
A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities

1
Department of Life Science, Gachon University, Seongnam 13120, Republic of Korea
2
Department of Health Sciences and Technology, GAIHST, Lee Gil Ya Cancer and Diabetes Institute, Incheon 21999, Republic of Korea
*
Author to whom correspondence should be addressed.
Cells 2023, 12(8), 1110; https://doi.org/10.3390/cells12081110
Submission received: 31 January 2023
/
Revised: 3 April 2023
/
Accepted: 5 April 2023
/
Published: 8 April 2023
(This article belongs to the Special Issue From Mechanisms to Therapeutics: Wnt Signaling in Cancer)
Abstract
:Aberrant Wnt signaling activation is frequently observed in many cancers. The mutation acquisition of Wnt signaling leads to tumorigenesis, whereas the inhibition of Wnt signaling robustly suppresses tumor development in various in vivo models. Based on the excellent preclinical effect of targeting Wnt signaling, over the past 40 years, numerous Wnt-targeted therapies have been investigated for cancer treatment. However, Wnt signaling-targeting drugs are still not clinically available. A major obstacle to Wnt targeting is the concomitant side effects during treatment due to the pleiotropic role of Wnt signaling in development, tissue homeostasis, and stem cells. Additionally, the complexity of the Wnt signaling cascades across different cancer contexts hinders the development of optimized targeted therapies. Although the therapeutic targeting of Wnt signaling remains challenging, alternative strategies have been continuously developed alongside technological advances. In this review, we give an overview of current Wnt targeting strategies and discuss recent promising trials that have the potential to be clinically realized based on their mechanism of action. Furthermore, we highlight new waves of Wnt targeting that combine recently developed technologies such as PROTAC/molecular glue, antibody–drug conjugates (ADC), and anti-sense oligonucleotides (ASO), which may provide us with new opportunities to target ‘undruggable’ Wnt signaling.
1. Wnt Signaling Pathway
In the early 1980s, Wnt signaling was first discovered when wingless, an essential developmental gene of Drosophila, was found [1]. Concurrently, Int-1, a mammalian wingless homolog, was identified as a driver gene for malignancy when its transcriptional activation was induced by the murine mammary tumor virus [2]. Through wingless and Int-1, a portmanteau name of ‘Wnt’ was created. Over the four decades since its discovery, Wnt-mediated signaling has been extensively studied, revealing its evolutionarily conserved roles in regulating diverse cellular processes, including embryonic development, tissue homeostasis, and cell fate determination [3].
Wnt signaling consists of Wnt ligands, frizzled receptors (FZD family), co-receptors, β-catenin destruction complexes, β-catenin/transcriptional partners, and other modulating components [3]. Wnt is a secreted ligand and mediates autocrine and paracrine signal transduction through its receptors and downstream effectors [4]. The intracellular downstream signaling of Wnt ligands/receptors is broadly divided into β-catenin-dependent signaling (also referred to as canonical Wnt signaling) and β-catenin-independent signaling (also referred to as non-canonical Wnt signaling) according to its dependence on β-catenin, a central effector of Wnt signaling (Figure 1). The type of Wnt ligands and their corresponding receptors/co-receptors in β-catenin-dependent and -independent Wnt signaling vary in each physiological context. For instance, among the 19 human Wnt ligands, Wnt3a is mainly involved in the β-catenin-dependent Wnt pathway. On the other hand, Wnt5a is predominantly associated with the β-catenin-independent Wnt pathway. However, in some cases, Wnt5A often acts the opposite way [5]. Of human frizzled (FZD) receptors, FZD1 and FZD7 are mainly related to the canonical Wnt pathway. In contrast, FZD2 and FZD6 are implicated with the non-canonical Wnt pathway [5]. However, due to the redundancy and complexity of Wnt signaling, the precise pairs of working Wnt ligands/receptors are still elusive in many biological contexts.
Herein, we briefly describe Wnt signaling transduction, the understanding of which plays a key role in the development of targeting strategies (Figure 1).
1.1. Wnt Ligands and Receptors
So far, 19 secreted Wnt ligands and more than 18 Wnt receptors/co-receptors have been identified in the mammalian system [4]. Different combinations of Wnt ligands and their receptors/co-receptors operate in various physiological contexts [6]. Wnt ligands are lipid-modified glycoproteins. The extracellular transport of Wnt requires palmitoylation, a lipid modification mediated by a protein-serine O-palmitoyltransferase, porcupine (PORCN) [7]. Palmitoylated Wnt ligands bind to Wntless and are transported from the Golgi apparatus to the cell membrane for secretion. Secreted Wnts are recognized by the FZD receptor family [3,8]. These G-protein coupled FZD receptors act as primary Wnt receptors and transduce the Wnt signaling intracellularly [8,9]. There are ten FZD receptors in humans. In addition, low-density lipoprotein receptor-related proteins 5 and 6 (LRP5/6) bind FZD and act as typical co-receptors [10]. Additionally, diverse families of Wnt signaling modulators exist. Ring finger protein 43 (RNF43) or zinc and ring finger 3 (ZNFR3) are transmembrane E3 ligases that act as negative regulators of Wnt signaling by inducing the lysosomal degradation of FZD receptors. Conversely, secreted ligands, R-spondins (RSPOs 1-4), serve as Wnt signaling enhancers [11,12]. R-spondins bind to the complex of leucine-rich repeat-containing G protein-coupled receptor 4-6 (LGR4-6) and RNF43/ZNFR3. This interaction prevents the RNF43/ZNFR3-mediated lysosomal degradation of FZD and maintains Wnt signaling transduction [13]. The non-Wnt protein, Norrie Disease Protein (Norrin), also triggers the β-catenin-dependent Wnt pathway [14,15]. Though Norrin is a cystine-knot-like growth factor, it activates Wnt signaling by interacting with FZD4, Lrp5/6, and Tetraspanin-12 [14,15]. Several Wnts, receptors, and co-receptors, and various Wnt modulators operate convergently and divergently. It is also possible that additional unidentified Wnt co-receptors and modulators exist. The complexity of the Wnt signaling cascade makes it challenging to understand the exact responses elicited by the specific targeting of the Wnt signaling steps, which is one of the major difficulties in developing Wnt signaling targeting strategies.
1.2. β-Catenin-Dependent Signaling
The β-catenin-dependent Wnt signaling pathway is more elucidated and established compared to the β-catenin-independent pathway (Figure 1). β-catenin is an armadillo repeat protein associated with the cytoplasmic domain of cadherins, cell–cell junction proteins [16,17]. Intracellular levels of non-E-cadherin-bound cytoplasmic β-catenin are very low without Wnt ligand stimulation [18]. The β-catenin destruction complex tightly controls levels of cytoplasm β-catenin. The destruction complex consists of axis inhibitor (AXIN), adenomatous polyposis coli (APC), casein kinase 1 (CK1), glycogen synthase kinase 3β (GSK3β), and β-transducin repeat-containing protein (βTrCP) [10]. In the absence of a Wnt ligand (Wnt-off status), the GSK3β and CK1 of the destruction complex phosphorylate β-catenin at Ser45 (CK1) and Thr41/Ser37/Ser33 (GSK3β) residues, respectively. Phosphorylated β-catenin is sequentially ubiquitinated by the E3 ligase βTrCP and degraded by a ubiquitin-mediated proteasomal system [3]. Upon Wnt ligand stimulation (Wnt-on status), AXIN binds to the phosphorylated cytoplasmic tail of LRP5/6 and the FZD receptor adapter Disheveled (Dvl) [3,8]. This protein interaction sequesters the β-catenin destruction complex from the cytosol to the cell membrane and prevents the destruction complex-mediated degradation of β-catenin. Accumulated β-catenin in the cytoplasm translocates to the nucleus and forms a complex with the transcription factor T cell factor (TCF) or lymphoid enhancer-binding factor 1 (LEF1), replacing Groucho and CtBP corepressors [8,19,20]. β-catenin/transcription factor complexes bind to a co-activator (CBP, p300) for the transcriptional activation of a variety of downstream genes, including c-Myc, Axin2, CCND1, and CD44 [20,21].
1.3. β-Catenin-Independent Signaling
In β-catenin-independent signaling, the intracellular downstream signaling of the Wnt/receptor interactions is mediated not by β-catenin but by various cellular signaling modules, such as RAC1-JNK, RHOA-ROCK, and PLC-IP3-Ca2+ (Figure 1). Well-characterized physiological contexts of β-catenin-independent signaling include the planar cell polarity (PCP) pathway and Wnt/Ca2+ signaling [19]. In Wnt/PCP signaling, binding Wnt ligands to FZDs activates the small GTPases RHOA and RAC1 [22]. Consequently, these signals trigger the activation of Rho-associated protein kinase (ROCK) and JUN N-terminal kinase (JNK), respectively [22]. The Wnt/PCP signaling directs the cell polarity to induce cell asymmetry, which is crucial for various developmental processes. In the Wnt/Ca2+ pathway, Wnt ligand–FZD formation activates phospholipase C (PLC), which releases Ca2+ from the intracellular stores. The raised levels of Ca2+ induce the activation of protein kinase C, protein kinase II, and calcineurin [23,24]. Although many studies have been conducted in β-catenin-independent signaling, new Wnt ligands, unknown modulators, and cytosolic signaling mediators are constantly being discovered. Thus, the whole map of non-canonical Wnt signaling is still unclear and is different in each biological context.
2. Alterations of Wnt Signaling in Cancers
Aberrant Wnt signaling has been implicated in the tumorigenesis of various cancers (Figure 1). For example, genetic mutations of Wnt signaling in colorectal cancer (CRC) are observed in more than 90% of patients and are well-known cancer-driving alterations [25,26]. Genetic alterations in Wnt signaling primarily occur through gene mutation in APC, ZNRF3, CTNNB1 (encodes β-catenin), AXIN1, and RSPOs [3]. In addition, overexpression, downregulation, and copy number changes have also been observed in the genes of Wnt signaling. These genetic alterations of patterns, frequency, and affected genes vary by cancer type [26] (Figure 1). Notably, in most cancers, alterations in Wnt signaling are mainly related to the direction of Wnt signaling activation. Additionally, importantly, the suppression of the hyperactivation of Wnt signaling has shown excellent tumor suppression effects in many preclinical models.
2.1. Driver Alterations of Wnt Signaling in Cancers
In colorectal cancer (CRC), the loss-of-mutation of APC and the activating mutation of CTNNB1 are frequently observed [26,27]. The mutation frequency of APC is about 60~80%, and that of CTNNB1 is about 5~10% [25]. The mutation of APC and CTNNB1 leads to excessive activation of Wnt signaling by inducing the stabilization of β-catenin, which initiates intestinal tumors [26]. In addition, about 8~13% mutations of RNF43 were found in CRC patients [28]. Mutations in ZNRF3/RNF43 render cancer hypersensitive to Wnt ligands, leading to the hyperactivation of Wnt signaling and promotion of tumorigenesis [28].
In gastric cancer (GC), 13~22% APC mutations are detected [4,27]. APC mutation is sufficient to initiate gastric tumors in in vivo models [27]. Thus, the driver role of Wnt signaling mutations is demonstrated. Mutations in RNF43, another Wnt signaling component, were observed in 4.3% of the MSS (microsatellite stable) subtype (~20% of GC patients) and 54.6% of the MSI (microsatellite instable) subtype (~80% of patients) and were considered key driver mutations [29,30]. In addition to mutations in Wnt signaling, the overexpression of Wnt1 and Wnt6 has been observed in gastric cancer [31,32]. The overexpression of Wnt1 or a combination of prostaglandin pathways induces preneoplastic lesions or invasive gastric cancer, respectively, in mouse models [33]. Indeed, Wnt signaling activations were also observed in two major predisposed causes of gastric cancer development, CDH1 mutation (encodes E-cadherin) and Helicobacter pylori infections [34,35,36]. These results imply that Wnt signaling is involved both in tumor-driving and -promoting roles in gastric cancers.
In hepatocellular carcinoma (HCC), almost 50% of mutations in the Wnt signaling pathway are activation mutations [37]. Approximately 20~25% of CTNNB1, encoding β-catenin, ~10% of AXIN1, and ~3% of AXIN2, are mutated [37,38]. Additionally, FZD3, FZD6, and FZD7 receptors, Wnt 3, Wnt4, Wnt 5A ligands, and the modulator RSPO2 are frequently overexpressed or amplified in HCC [39]. These genetic alterations in Wnt signaling are closely associated with the progression of hepatocarcinoma (HCC). Although the activating mutation of CTNNB1, the most frequent mutation in HCC, does not induce a tumor, the loss-of-APC or Axin1 mutation, both less common in HCC, are able to do so [40,41].
2.2. Cancer-Promoting Alterations of Wnt Signaling in Cancers
In addition to the oncogenic role of Wnt signaling, many genetic alterations in the Wnt signaling of specific types of cancers do not drive tumorigenesis but play a tumor-promoting role during cancer progression [39,42]. Additionally, Wnt signaling supporting tumor niches is essential for promoting various primary and metastatic cancers [43,44].
In lung cancer, the mutation of CTNNB1 and APC genes is not common [45,46]. However, the hyperactivation of Wnt signaling is associated with tumor formation, relapse, and poor prognosis [35,47]. In small-cell lung cancer (SCLC), the mutation in APC and CHD8, which inhibits transcription mediated by CTNNB1, is related to the relapse of SCLC [48]. In non-small cell lung cancer (NSCLC), Wnt ligands (Wnt1, Wnt2, Wnt3, and Wnt5a) and other signaling modules (FZD8, PORCN, and TCF-4) are overexpressed [49]. A recent elegant study shows that the Wnt-producing niche is essential for lung adenocarcinoma tumorigenesis [38]. Moreover, this study shows that the pharmacological inhibition of the Wnt niche by the PORCN inhibitor LGK974 suppresses lung tumor growth in in vivo mouse models.
Pancreatic ductal acinar cell carcinoma (PDAC) exhibits approximately 4~10% mutations in RNF43 and <1% mutations in APC and CTNNB1 [27]. Although genetic alterations in Wnt signaling are rarely detected in PDAC, many studies have suggested that the activation of Wnt signaling is involved in pancreatic tumorigenesis [42,50]. Indeed, the inhibition of the interaction between FZD and Wnt ligands prevents tumorigenesis. In addition, in patient-derived PDAC cell lines harboring RNF43 mutations, the inhibition of porcupine and FZD5 suppresses the growth of PDAC cells [42,51].
3. Targeting Strategies for Wnt Signaling in Cancer
Since aberrant Wnt activation initiates or promotes tumorigenesis, Wnt-targeting strategies in cancer therapy are directed toward the downregulation or restoration of overactivated Wnt signaling. Current Wnt targeting strategies can be classified into four categories according to the target location in Wnt signaling: targeting Wnt ligands, targeting Wnt receptors, targeting destruction complex, and targeting β-catenin/transcriptional factors. Extracellular Wnt ligands and receptors are good targets for specific antibodies. However, intracellular signaling components, such as the destruction complex and β-catenin/transcriptional factors, cannot be targeted with antibodies. Only small chemicals and peptides are available to target those. Thus, the enzymatic domains of Wnt signaling components are considered key targets of intracellular components.
In this section, we describe current Wnt targeting strategies as well as recent updates, with meaningful clinical trials involving each strategy (Table 1 and Table 2, and Figure 2).
3.1. Targeting Wnt Ligands
Targeting cancer-specific Wnt ligands in certain cancers may be an excellent way to increase the specificity of cancer treatment. However, Wnt ligands often act redundantly, and which Wnt ligands are essential is not fully understood in each cancer context [5]. Additionally, in most cases, targeting Wnt ligands may be less helpful. This is because mutations in Wnt signaling mainly occur downstream of Wnt ligands [27]. Therefore, targeting Wnt ligands is likely more beneficial in cases where Wnt acts as a component of a cancer-propagating niche.
Indeed, the PORCN inhibitor LGK974 (also referred to as WNT974) prevents the secretion of Wnt by inhibiting the palmitoylation of Wnt and shows dramatic suppression of lung tumorigenesis and metastatic CRC progression [43,54]. In metastatic colorectal cancer, WNT974 with a combination of LGX818 and cetuximab was used in a clinical trial (NCT02278133) (Table 1 and Figure 2). Other PORCN inhibitors, RXC004 (NCT03447470, NCT04907539, and NCT04907851) and ETC-159 (NCT02521844) have also been used in clinical trials [57,59] (Table 1 and Figure 2). RXC004 caused the suppression of tumor growth in pancreatic cancer xenograft and gastric cancer PDX models [57]. ETC-159 inhibits the secretion and activity of all Wnt ligands [59].
Soluble Wnt modulators could also be a good target for Wnt-targeted therapy. Soluble FZD-related proteins (SFRP) restrain the Wnt signaling pathway. SFRP has a cysteine-rich domain (CRD) homologous to FZD-CRD. This domain acts as an SFRP to bind competitively to Wnt ligands in order to inhibit Wnt signaling [129,130]. V3Nter, an SFRP-derived polypeptide, binds to Wnt3A and inhibits Wnt signaling in CRC [129]. However, Wnt-targeted therapeutics using SFRP have not been tested in clinical trials.
The targeting of another soluble Wnt modulator, dickkopf-related protein family (DKK), has proved to be more promising in clinical trials. DKKs are composed of five types, of which dickkopf-1, DKK1, binds to LRP5/6 and was initially considered to inhibit canonical Wnt signaling [4,19,131]. However, several studies have since reported that DKK1 is also related to activating the non-canonical Wnt pathway [132,133,134,135]. Although the mechanism of DKK1 in regulating Wnt signaling is unclear, importantly, DKK1 overexpression has been observed in many types of cancer [136], and the inhibition of DKK1 suppresses tumorigenesis in several cancers [137,138,139,140]. Therefore, DKK1 is emerging as an important target for cancer therapy. DKN-01 is a humanized antibody that binds to DKK1, inhibiting cancer progression [68]. Phase 1 and 2 clinical trials are currently underway for monotherapy and combination therapy of DKN-01 in various cancer types, such as colorectal, gastric, endometrial, and liver cancer (NCT05480306, NCT04057365, NCT03395080, and NCT03645980) (Table 1 and Figure 2). As many clinical trials for DKN-01 are underway, we can hopefully expect the first Wnt-targeted therapy targeting DKK1 to receive FDA (Food and Drug Administration) approval soon.
3.2. Targeting Wnt Receptors
Targeting Wnt receptors is challenging in cancers harboring mutations downstream of Wnt signaling components. However, cell surface receptors are relatively easy to develop antibodies for, making them attractive drug targets. Additionally, antibodies targeting cancer-specific Wnt receptors could give more specificity with fewer side effects [141]. Furthermore, Wnt receptor-specific antibodies can be applied to Wnt niches in monotherapy and combination therapy with existing anticancer drugs. Here, we introduce cases using receptor-specific antibodies or clinical trials that may be more feasible for therapeutic application (Table 1).
A fair number of FZD-targeting antibodies are currently undergoing clinical trials (Table 1). A monoclonal antibody (mAB), OMP-18R5, binds to five FZD receptors (FZD1, FZD2, FZD5, FZD7, and FZD8) and blocks the canonical Wnt pathway [65]. This OMP-18R5 mAB inhibits tumor growth in colon, breast, and pancreatic cancer cells [65]. OMP-18R5 is being tested in a phase 1 clinical trial (Table 1 and Figure 2). OTSA-101 is a mAB targeting FZD10 and is in a phase 1 clinical trial for sarcoma [66] (Table 1 and Figure 2). An Fc fusion protein, OMP-54F28, has an extracellular N-terminal cysteine-rich domain (CRD) of FZD8 [60]. By acting as a competitor of FZD8, OMP-54F28 inhibits Wnt ligand signaling. Testing OMP-54F28 is in a phase 1 clinical trial for various solid tumors, including liver, ovarian, and pancreatic cancers (Table 1 and Figure 2).
Naturally secreted R-spondins induce the accumulation of FZD receptors and enhance Wnt signaling. Thus, inhibiting natural or cancer-specific RSPOs could be a therapeutic target for Wnt signaling. OMP-131R10 (Rosmantuzumab) is a monoclonal antibody targeting R-spondin 3 (RSPO3) that has now completed a phase 1 clinical trial in advanced solid or relapsed tumors (NCT02482441) [52] (Table 1 and Figure 2).
3.3. Targeting the β-Catenin Destruction Complex
The β-catenin destruction complex, consisting of APC, CK1, AXIN, and GSK3β, plays a vital role in controlling the concentration of cytosol and nuclear β-catenin [142]. Loss-of-function mutations in the destruction complex components are found in multiple cancers [27]. Thus, the restoration of the destruction complex or enhancement of its function is required for cancer therapy [19].
CK1 and GSK3β phosphorylate β-catenin, which subsequently induces the ubiquitination and proteasomal degradation of β-catenin [4]. Thus, activating CK1 and GSK3β could enhance the function of the destruction complex. Pyrvinium is an FDA-approved drug used initially as an effective anthelmintic against pinworms [143]. Later, people found that Pyrvinium inhibits Wnt signaling by enhancing CK1 kinase activity [74,144] (Table 1 and Figure 2). SSTC3 is a preclinical small-molecule activator of CK1, which has better pharmacokinetics than existing CK1-activating drugs [106]. CCT031374 is a preclinical activator of GSK3β that may act as a Wnt signaling inhibitor by inducing the degradation of wild-type β-catenin [107] (Table 2).
Sulindac is an FDA-approved non-steroidal anti-inflammatory drug [145]. Later, Sulindac exhibited inhibition of the Wnt/β-catenin pathway by binding to the PDZ domain of Dvl, blocking the destruction complex at the cell membrane [73]. Other small chemicals bind to the PDZ domain of Dvl and are also undergoing preclinical tests [99,100,101,102,103,104,105] (Table 2).
Another way to enhance the destruction of complex-mediated β-catenin degradation is to stabilize the AXIN. Tankyrase belongs to the poly ADP-ribose polymerase family and induces the proteasomal degradation of AXIN [146,147]. Thus, tankyrase inhibition significantly increases the stability of the AXIN protein [147]. Various tankyrase inhibitors, including IWR-1, G007-LK, JW55, JW74, and XAV939, have been evaluated in preclinical studies [117,118,119,120] (Table 2). E7449, an inhibitor of Poly (ADP-ribose) Polymerases (PARP) 1/2 and tankyrase (TNKS) 1/2 is undergoing phase 1 and 2 clinical trials for advanced solid tumors (NCT03878849 and NCT01618136) [148] (Table 1 and Figure 2).
H+-ATPase (v-ATPase) mediates the vesicular acidification of lysosomes. This acidification of lysosomes is required for the lysosomal degradation of Wnt receptors/co-receptors and APC, which hyperactivates Wnt/β-catenin signaling [148,149]. Thus, the treatment of v-ATPase inhibitors led to the suppression of Wnt/β-catenin signaling and tumorigenesis in several contexts [148]. A substantial number of v-ATPase inhibitors are undergoing phase 2 clinical trials (Table 1 and Figure 2).
Targeting the intracellular protein complex is quite challenging. Moreover, since most Wnt-associated cancers carry APC mutations, enhancing the destruction complex’s function is impossible. In addition, enhancing destruction complexes via v-ATPase inhibitors can lead to a broad range of potential side effects induced by pan-lysosomal inhibition.
3.4. Targeting β-Catenin and Its Transcription Partners
β-catenin acts as a central transcriptional activator by forming a complex with its transcriptional partner, TCF/LEF. β-catenin also recruits co-activators such as cAMP response element binding protein (CBP) and p300 [20,21]. The interaction of these factors is essential for the full activation of β-catenin transcriptional responses. Thus, blocking these interactions could be a strategy for inhibiting Wnt signaling.
ICG-001 and its analog PRI-724 interfere with the interaction between β-catenin and CBP [80,81]. Indeed, PRI-724 blocked β-catenin/CBP interaction in a liver fibrosis model [81]. The testing of PRI-724 in advanced solid cancers is currently ongoing and phase 1 clinical trials have been completed (Table 1 and Figure 2). The stabilized α helix of BCL9 (SAH-BCL9) hinders the interaction between β-catenin and BCL9, a transcriptional coactivator [126]. Vitamin D3 (1α,25-dehydroxyvitamin D3) also disrupts the interaction between β-catenin and TCF-4. The vitamin D receptor binds its ligand and competes with TCF-4 to bind the β-catenin [150]. Several clinical trials using vitamin D3 as a supplement are underway in phase I–III clinical trials combined with standard chemotherapy in multiple cancers (NCT02726113, NCT04166253, NCT02603757, and NCT01150877) (Table 1 and Figure 2).
Theoretically, interfering with the binding of β-catenin to its transcriptional partner might be an excellent strategy. It would also be ideal if we could specifically inhibit the mutant beta-catenin. However, it is tremendously difficult to understand the specific interactions between proteins. Moreover, finding chemicals that prevent such interactions is another obstacle. Although there are many limitations, several small-molecule compounds are undergoing pre- or clinical tests (Table 1 and Table 2), and new small-molecule compounds are continuously emerging.
3.5. Current Caveats of Targeting Wnt Signaling
Despite ongoing clinical trials, devastating adverse effects on tissue homeostasis and regeneration occur while targeting Wnt signaling. Therefore, Wnt signaling-targeted therapy holds excellent promise but carries high risks in cancer treatment [151]. In this context, potent inhibition of general Wnt signaling might not be a smart method. Lower doses and combination therapy with other anti-cancer therapies may provide an alternative and rational approach to Wnt-targeted therapies. Additionally, targeting Wnt-supporting cancer niches may be more beneficial than Wnt-targeted monotherapy.
As has been mentioned elsewhere, targeting Wnt ligands and receptors against antibodies can give specificity to cancer therapy and cause fewer side effects compared to chemical drugs [152]. However, it is difficult to identify specific ligands and receptors in each cancer context [141]. Moreover, these strategies cannot be effective for most Wnt-mutated cancers that carry APC or CTNNB1 mutations downstream of Wnt ligands and receptors. Similarly, targeting intracellular destruction complexes is also not very beneficial for these Wnt mutant cancers.
Although targeting mutant or nuclear β-catenin is theoretically ideal, finding small chemical inhibitors that prevent β-catenin interactions is extremely challenging.
4. New Targeting Strategies for Wnt Signaling in Cancer
Though many limitations exist in Wnt targeting fields, new trials are continuously ongoing. Moreover, clinical trials are underway for promising Wnt signaling-targeted drugs, such as DKK1 and FZD-targeting antibodies. Thus, hope still exists to develop the first Wnt-targeting drug. Furthermore, new advanced technologies are being developed to turn ‘undruggable’ proteins into ‘druggable’ proteins. Here, we introduce a new wave of Wnt-targeting strategies that makes use of the latest cutting-edge technologies.
4.1. PROTAC/Molecular Glue-Based Wnt Signaling Targeting
4.1.1. PROTAC
PROteolysis TArgeting Chimera (PROTAC) is a bifunctional molecule composed of an E3 ligase, a protein of interest (POI), and a linker capable of linking two ligands [153].
The E3 ligase ubiquitinates POIs that interact with PROTAC, and the target protein is eventually degraded by the proteasome [154]. PROTAC was first proposed in 2003. However, little progress was made for a long time due to the limited number of binders and poor intracellular delivery [154]. In recent years, PROTAC has started to make remarkable progress thanks to various cutting-edge technologies, such as new ligands that bind with E3 ligases, advanced linker technology, and accumulated chemical design knowledge [154]. In particular, the discovery of small ligands for the E3 ligase cereblon (CRBN) and Von Hippel–Lindau disease (VHL) provided great insights into the design of PROTAC and revolutionized the PROTAC field [153,154,155]. As a result, PROTAC is currently attracting significant attention and being actively studied as an efficient method of erasing target proteins.
In Wnt-targeted cancer therapy using PROTAC, β-catenin is an attractive intracellular target. xStAx-VHL is a PROTAC-targeting β-catenin consisting of xStAx and VHLL [153] (Figure 3 and Table 3). xStAx is a peptide that shows high similarity with the β-catenin-binding domain of AXIN. xStAx-VHL induces dose-dependent and lasting degradation of β-catenin in cell lines and APC−/− organoids. Additionally, xStAx-VHL inhibits the growth of CRC xenografts and APC Min/+-driven intestinal tumors [153].
Since PROTAC does not work based on equilibrium occupancy, the working doses of PROTAC are very low, i.e., nanomolar concentrations [155]. Thus, PROTACs are expected to have low toxicity and high selectivity compared to conventional inhibitors. For example, the BCL-XL-specific PROTAC DT2216 shows low toxicity in vitro and inhibits the growth of xenograft tumors [162]. Additionally, DT2216 shows target specificity to BCl-XL over all other BCL-2 family members (BCL-2, BCl-XL, MCL-1) [162].
4.1.2. Molecular Glue
A small molecule that induces or stabilizes the neo-interaction between two different proteins is called a ‘molecular glue.’ Some molecular glues, such as PROTAC, promote targets’ degradation by inducing new interactions between E3 ligase and target proteins. Additionally, molecular glue could complement PROTACs with far more advantages in terms of molecular weight than PROTACs [156]. In this context, a recent study reports β-catenin’s molecular glue. Usually, mutated β-catenin cannot bind to β-TrCP, a natural E3 ligase. However, β-catenin molecular glue NRX-252114 restores the interaction and induces the proteasome degradation of mutated β-catenin [156] (Figure 3 and Table 3).
4.1.3. Other Protein Degradation Technologies
Recently, various protein degradation technologies, such as PROTAB (proteolytic-targeting antibody), AUTOTAC (AUTOphagy-TArgeting Chimera), and LYTAC (lysosome-targeting chimeras), have been developed [163,164]. These new platforms could be used in Wnt-targeting strategies for cancer treatment in the near future.
Indeed, a recent study showed a new targeting strategy, PROTAB, that uses Wnt signaling components. PROTAB is an antibody that induces the proteolysis of extracellular target receptors by linking to transmembrane E3 ligase ZNRF3, a negative regulator of Wnt signaling [165]. A ZNRF3-HER2 PROTAB induces the degradation of HER2 in CRC cells (SW48) and tumors of the SW48 xenograft model [165]. Additionally, ZNRF3-HER2 PROTAB shows tumor-specific degradation in CRC organoids. Though this platform does not directly target Wnt signaling compartments, it might be suitable for targeting the membrane receptors of the Wnt pathway.
4.2. Antibody–Drug Conjugate (ADC)-Based Wnt Signaling Targeting
An antibody–drug conjugate (ADC) consists of a monoclonal antibody (mAB), a cytotoxic drug (payload or warhead), and a chemical linker [166]. ADCs induce the apoptosis of antibody-bounded antigen-expressing cells. ADCs may enable more precise and effective targeting and elimination of target cells, combining the advantages of monoclonal antibodies and cytotoxic drugs [159].
Septuximab vedotin (F7-ADC) is an ADC for targeting FZD7 [157] (Figure 3 and Table 3). It comprises a human FZD7 antibody and the microtubule-inhibiting drug monomethyl auristatin E (MMAE). Ovarian serous cystadenocarcinoma overexpresses the Wnt receptor FZD7 [157]. Thus, FZD7 could be a tumor-specific antigen in ovarian serous cystadenocarcinoma. Indeed, Septuximab vedotin (F7-ADC) kills ovarian cancer cells without toxicity in vitro and in vivo.
PTK7, involving both Wnt and VEGF signaling, is associated with cancer drive and repression [158]. In the Wnt pathway, PTK7 is a co-receptor of Wnt ligands. WNT2A binds to the dimer of FZD7 and PTK7. The interaction of WNT2A, FZD7, and PTK7 inhibits the canonical Wnt pathway, while the interaction of WNT5A, ROR2, and PTK7 activates Wnt/PCP pathway. Targeting PTK7 could be suitable for specifically targeting Wnt responder CSCs in cancers. PF-06647020 is a PTK7-targeted ADC [158] (Figure 3 and Table 3). PF-06647020 delivered anti-cancer drugs effectively and has shown anti-cancer effects in various cancer cell lines and PDXs (NSCLC, OVAC, and TNBC). A phase 1 clinical trial of PF-06647020 has been completed in advanced solid tumors.
LGR5-targeted ADCs, LGR5–MC-vc-PAB–MMAE and LGR5–NMS818, were recently developed. These LGR5-targeted ADCs target the LGR5-expressing population of tumor-initiating cells or cancer stem cells (CSCs) [159] (Figure 3 and Table 3). These cells are highly responsive to Wnt signaling and are essential for tumor progression [167,168,169]. Although these ADCs do not directly inhibit the Wnt pathway, they kill Wnt responder cells such as CSCs. Moreover, these LGR5-targeted ADCs can dramatically restrain tumor growth and recurrence in vivo.
Currently, there are still difficulties in applying ADCs to cancer treatment. Most approved ADCs have side effects such as hematotoxicity [170]. Moreover, they have heavier molecular weights than conventional cytotoxic drugs, which causes poor delivery efficiency of drugs to tumors [170]. Despite these limitations, ADCs have substantial potential and are of great interest as an excellent method of targeting tumors specifically.
4.3. Oligonucleotide-Based Wnt Signaling Targeting
Oligonucleotides are emerging therapeutics that use small nucleotides (15–30 bp) with chemical modifications mainly to regulate the target gene’s transcription [171]. These small oligonucleotide therapeutics come in various forms, such as MicroRNAs (miRNA), antisense oligonucleotides (ASOs), and short-interfering RNAs (siRNAs). In addition, numerous new types of oligonucleotides, modifications, and delivery systems are continuously being developed. Thus, oligonucleotide therapeutics are expected to be applicable to cancer treatment soon, which may involve targeting Wnt signaling.
4.3.1. MicroRNAs and siRNAs
miRNAs and siRNAs consist of short sequences to target RNA through partial base pairing [172]. In prostate cancer, miR-15a and miR-16-1 are reported as tumor suppressors and downregulate Wnt3a in prostate cancer cells [173]. TargomiRs are minicells that contain miR-16-based mimic microRNA, which has 23 base pairs, and target EGFR and Wnt3a on the cell surface [174]. TargomiRs have completed clinical trials (Phase 1, NCT02369198) for the treatment of recurrent malignant pleural mesothelioma and non-small cell lung cancer [174]. Myc is a central effector of the β-catenin-dependent Wnt pathway [175]. DCR-Myc is an anti-Myc DsiRNA (Dicer-substrate small interfering RNA) that inhibits tumor growth in vivo [176]. These drugs recently completed clinical trials in hepatocellular carcinoma (Phase 1b/2, NCT02314052) and solid tumors (Phase 1, NCT02110563). Though these miRNAs and siRNAs have been tested in clinics, oligonucleotide therapies have potential hurdles, off-target effects, and inefficient delivery [177]. Additionally, these miRNAs and siRNAs do not directly target Wnt-signaling compartments.
4.3.2. Antisense Oligonucleotides
Antisense oligonucleotides (ASOs) are single-stranded DNA analogs that consist of 16–22 bases [160]. ASO binds to the target RNA sequence and controls its expression via various mechanisms (Figure 3). Recently, ribose substitutions such as 2′-O-methoxyethyl (2′-MOE) and locked nucleic acid (LNA) have improved the stability and accuracy of target binding [178]. Importantly, various antisense oligonucleotides (ASO) have recently gained FDA approval [171,179,180,181].
Several ASOs that target the Wnt pathway indirectly or directly are undergoing preclinical studies. LNA-modified ASO is reported as an indirect targeting ASO for the Wnt pathway (Figure 3 and Table 3). The long non-coding RNA (lncRNA) AC10401.1 is highly expressed in head and neck squamous cell carcinoma (HNSCC) and is related to poor prognosis in HNSCC patients. It acts as a competing endogenous RNA to miR-6817-3d in the cytoplasm and increases the stability of Wnt2B, which activates the canonical Wnt pathway [160]. Furthermore, a combination of LNA-modified ASO and a Wnt signaling inhibitor restrain AC10401.1 and inhibit cancer cell growth in cell-line and patient-derived xenograft models. This result implies that LNA-ASO can potentially suppress tumorigenesis by targeting Wnt signaling.
β-catenin is directly targeted by 2’-O-methoxyethyl chimeric ASO. Treatment with this β-catenin-targeted ASO showed a specific decrease in β-catenin expression in liver and white adipose tissue in high-fat-fed C57BL/6 mice [161] (Figure 3 and Table 3). These data show that the specific targeting of β-catenin is feasible in in vivo models. Although ASOs directly targeting Wnt signaling have not been tested, these results will soon emerge in preclinical and clinical cancer models.
5. Conclusions and Future Perspectives
Despite the contribution of Wnt signaling to tumorigenesis being obvious and Wnt signaling inhibition having shown significant effects in preclinical models, no Wnt signaling-targeted drugs have proved clinically successful in cancer or other diseases [4,6]. The main reason may be that Wnt signaling is responsible for a broad range of physiological regulations [8]. This feature could trigger diverse side effects as a consequence of treatment. Additionally, it may be because Wnt-targeted therapy itself does not have significant benefits over conventional anticancer drugs. Thus, recent Wnt targeting methods have focused on increasing treatment specificity, minimizing side effects, and combination therapies that make up for the low effectiveness of safe-dose Wnt-targeting drugs.
In addition to these efforts, combining the newest targeting strategies, such as PROTAC/molecular glue, ASOs, and ADCs, was recently introduced to Wnt-targeted therapy. Adopting these technologies and accumulating knowledge on mechanisms of action from past and current Wnt-targeting trials have allowed for a new wave of Wnt signaling-targeting therapies.
These new targeting technologies could enhance the specificity of drugs. PROTACs eliminate their targets; they do not inhibit them. ADC combines the advantages of antibodies and cytotoxic drugs, ameliorating conventional side effects. The design of ASOs is straightforward and assigns specificity toward targets. These three methods can help improve the limitations of existing targeting strategies. Additionally, numerous combinations of these new techniques are possible.
ASOs were first suggested in the early 2000s. However, the initial ASO system had limitations: poor cellular delivery, not being sufficiently stable under degradation by nuclease, and off-target effects. Additionally, some modified ASOs show high toxicity [182]. Overcoming these limitations requires time and the use of ASOs as therapeutic drugs. Nusinersen, an ASO drug, was approved in 2016 [183]. This drug has been enormously successful for spinal muscular atrophy patients. Additionally, many FDA-approved ASO drugs are used to treat rare diseases [179,180,181]. In cancer, two ASOs, danvatirsen and AZD5312 (NCT02144051), are currently undergoing clinical trials [184,185]. Danvatirsen degrades STAT3 mRNA via RNase H1 and AZD5312 degrades androgen receptor mRNA.
In conclusion, before the advent of third-generation ASOs and FDA-approved ASOs, it was assumed that ASOs could not be used for treatment. Twenty years ago, before the success of imatinib, the first FDA-approved kinase drug [186], people thought kinase drugs were impossible. Likewise, prior to Herceptin, antibodies were also considered unavailable for therapeutic use [187,188]. Therefore, the first success in Wnt-targeted therapeutics will be a significant milestone. Based on recent advanced technologies, PROTAC/molecular glue, ASO, and ADC, and accumulated knowledge of Wnt signaling, we cautiously expect that the first case of a new cancer treatment targeting Wnt signaling will appear in the near future.
Author Contributions
W.-J.P. and M.J.K. wrote and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Gachon University research fund of 2021 (GCU-202110420001) and the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (NRF-2022R1F1A1074582).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We apologize to the authors of the many original studies we were not able to cite due to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Yu, F.; Yu, C.; Li, F.; Zuo, Y.; Wang, Y.; Yao, L.; Wu, C.; Wang, C.; Ye, L. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 2021, 6, 307. [Google Scholar] [CrossRef]
- Jackstadt, R.; Hodder, M.C.; Sansom, O.J. WNT and β-Catenin in Cancer: Genes and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 177–196. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014, 346, 1248012. [Google Scholar] [CrossRef]
- Galli, L.M.; Zebarjadi, N.; Li, L.; Lingappa, V.R.; Burrus, L.W. Divergent effects of Porcupine and Wntless on WNT1 trafficking, secretion, and signaling. Exp. Cell Res. 2016, 347, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Schulte, G.; Bryja, V. The Frizzled family of unconventional G-protein-coupled receptors. Trends Pharmacol. Sci. 2007, 28, 518–525. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef]
- Zebisch, M.; Xu, Y.; Krastev, C.; MacDonald, B.T.; Chen, M.; Gilbert, R.J.; He, X.; Jones, E.Y. Structural and molecular basis of ZNRF3/RNF43 transmembrane ubiquitin ligase inhibition by the Wnt agonist R-spondin. Nat. Commun. 2013, 4, 2787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junge, H.J.; Yang, S.; Burton, J.B.; Paes, K.; Shu, X.; French, D.M.; Costa, M.; Rice, D.S.; Ye, W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell 2009, 139, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Q.; Wang, Y.; Dabdoub, A.; Smallwood, P.M.; Williams, J.; Woods, C.; Kelley, M.W.; Jiang, L.; Tasman, W.; Zhang, K.; et al. Vascular development in the retina and inner ear: Control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 2004, 116, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Morin, P.J. beta-catenin signaling and cancer. Bioessays 1999, 21, 1021–1030. [Google Scholar] [CrossRef]
- Yu, W.; Yang, L.; Li, T.; Zhang, Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front. Oncol. 2019, 9, 989. [Google Scholar] [CrossRef] [PubMed]
- Orsulic, S.; Huber, O.; Aberle, H.; Arnold, S.; Kemler, R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci. 1999, 112, 1237–1245. [Google Scholar] [CrossRef]
- Kim, M.J.; Huang, Y.; Park, J.I. Targeting Wnt Signaling for Gastrointestinal Cancer Therapy: Present and Evolving Views. Cancers 2020, 12, 3638. [Google Scholar] [CrossRef]
- Takemaru, K.I.; Moon, R.T. The transcriptional coactivator CBP interacts with beta-catenin to activate gene expression. J. Cell Biol. 2000, 149, 249–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, A.; Vleminckx, K.; Stemmler, M.P.; van Roy, F.; Kemler, R. The p300/CBP acetyltransferases function as transcriptional coactivators of beta-catenin in vertebrates. EMBO J. 2000, 19, 1839–1850. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Hocking, A.M.; Brown, J.D.; Moon, R.T. Mechanism and function of signal transduction by the Wnt/beta-catenin and Wnt/Ca2+ pathways. Oncogene 1999, 18, 7860–7872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, C.G.A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef]
- Li, S.; Lavrijsen, M.; Bakker, A.; Magierowski, M.; Magierowska, K.; Liu, P.; Wang, W.; Peppelenbosch, M.P.; Smits, R. Commonly observed RNF43 mutations retain functionality in attenuating Wnt/β-catenin signaling and unlikely confer Wnt-dependency onto colorectal cancers. Oncogene 2020, 39, 3458–3472. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Hahne, J.C.; Passalacqua, R.; Valeri, N. Microsatellite instability in gastric cancer: Molecular bases, clinical perspectives, and new treatment approaches. Cell. Mol. Life Sci. 2018, 75, 4151–4162. [Google Scholar] [CrossRef]
- Wang, W.; Xu, L.; Liu, P.; Jairam, K.; Yin, Y.; Chen, K.; Sprengers, D.; Peppelenbosch, M.P.; