Prostate Cancer Angiogenesis. Cancers 2019, 11, 956. [Google Scholar] [CrossRef] [PubMed]
- Vancauwenberghe, E.; Noyer, L.; Derouiche, S.; Lemonnier, L.; Gosset, P.; Sadofsky, L.R.; Mariot, P.; Warnier, M.; Bokhobza, A.; Slomianny, C.; et al. Activation of mutated TRPA1 ion channel by resveratrol in human prostate cancer associated fibroblasts (CAF). Mol. Carcinog. 2017, 56, 1851–1867. [Google Scholar] [CrossRef]
- Genova, T.; Grolez, G.P.; Camillo, C.; Bernardini, M.; Bokhobza, A.; Richard, E.; Scianna, M.; Lemonnier, L.; Valdembri, D.; Munaron, L.; et al. TRPM8 inhibits endothelial cell migration via a non-channel function by trapping the small GTPase Rap1. J. Cell Biol. 2017, 216, 2107–2130. [Google Scholar] [CrossRef]
- Grolez, G.P.; Chinigo, G.; Barras, A.; Hammadi, M.; Noyer, L.; Kondratska, K.; Bulk, E.; Oullier, T.; Marionneau-Lambot, S.; Le Mee, M.; et al. TRPM8 as an Anti-Tumoral Target in Prostate Cancer Growth and Metastasis Dissemination. Int. J. Mol. Sci. 2022, 23, 6672. [Google Scholar] [CrossRef] [PubMed]
- Soda, T.; Brunetti, V.; Berra-Romani, R.; Moccia, F. The Emerging Role of N-Methyl-D-Aspartate (NMDA) Receptors in the Cardiovascular System: Physiological Implications, Pathological Consequences, and Therapeutic Perspectives. Int. J. Mol. Sci. 2023, 24, 3914. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Scolari, F.; Vismara, M.; Brunetti, V.; Faris, P.; Terribile, G.; Sancini, G.; Berra-Romani, R.; Moccia, F. GABA(A) and GABA(B) Receptors Mediate GABA-Induced Intracellular Ca(2+) Signals in Human Brain Microvascular Endothelial Cells. Cells 2022, 11, 3860. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca(2+) signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef]
- Berrout, J.; Kyriakopoulou, E.; Moparthi, L.; Hogea, A.S.; Berrout, L.; Ivan, C.; Lorger, M.; Boyle, J.; Peers, C.; Muench, S.; et al. TRPA1-FGFR2 binding event is a regulatory oncogenic driver modulated by miRNA-142-3p. Nat. Commun. 2017, 8, 947. [Google Scholar] [CrossRef]
- Louhivuori, L.M.; Bart, G.; Larsson, K.P.; Louhivuori, V.; Nasman, J.; Nordstrom, T.; Koivisto, A.P.; Akerman, K.E. Differentiation dependent expression of TRPA1 and TRPM8 channels in IMR-32 human neuroblastoma cells. J. Cell. Physiol. 2009, 221, 67–74. [Google Scholar] [CrossRef]
- Bansaghi, S.; Golenar, T.; Madesh, M.; Csordas, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnoczky, G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef]
- Joseph, S.K.; Booth, D.M.; Young, M.P.; Hajnoczky, G. Redox regulation of ER and mitochondrial Ca(2+) signaling in cell survival and death. Cell Calcium 2019, 79, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an Emerging Molecular Target to Promote Therapeutic Angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Martinotti, S.; Laforenza, U.; Patrone, M.; Moccia, F.; Ranzato, E. Honey-Mediated Wound Healing: H(2)O(2) Entry through AQP3 Determines Extracellular Ca(2+) Influx. Int. J. Mol. Sci. 2019, 20, 764. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Ronchi, C.; Lodola, F. Optical excitation of organic semiconductors as a highly selective strategy to induce vascular regeneration and tissue repair. Vascul. Pharmacol. 2022, 144, 106998. [Google Scholar] [CrossRef]
- Booth, D.M.; Varnai, P.; Joseph, S.K.; Hajnoczky, G. Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Mol. Cell 2021, 81, 3866–3876. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Varnai, P.; Hajnoczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef]
- Wang, S.; Lv, J.; Pang, Y.; Hu, S.; Lin, Y.; Li, M. Ion channel-targeting near-infrared photothermal switch with synergistic effect for specific cancer therapy. J. Mater. Chem. B 2022, 10, 748–756. [Google Scholar] [CrossRef]
