
Inducible MLL-AF9 Expression Drives an AML Program during Human Pluripotent Stem Cell-Derived Hematopoietic Differentiation
 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. iMLL-AF9 Generation
- -
- MLL-AF9_fwd_PCR: GGACTTTTCACTTCAAGAATCTTTTCTTTTGG;
- -
- MLL-AF9_rev_PCR: CCAAAAGAAAAGATTCTTGAAGTGAAAAGTCC.
2.2. Cell Culture
2.3. Myelomonocytic Differentiation
2.4. RNA Extraction and Real-Time PCR
- -
- MLL-AF9_fwd_qPCR: CGA AGA CGA AGA CGA GGC GG;
- -
- MLL-AF9_rev_qPCR: AGA CAC ATT CTG CAG CAG ATC GTG;
- -
- AAVS1_fwd: CAG TTA CAT TGG ATC CCT GCA GGC.
2.5. Western Blot
2.6. Flow Cytometric Analysis
2.7. Publicly Available Datasets
2.8. RNA Sequencing
2.9. Assay for Transposase-Accessible Chromatin Using Sequencing
2.10. ANalysis Algorithm for Networks Specified by Enhancers (ANANSE)
2.11. Peak Calling
2.12. Comparative Bulk RNA Sequencing Analysis
2.13. Single-Cell RNA Sequencing
3. Results
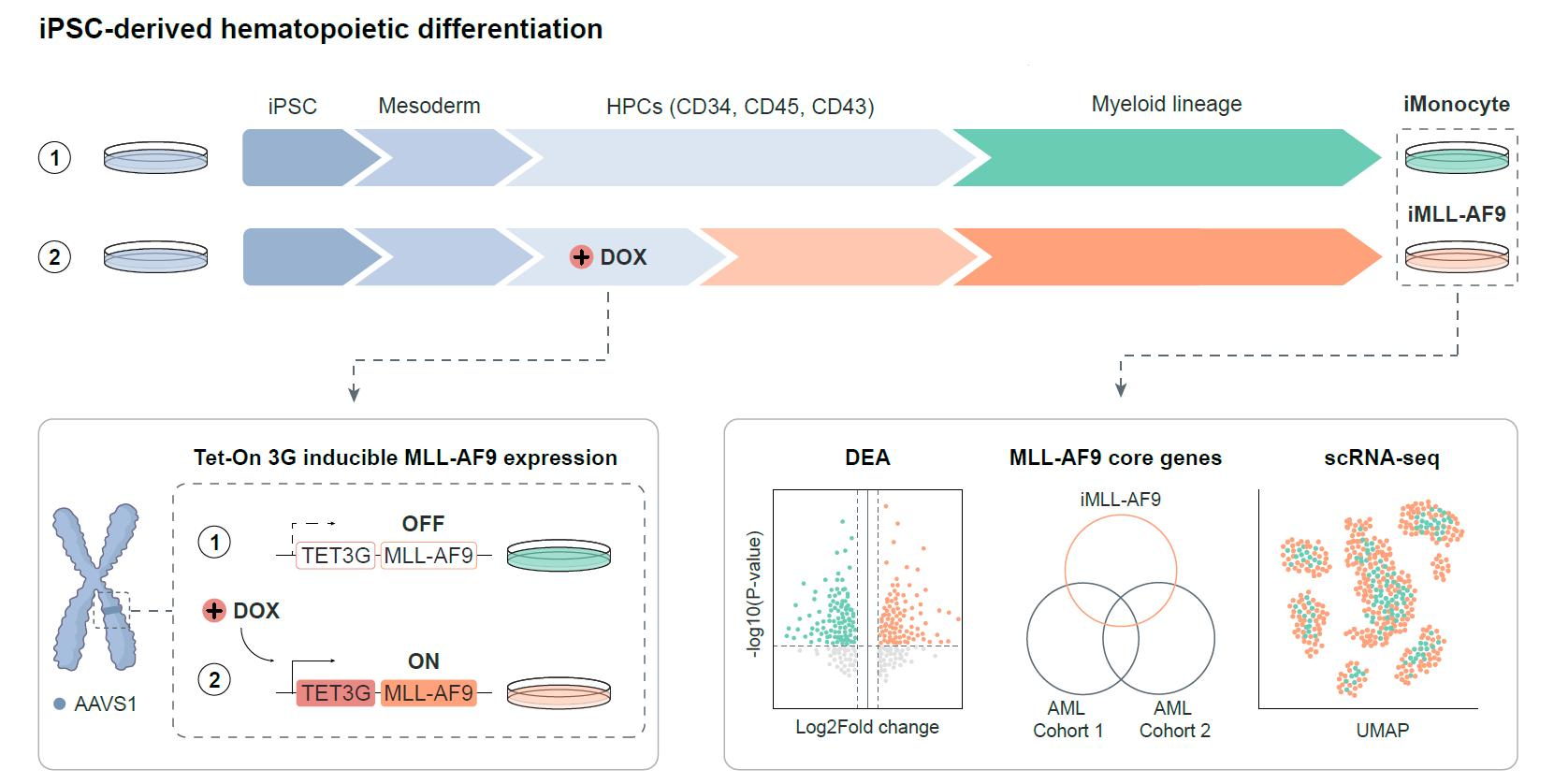
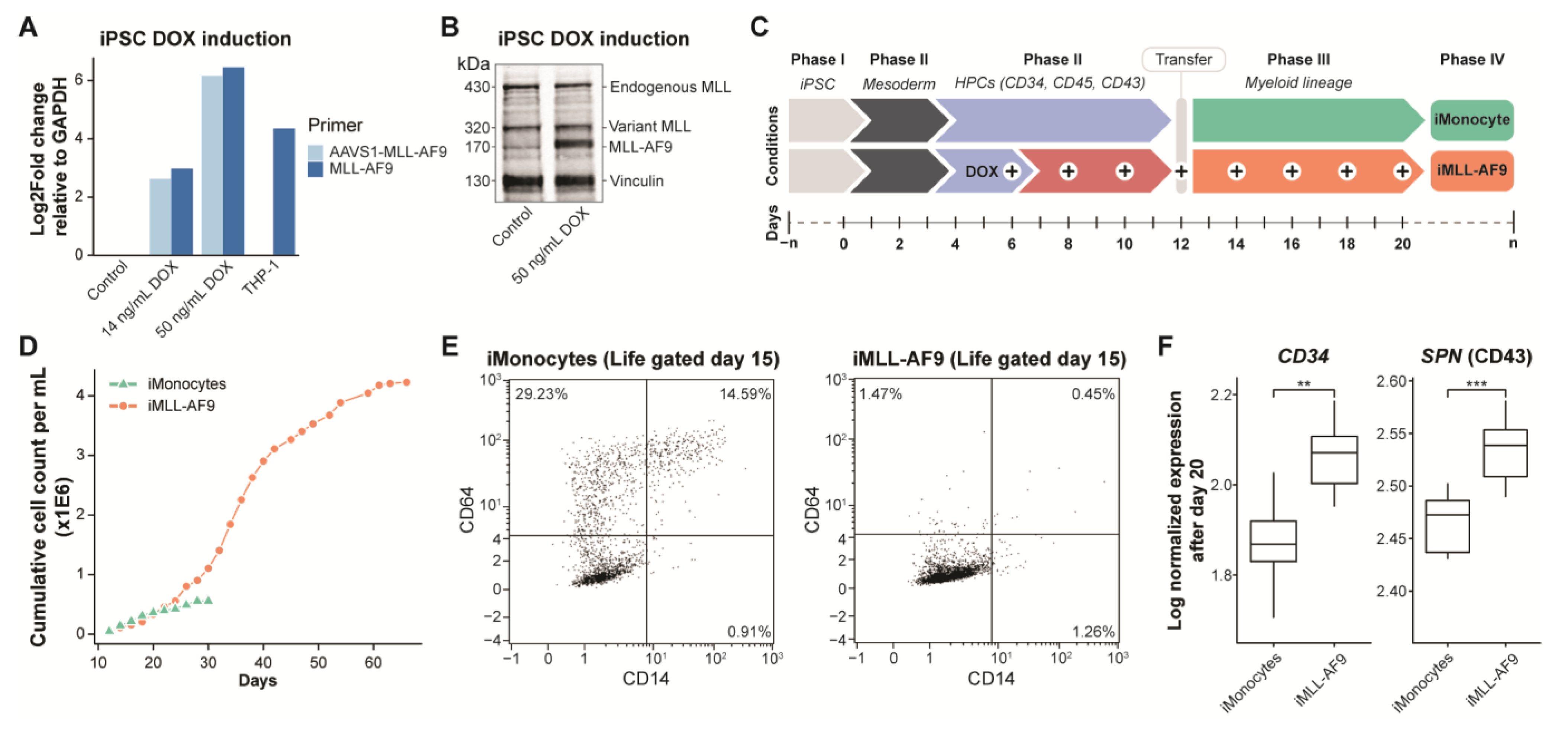
3.1. Generation of an Inducible MLL-AF9 Human Pluripotent Stem Cell Model
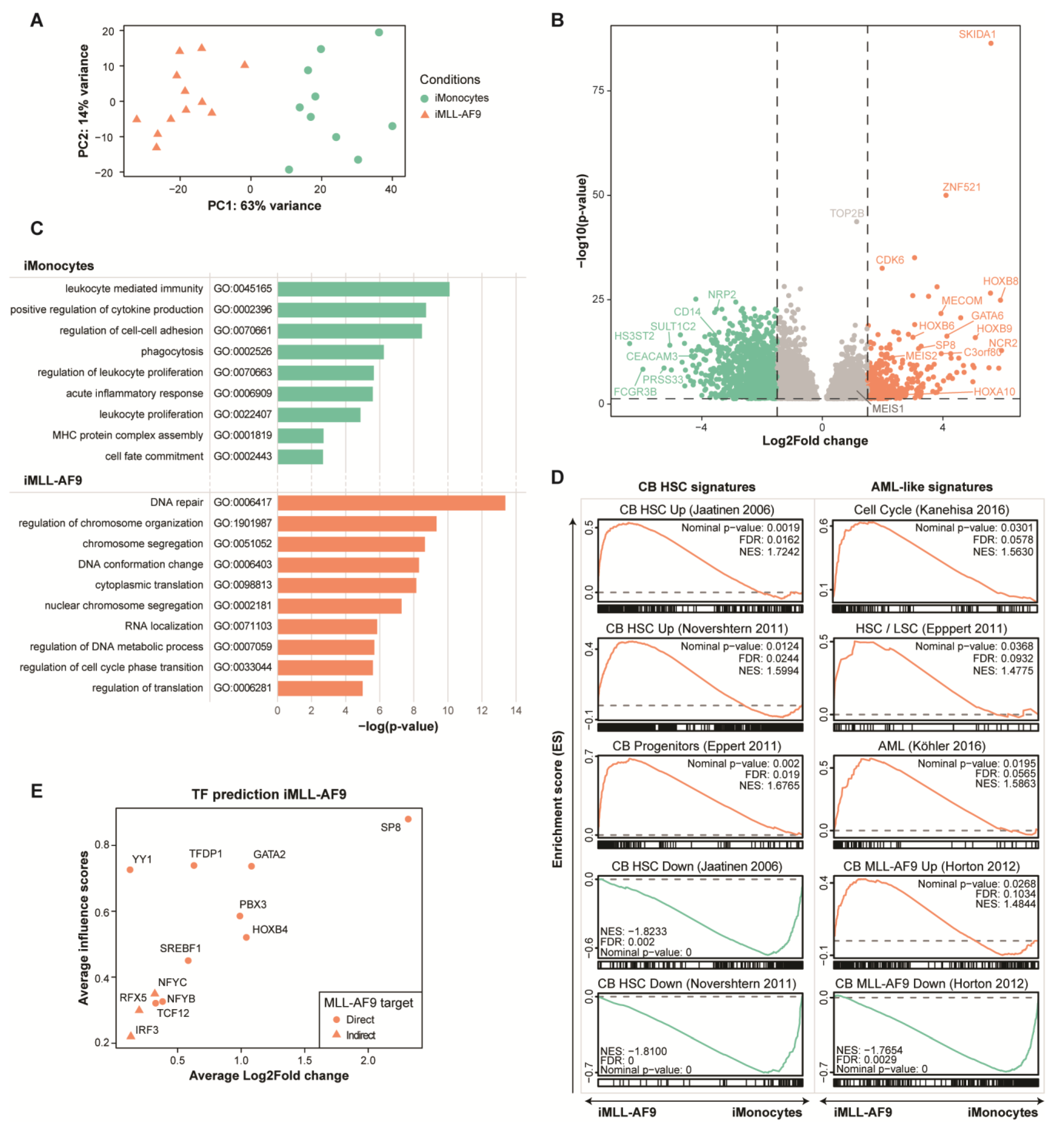
3.2. MLL-AF9-Induced Early Hematopoietic Progenitor Cells Express Cord Blood and Leukemia Related Genes
3.3. Identifying Key Transcription Factors Driving MLL-AF9-Induced Early Hematopoietic Progenitor Cells
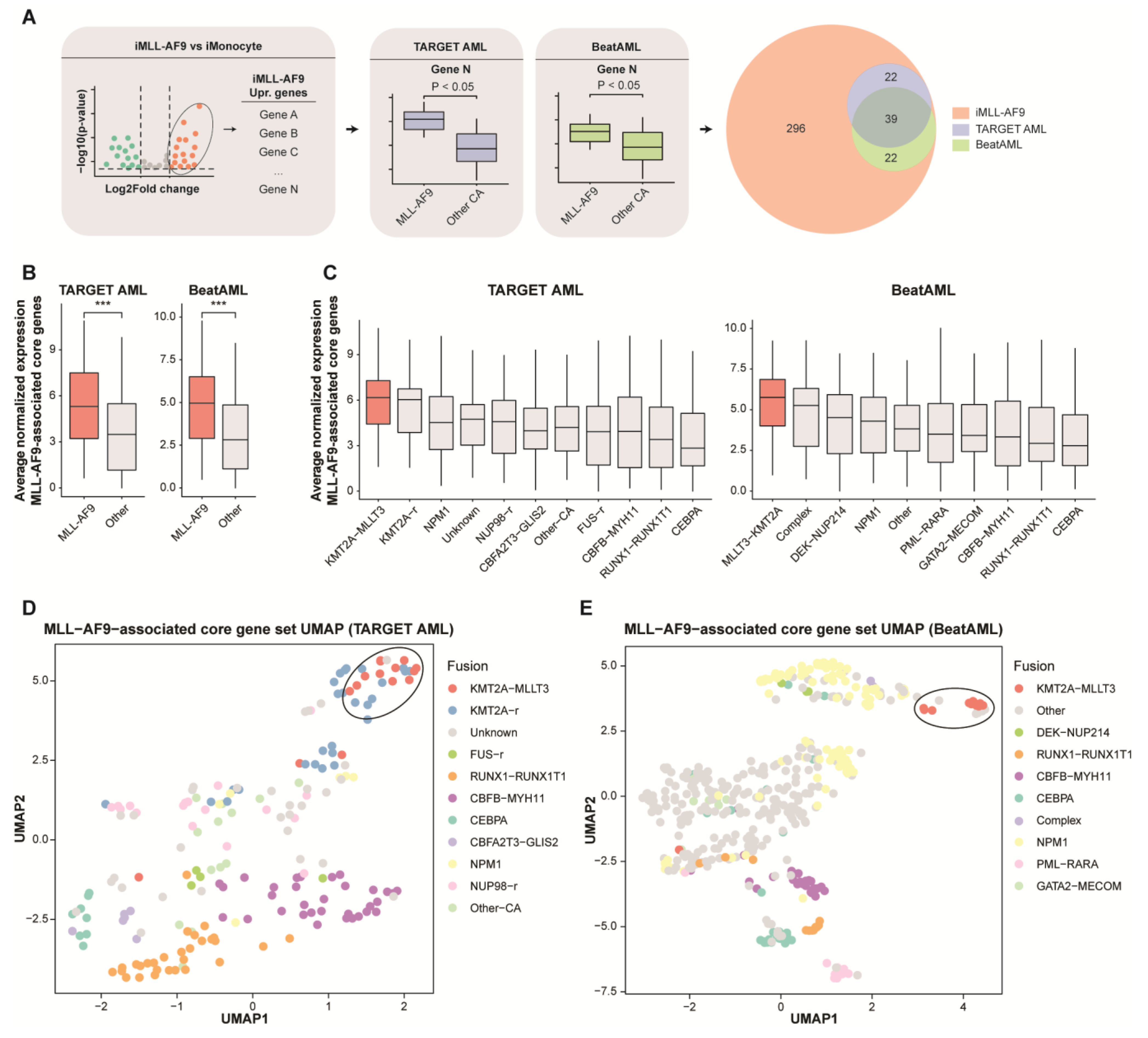
3.4. Comparative Gene Expression Profiling Reveals MLL-AF9-Associated Core Genes in Primary AML
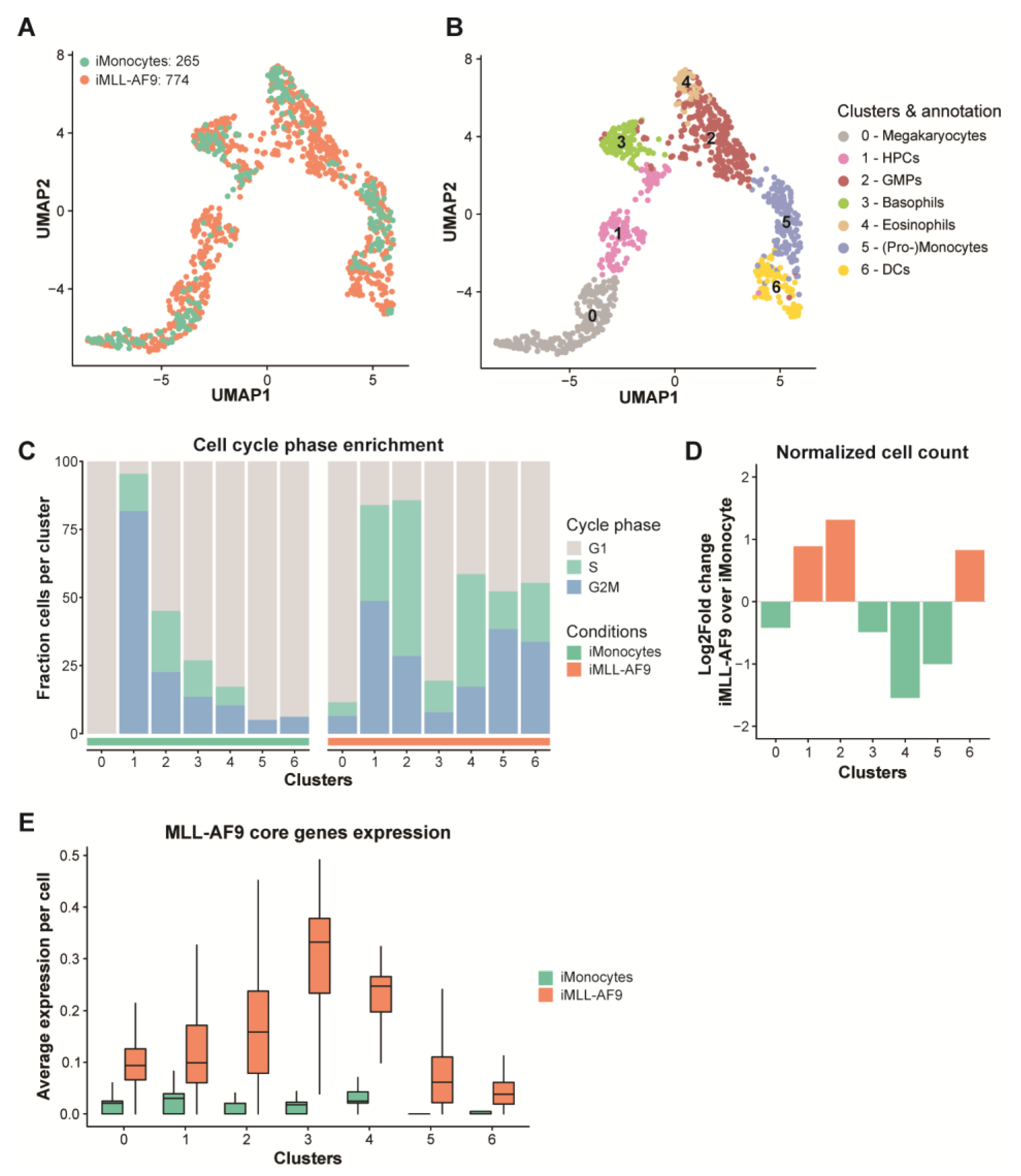
3.5. MLL-AF9 Expression Affects Distinct Cellular Populations during Early Hematopoietic Development
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Look, A.T. Oncogenic Transcription Factors in the Human Acute Leukemias. Science 1997, 278, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.H.A.; Stunnenberg, H.G. The Molecular Signature of Oncofusion Proteins in Acute Myeloid Leukemia. FEBS Lett. 2010, 584, 2662–2669. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Armstrong, S.A. MLL Translocations, Histone Modifications and Leukaemia Stem-Cell Development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Muntean, A.G.; Hess, J.L. The Pathogenesis of Mixed-Lineage Leukemia. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 283–301. [Google Scholar] [CrossRef]
- Kotani, S.; Yoda, A.; Kon, A.; Kataoka, K.; Ochi, Y.; Shiozawa, Y.; Hirsch, C.; Takeda, J.; Ueno, H.; Yoshizato, T.; et al. Molecular Pathogenesis of Disease Progression in MLL-Rearranged AML. Leukemia 2018, 33, 612–624. [Google Scholar] [CrossRef]
- Horton, S.J.; Jaques, J.; Woolthuis, C.; van Dijk, J.; Mesuraca, M.; Huls, G.; Morrone, G.; Vellenga, E.; Schuringa, J.J. MLL–AF9-Mediated Immortalization of Human Hematopoietic Cells along Different Lineages Changes during Ontogeny. Leukemia 2012, 27, 1116–1126. [Google Scholar] [CrossRef]
- Chen, X.; Burkhardt, D.B.; Hartman, A.A.; Hu, X.; Eastman, A.E.; Sun, C.; Wang, X.; Zhong, M.; Krishnaswamy, S.; Guo, S. MLL-AF9 Initiates Transformation from Fast-Proliferating Myeloid Progenitors. Nat. Commun. 2019, 10, 5767. [Google Scholar] [CrossRef] [PubMed]
- Canu, G.; Ruhrberg, C. First blood: The endothelial origins of hematopoietic progenitors. Angiogenesis 2021, 24, 199–211. [Google Scholar] [CrossRef]
- Tijchon, E.; Yi, G.; Mandoli, A.; Smits, J.G.A.; Ferrari, F.; Heuts, B.M.H.; Wijnen, F.; Kim, B.; Janssen-Megens, E.M.; Schuringa, J.J.; et al. The Acute Myeloid Leukemia Associated AML1-ETO Fusion Protein Alters the Transcriptome and Cellular Progression in a Single-Oncogene Expressing in Vitro Induced Pluripotent Stem Cell Based Granulocyte Differentiation Model. PLoS ONE 2019, 14, e0226435. [Google Scholar] [CrossRef]
- Hansen, M.; Varga, E.; Wüst, T.; Brouwer, N.; Beauchemin, H.; Mellink, C.; van der Kevie-Kersemaekers, A.M.; Möröy, T.; van der Reijden, B.; von Lindern, M.; et al. Generation and Characterization of Human IPSC Line MML-6838-Cl2 from Mobilized Peripheral Blood Derived Megakaryoblasts. Stem Cell Res. 2017, 18, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Mandoli, A.; Singh, A.A.; Prange, K.H.M.; Tijchon, E.; Oerlemans, M.; Dirks, R.; Ter Huurne, M.; Wierenga, A.T.J.; Janssen-Megens, E.M.; Berentsen, K.; et al. The Hematopoietic Transcription Factors RUNX1 and ERG Prevent AML1-ETO Oncogene Overexpression and Onset of the Apoptosis Program in t(8;21) AMLs. Cell Rep 2016, 17, 2087–2100. [Google Scholar] [CrossRef]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional Genomic Landscape of Acute Myeloid Leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Olsen, S.N.; Godfrey, L.; Healy, J.P.; Choi, Y.A.; Kai, Y.; Hatton, C.; Perner, F.; Haarer, E.L.; Nabet, B.; Yuan, G.C.; et al. MLL::AF9 Degradation Induces Rapid Changes in Transcriptional Elongation and Subsequent Loss of an Active Chromatin Landscape. Mol. Cell 2022, 82, 1140–1155.e11. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally Applicable Gene Set Enrichment for Pathway Analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef]
- Xu, Q.; Georgiou, G.; Frölich, S.; van der Sande, M.; Veenstra, G.J.C.; Zhou, H.; van Heeringen, S.J. ANANSE: An Enhancer Network-Based Computational Approach for Predicting Key Transcription Factors in Cell Fate Determination. Nucleic Acids Res. 2021, 49, 7966–7985. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT Improves Functional Interpretation of Cis-Regulatory Regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef]
- Zhu, L.J.; Gazin, C.; Lawson, N.D.; Pagès, H.; Lin, S.M.; Lapointe, D.S.; Green, M.R. ChIPpeakAnno: A Bioconductor Package to Annotate ChIP-Seq and ChIP-Chip Data. BMC Bioinform. 2010, 11, 237. [Google Scholar] [CrossRef]
- Ahlmann-Eltze, C.; Patil, I. Ggsignif: R Package for Displaying Significance Brackets for “Ggplot2”. PsyArXiv 2021. [Google Scholar] [CrossRef]
- Galili, T. Dendextend: An R Package for Visualizing, Adjusting and Comparing Trees of Hierarchical Clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Gerlach, J.P.; van Buggenum, J.A.G.; Tanis, S.E.J.; Hogeweg, M.; Heuts, B.M.H.; Muraro, M.J.; Elze, L.; Rivello, F.; Rakszewska, A.; van Oudenaarden, A.; et al. Combined Quantification of Intracellular (Phospho-)Proteins and Transcriptomics from Fixed Single Cells. Sci. Rep. 2019, 9, 1469. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Linderman, G.C.; Zhao, J.; Roulis, M.; Bielecki, P.; Flavell, R.A.; Nadler, B.; Kluger, Y. Zero-Preserving Imputation of Single-Cell RNA-Seq Data. Nat. Commun. 2022, 13, 192. [Google Scholar] [CrossRef]
- Kumar, A.R.; Hudson, W.A.; Chen, W.; Nishiuchi, R.; Yao, Q.; Kersey, J.H. Hoxa9 Influences the Phenotype but Not the Incidence of Mll-AF9 Fusion Gene Leukemia. Blood 2004, 103, 1823–1828. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Iwasaki, M.; Somervaille, T.C.P.; So, C.W.E.; Cleary, M.L. Meis1 Is an Essential and Rate-Limiting Regulator of MLL Leukemia Stem Cell Potential. Genes Dev. 2007, 21, 2762. [Google Scholar] [CrossRef]
- Germano, G.; Morello, G.; Aveic, S.; Pinazza, M.; Minuzzo, S.; Frasson, C.; Persano, L.; Bonvini, P.; Viola, G.; Bresolin, S.; et al. ZNF521 Sustains the Differentiation Block in MLL-Rearranged Acute Myeloid Leukemia. Oncotarget 2017, 8, 26129–26141. [Google Scholar] [CrossRef]
- Placke, T.; Faber, K.; Nonami, A.; Putwain, S.L.; Salih, H.R.; Heidel, F.H.; Krämer, A.; Root, D.E.; Barbie, D.A.; Krivtsov, A.v.; et al. Requirement for CDK6 in MLL-Rearranged Acute Myeloid Leukemia. Blood 2014, 124, 13. [Google Scholar] [CrossRef]
- Ziegler-Heitbrock, H.W.L.; Ulevitch, R.J. CD14: Cell Surface Receptor and Differentiation Marker. Immunol. Today 1993, 14, 121–125. [Google Scholar] [CrossRef]
- Skubitz, K.M.; Campbell, K.D.; Skubitz, A.P.N. CD66a, CD66b, CD66c, and CD66d Each Independently Stimulate Neutrophils. J. Leukoc. Biol. 1996, 60, 106–117. [Google Scholar] [CrossRef]
- Sachs, U.J.; Reil, A.; Simtong, P.; Santoso, S. CD16b: Primary Structure of Human Neutrophil Antigen Epitopes and Their Functional Consequences. Blood 2016, 128, 3680. [Google Scholar] [CrossRef]
- Jaatinen, T.; Hemmoranta, H.; Hautaniemi, S.; Niemi, J.; Nicorici, D.; Laine, J.; Yli-Harja, O.; Partanen, J. Global Gene Expression Profile of Human Cord Blood–Derived CD133+ Cells. Stem Cells 2006, 24, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Novershtern, N.; Subramanian, A.; Lawton, L.N.; Mak, R.H.; Haining, W.N.; McConkey, M.E.; Habib, N.; Yosef, N.; Chang, C.Y.; Shay, T.; et al. Densely Interconnected Transcriptional Circuits Control Cell States in Human Hematopoiesis. Cell 2011, 144, 296–309. [Google Scholar] [CrossRef]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem Cell Gene Expression Programs Influence Clinical Outcome in Human Leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Pineault, N.; Helgason, C.D.; Lawrence, H.J.; Humphries, R.K. Differential Expression of Hox, Meis1, and Pbx1 Genes in Primitive Cells throughout Murine Hematopoietic Ontogeny. Exp. Hematol. 2002, 30, 49–57. [Google Scholar] [CrossRef]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef]
- Hammal, F.; de Langen, P.; Bergon, A.; Lopez, F.; Ballester, B. ReMap 2022: A Database of Human, Mouse, Drosophila and Arabidopsis Regulatory Regions from an Integrative Analysis of DNA-Binding Sequencing Experiments. Nucleic Acids Res. 2022, 50, D316–D325. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Li, Y.; Arnovitz, S.; Chen, P.; Huang, H.; Jiang, X.; Hong, G.M.; Kunjamma, R.B.; Ren, H.; et al. PBX3 Is an Important Cofactor of HOXA9 in Leukemogenesis. Blood 2013, 121, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chu, Y.; Wang, L.; Chen, X.; Chen, Y.; Cheng, H.; Zhang, L.; Zhou, Y.; Yang, F.C.; Cheng, T.; et al. PBX3 Is Essential for Leukemia Stem Cell Maintenance in MLL-Rearranged Leukemia. Int. J. Cancer 2017, 141, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Thorsteinsdottir, U.; Sauvageau, G.; Hough, M.R.; Dragowska, W.; Lansdorp, P.M.; Lawrence, H.J.; Largman, C.; Humphries, R.K. Overexpression of HOXA10 in Murine Hematopoietic Cells Perturbs Both Myeloid and Lymphoid Differentiation and Leads to Acute Myeloid Leukemia. Mol. Cell Biol. 1997, 17, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Coukos, G.; Benencia, F.; Buckanovich, R.J.; Conejo-Garcia, J.R. The Role of Dendritic Cell Precursors in Tumour Vasculogenesis. Br. J. Cancer 2005, 92, 1182–1187. [Google Scholar] [CrossRef]
- Lopes, B.A.; Poubel, C.P.; Teixeira, C.E.; Caye-Eude, A.; Cavé, H.; Meyer, C.; Marschalek, R.; Boroni, M.; Emerenciano, M. Novel Diagnostic and Therapeutic Options for KMT2A-Rearranged Acute Leukemias. Front Pharm. 2022, 13, 749472. [Google Scholar] [CrossRef]
- Bhagwan, J.R.; Denning, C.; Collins, E.; Mosqueira, D.; Bakar, M.; Johnson, B.B.; Thompson, A.; Smith, J.G.W. Variable Expression and Silencing of CRISPR-Cas9 Targeted Transgenes Identifies the AAVS1 Locus as Not an Entirely Safe Harbour. F1000Research 2020, 8, 1911. [Google Scholar] [CrossRef]
- Klatt, D.; Cheng, E.; Hoffmann, D.; Santilli, G.; Thrasher, A.J.; Brendel, C.; Schambach, A. Differential Transgene Silencing of Myeloid-Specific Promoters in the AAVS1 Safe Harbor Locus of Induced Pluripotent Stem Cell-Derived Myeloid Cells. Hum. Gene 2020, 31, 199. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from Committed Progenitor to Leukaemia Stem Cell Initiated by MLL–AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heuts, B.M.H.; Arza-Apalategi, S.; Alkema, S.G.; Tijchon, E.; Jussen, L.; Bergevoet, S.M.; van der Reijden, B.A.; Martens, J.H.A. Inducible MLL-AF9 Expression Drives an AML Program during Human Pluripotent Stem Cell-Derived Hematopoietic Differentiation. Cells 2023, 12, 1195. https://doi.org/10.3390/cells12081195
Heuts BMH, Arza-Apalategi S, Alkema SG, Tijchon E, Jussen L, Bergevoet SM, van der Reijden BA, Martens JHA. Inducible MLL-AF9 Expression Drives an AML Program during Human Pluripotent Stem Cell-Derived Hematopoietic Differentiation. Cells. 2023; 12(8):1195. https://doi.org/10.3390/cells12081195
Chicago/Turabian StyleHeuts, Branco M. H., Saioa Arza-Apalategi, Sinne G. Alkema, Esther Tijchon, Laura Jussen, Saskia M. Bergevoet, Bert A. van der Reijden, and Joost H. A. Martens. 2023. "Inducible MLL-AF9 Expression Drives an AML Program during Human Pluripotent Stem Cell-Derived Hematopoietic Differentiation" Cells 12, no. 8: 1195. https://doi.org/10.3390/cells12081195