- Nardin, C.; Peres, C.; Mazzarda, F.; Ziraldo, G.; Salvatore, A.M.; Mammano, F. Photosensitizer Activation Drives Apoptosis by Interorganellar Ca(2+) Transfer and Superoxide Production in Bystander Cancer Cells. Cells 2019, 8, 1175. [Google Scholar] [CrossRef]
- Hausmann, D.; Hoffmann, D.C.; Venkataramani, V.; Jung, E.; Horschitz, S.; Tetzlaff, S.K.; Jabali, A.; Hai, L.; Kessler, T.; Azorin, D.D.; et al. Autonomous rhythmic activity in glioma networks drives brain tumour growth. Nature 2023, 613, 179–186. [Google Scholar] [CrossRef]
- Rosa, N.; Ivanova, H.; Wagner, L.E., 2nd; Kale, J.; La Rovere, R.; Welkenhuyzen, K.; Louros, N.; Karamanou, S.; Shabardina, V.; Lemmens, I.; et al. Bcl-xL acts as an inhibitor of IP(3)R channels, thereby antagonizing Ca(2+)-driven apoptosis. Cell Death Differ. 2022, 29, 788–805. [Google Scholar] [CrossRef] [PubMed]
- Adeshakin, F.O.; Adeshakin, A.O.; Afolabi, L.O.; Yan, D.; Zhang, G.; Wan, X. Mechanisms for Modulating Anoikis Resistance in Cancer and the Relevance of Metabolic Reprogramming. Front. Oncol. 2021, 11, 626577. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 2015, 8, ra2. [Google Scholar] [CrossRef]
- Oehler, B.; Kloka, J.; Mohammadi, M.; Ben-Kraiem, A.; Rittner, H.L. D-4F, an ApoA-I mimetic peptide ameliorating TRPA1-mediated nocifensive behaviour in a model of neurogenic inflammation. Mol. Pain 2020, 16, 1744806920903848. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K(+) (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca(2+) signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell. Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Reyes, J.; Baksh, S.; Undem, C.; Caterina, M.; Pearse, D.B.; Shimoda, L.A. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L1467–L1477. [Google Scholar] [CrossRef]
- Pandey, A.; Tripathi, S.C.; Mai, J.; Hanash, S.M.; Shen, H.; Mitra, S.; Rostomily, R.C. Combinatorial Effect of PLK1 Inhibition with Temozolomide and Radiation in Glioblastoma. Cancers 2021, 13, 5114. [Google Scholar] [CrossRef]
- Thomas, A.; Tanaka, M.; Trepel, J.; Reinhold, W.C.; Rajapakse, V.N.; Pommier, Y. Temozolomide in the Era of Precision Medicine. Cancer Res. 2017, 77, 823–826. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Ozkal, B.; Ovey, I.S. Selenium enhances TRPA1 channel-mediated activity of temozolomide in SH-SY5Y neuroblastoma cells. Childs Nerv. Syst. 2020, 36, 1283–1292. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Induces Intracellular Ca(2+) Release through the Two-Pore Channel TPC1 in Metastatic Colorectal Cancer Cells. Cancers 2019, 11, 542. [Google Scholar] [CrossRef] [PubMed]
- Farfariello, V.; Gordienko, D.V.; Mesilmany, L.; Touil, Y.; Germain, E.; Fliniaux, I.; Desruelles, E.; Gkika, D.; Roudbaraki, M.; Shapovalov, G.; et al. TRPC3 shapes the ER-mitochondria Ca(2+) transfer characterizing tumour-promoting senescence. Nat. Commun. 2022, 13, 956. [Google Scholar] [CrossRef]
- Ziegler, D.V.; Vindrieux, D.; Goehrig, D.; Jaber, S.; Collin, G.; Griveau, A.; Wiel, C.; Bendridi, N.; Djebali, S.; Farfariello, V.; et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat. Commun. 2021, 12, 720. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, M.; Bittremieux, M.; Morciano, G.; Giorgi, C.; Pinton, P.; Parys, J.B.; Bultynck, G. Emerging molecular mechanisms in chemotherapy: Ca(2+) signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 2018, 9, 334. [Google Scholar] [CrossRef]
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 2018, 20, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Astesana, V.; Faris, P.; Ferrari, B.; Siciliani, S.; Lim, D.; Biggiogera, M.; De Pascali, S.A.; Fanizzi, F.P.; Roda, E.; Moccia, F.; et al. [Pt(O,O′-acac)(gamma-acac)(DMS)]: Alternative Strategies to Overcome Cisplatin-Induced Side Effects and Resistance in T98G Glioma Cells. Cell. Mol. Neurobiol. 2020, 41, 563–587. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Kondratskyi, A.; Bidaux, G.; Noyer, L.; Vancauwenberghe, E.; Farfariello, V.; Toillon, R.A.; Roudbaraki, M.; Tierny, D.; Bonnal, J.L.; et al. Co-targeting Mitochondrial Ca(2+) Homeostasis and Autophagy Enhances Cancer Cells’ Chemosensitivity. iScience 2020, 23, 101263. [Google Scholar] [CrossRef] [PubMed]
- Tandl, D.; Sponagel, T.; Alansary, D.; Fuck, S.; Smit, T.; Hehlgans, S.; Jakob, B.; Fournier, C.; Niemeyer, B.A.; Rodel, F.; et al. X-ray irradiation triggers immune response in human T-lymphocytes via store-operated Ca2+ entry and NFAT activation. J. Gen. Physiol. 2022, 154, e202112865. [Google Scholar] [CrossRef]
- Lu, F.; Sun, J.; Zheng, Q.; Li, J.; Hu, Y.; Yu, P.; He, H.; Zhao, Y.; Wang, X.; Yang, S.; et al. Imaging elemental events of store-operated Ca(2+) entry in invading cancer cells with plasmalemmal targeted sensors. J. Cell Sci. 2019, 132, 224923. [Google Scholar] [CrossRef]
- Lee, D.; Hong, J.H. Activated PyK2 and Its Associated Molecules Transduce Cellular Signaling from the Cancerous Milieu for Cancer Metastasis. Int. J. Mol. Sci. 2022, 23, 15475. [Google Scholar] [CrossRef]
- Chen, Y.F.; Chiu, W.T.; Chen, Y.T.; Lin, P.Y.; Huang, H.J.; Chou, C.Y.; Chang, H.C.; Tang, M.J.; Shen, M.R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef]
- Vismara, M.; Negri, S.; Scolari, F.; Brunetti, V.; Trivigno, S.M.G.; Faris, P.; Galgano, L.; Soda, T.; Berra-Romani, R.; Canobbio, I.; et al. Platelet-Derived Extracellular Vesicles Stimulate Migration through Partial Remodelling of the Ca(2+) Handling Machinery in MDA-MB-231 Breast Cancer Cells. Cells 2022, 11, 3120. [Google Scholar] [CrossRef]
- Rhythmic Ca2+ Communication Promotes Glioma Cell Proliferation. Cancer Discov. 2023, 13, 259. [CrossRef]
- Alharbi, A.; Zhang, Y.; Parrington, J. Deciphering the Role of Ca(2+) Signalling in Cancer Metastasis: From the Bench to the Bedside. Cancers 2021, 13, 179. [Google Scholar] [CrossRef]
- Sharma, A.; Ramena, G.T.; Elble, R.C. Advances in Intracellular Calcium Signaling Reveal Untapped Targets for Cancer Therapy. Biomedicines 2021, 9, 1077. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Sun, H.Y.; Lau, C.P.; Tse, H.F.; Lee, H.C.; Li, G.R. Cyclic ADP ribose is a novel regulator of intracellular Ca2+ oscillations in human bone marrow mesenchymal stem cells. J. Cell. Mol. Med. 2011, 15, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Balducci, V.; Faris, P.; Balbi, C.; Costa, A.; Negri, S.; Rosti, V.; Bollini, S.; Moccia, F. The human amniotic fluid stem cell secretome triggers intracellular Ca(2+) oscillations, NF-kappaB nuclear translocation and tube formation in human endothelial colony-forming cells. J. Cell. Mol. Med. 2021, 25, 8074–8086. [Google Scholar] [CrossRef] [PubMed]
- Galione, A.; Davis, L.C.; Martucci, L.L.; Morgan, A.J. NAADP-Mediated Ca(2+) Signalling. Handb. Exp. Pharmacol. 2022, 1–32. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca(2+) oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca(2+) release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef]
- Li, B.; Ren, S.; Gao, D.; Li, N.; Wu, M.; Yuan, H.; Zhou, M.; Xing, C. Photothermal Conjugated Polymer Nanoparticles for Suppressing Breast Tumor Growth by Regulating TRPA1 Ion Channels. Adv. Healthc. Mater. 2022, 11, e2102506. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.; Khare, P.; Kondepudi, K.K.; Bishnoi, M. TRPA1: Pharmacology, natural activators and role in obesity prevention. Eur. J. Pharmacol. 2021, 912, 174553. [Google Scholar] [CrossRef]
- Munaron, L.; Avanzato, D.; Moccia, F.; Mancardi, D. Hydrogen sulfide as a regulator of calcium channels. Cell Calcium 2013, 53, 77–84. [Google Scholar] [CrossRef]
- Liu, C.; Reese, R.; Vu, S.; Rouge, L.; Shields, S.D.; Kakiuchi-Kiyota, S.; Chen, H.; Johnson, K.; Shi, Y.P.; Chernov-Rogan, T.; et al. A Non-covalent Ligand Reveals Biased Agonism of the TRPA1 Ion Channel. Neuron 2021, 109, 273–284.e274. [Google Scholar] [CrossRef]
- Lin King, J.V.; Emrick, J.J.; Kelly, M.J.S.; Herzig, V.; King, G.F.; Medzihradszky, K.F.; Julius, D. A Cell-Penetrating Scorpion Toxin Enables Mode-Specific Modulation of TRPA1 and Pain. Cell 2019, 178, 1362–1374.e1316. [Google Scholar] [CrossRef]
- Hu, H.; Tian, J.; Zhu, Y.; Wang, C.; Xiao, R.; Herz, J.M.; Wood, J.D.; Zhu, M.X. Activation of TRPA1 channels by fenamate nonsteroidal anti-inflammatory drugs. Pflug. Arch. 2010, 459, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.M.; Movahed, P.; Hinman, A.; Axelsson, H.E.; Sterner, O.; Hogestatt, E.D.; Julius, D.; Jordt, S.E.; Zygmunt, P.M. Pungent products from garlic activate the sensory ion channel TRPA1. Proc. Natl. Acad. Sci. USA 2005, 102, 12248–12252. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.A.; Gentry, C.; Bevan, S. TRPA1 has a key role in the somatic pro-nociceptive actions of hydrogen sulfide. PLoS ONE 2012, 7, e46917. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Iwasaki, Y.; Narukawa, M.; Iitsuka, Y.; Fukao, T.; Seki, T.; Ariga, T.; Watanabe, T. Diallyl sulfides in garlic activate both TRPA1 and TRPV1. Biochem. Biophys. Res. Commun. 2009, 382, 545–548. [Google Scholar] [CrossRef]
- Bautista, D.M.; Jordt, S.E.; Nikai, T.; Tsuruda, P.R.; Read, A.J.; Poblete, J.; Yamoah, E.N.; Basbaum, A.I.; Julius, D. TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 2006, 124, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Mio, K.; Shiraishi, T.; Kurokawa, T.; Otsuka, S.; Mori, Y.; Uesugi, M. A Potent and Site-Selective Agonist of TRPA1. J. Am. Chem. Soc. 2015, 137, 15859–15864. [Google Scholar] [CrossRef]
- Bessac, B.F.; Jordt, S.E. Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology 2008, 23, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Clark, T.E.; McAlexander, M.A.; Nassenstein, C.; Sheardown, S.A.; Wilson, S.; Thornton, J.; Carr, M.J.; Undem, B.J. Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J. Physiol. 2008, 586, 3447–3459. [Google Scholar] [CrossRef]
- Bianchi, B.R.; Zhang, X.F.; Reilly, R.M.; Kym, P.R.; Yao, B.B.; Chen, J. Species comparison and pharmacological characterization of human, monkey, rat, and mouse TRPA1 channels. J. Pharmacol. Exp. Ther. 2012, 341, 360–368. [Google Scholar] [CrossRef]
- Lee, S.P.; Buber, M.T.; Yang, Q.; Cerne, R.; Cortes, R.Y.; Sprous, D.G.; Bryant, R.W. Thymol and related alkyl phenols activate the hTRPA1 channel. Br. J. Pharmacol. 2008, 153, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Leffler, A.; Lattrell, A.; Kronewald, S.; Niedermirtl, F.; Nau, C. Activation of TRPA1 by membrane permeable local anesthetics. Mol. Pain 2011, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, R.; Kashio, M.; Tominaga, M. Propofol-induced pain sensation involves multiple mechanisms in sensory neurons. Pflug. Arch. 2015, 467, 2011–2020. [Google Scholar] [CrossRef] [PubMed]
- Tasinov, O.; Dincheva, I.; Badjakov, I.; Kiselova-Kaneva, Y.; Galunska, B.; Nogueiras, R.; Ivanova, D. Phytochemical Composition, Anti-Inflammatory and ER Stress-Reducing Potential of Sambucus ebulus L. Fruit Extract. Plants 2021, 10, 2446. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M.; Siemens, J.; Deranian, K.L.; Zhao, M.; Hayward, N.J.; Chong, J.A.; Julius, D.; Moran, M.M.; et al. TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA 2007, 104, 13525–13530. [Google Scholar] [CrossRef]
- Wei, H.; Chapman, H.; Saarnilehto, M.; Kuokkanen, K.; Koivisto, A.; Pertovaara, A. Roles of cutaneous versus spinal TRPA1 channels in mechanical hypersensitivity in the diabetic or mustard oil-treated non-diabetic rat. Neuropharmacology 2010, 58, 578–584. [Google Scholar] [CrossRef]
- Defalco, J.; Steiger, D.; Gustafson, A.; Emerling, D.E.; Kelly, M.G.; Duncton, M.A. Oxime derivatives related to AP18: Agonists and antagonists of the TRPA1 receptor. Bioorg. Med. Chem. Lett. 2010, 20, 276–279. [Google Scholar] [CrossRef]
- McGaraughty, S.; Chu, K.L.; Perner, R.J.; Didomenico, S.; Kort, M.E.; Kym, P.R. TRPA1 modulation of spontaneous and mechanically evoked firing of spinal neurons in uninjured, osteoarthritic, and inflamed rats. Mol. Pain 2010, 6, 14. [Google Scholar] [CrossRef]
- Copeland, K.W.; Boezio, A.A.; Cheung, E.; Lee, J.; Olivieri, P.; Schenkel, L.B.; Wan, Q.; Wang, W.; Wells, M.C.; Youngblood, B.; et al. Development of novel azabenzofuran TRPA1 antagonists as in vivo tools. Bioorg. Med. Chem. Lett. 2014, 24, 3464–3468. [Google Scholar] [CrossRef]
- Rooney, L.; Vidal, A.; D’Souza, A.M.; Devereux, N.; Masick, B.; Boissel, V.; West, R.; Head, V.; Stringer, R.; Lao, J.; et al. Discovery, optimization, and biological evaluation of 5-(2-(trifluoromethyl)phenyl)indazoles as a novel class of transient receptor potential A1 (TRPA1) antagonists. J. Med. Chem. 2014, 57, 5129–5140. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agonist | Source | Chemical Nature | EC50 | Reference |
|---|---|---|---|---|
| Allyl isothiocyanate (AITC) | Mustard | Electrophilic | 64 ± 3 µM | [73] |
| Cinnamaldehyde | Cinnamon | Electrophilic | 400 ± 40 µM | [151] |
| Allicin | Garlic | Electrophilic | 7.5 ± 0.4 µM | [152] |
| Hydrogen sulphide | Garlic | Electrophilic | 1.8 ± 0.08 mM NaHS (mouse TRPA1) | [153] |
| Diallyl sulfide | Garlic | Electrophilic | 254 | [154] |
| Acrolein | Air pollutant | Electrophilic | 5 ± 1 µM | [155] |
| JT-010 | Synthetic | Electrophilic | 0.047 µM | [156] |
| H2O2 | ROS | Electrophilic | 290 ± 90 µM | [157] |
| 4-HNE | ROS | Electrophilic | 5 µM | [158] |
| PGJ2 | ROS | Electrophilic | 5.6 µM (mouse TRPA1) | [65] |
| 4-oxononenal (4-ONE) | ROS | Electrophilic | 5.8 µM | [158] |
| 4-hydroxyhexenal (4-HHE) | ROS | Electrophilic | ≥4.3 µM | [158] |
| Menthol | Mint | Non-electrophilic | 278 ± 30 µM | [159] |
| Thymol | Thyme | Non-electrophilic | 127 µM | [160] |
| Carvacrol | Thyme | Non-electrophilic | 7 µM | [160] |
| Lidocaine | Anesthetic | Non-electrophilic | 24.000 ± 600 µM | [161] |
| Propofol | Anesthetic | Non-electrophilic | 17 µM | [162] |
| WaTx | Synthetic | Non-electrophilic | 16 µM | [150] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moccia, F.; Montagna, D. Transient Receptor Potential Ankyrin 1 (TRPA1) Channel as a Sensor of Oxidative Stress in Cancer Cells. Cells 2023, 12, 1261. https://doi.org/10.3390/cells12091261
Moccia F, Montagna D. Transient Receptor Potential Ankyrin 1 (TRPA1) Channel as a Sensor of Oxidative Stress in Cancer Cells. Cells. 2023; 12(9):1261. https://doi.org/10.3390/cells12091261
Chicago/Turabian StyleMoccia, Francesco, and Daniela Montagna. 2023. "Transient Receptor Potential Ankyrin 1 (TRPA1) Channel as a Sensor of Oxidative Stress in Cancer Cells" Cells 12, no. 9: 1261. https://doi.org/10.3390/cells12091261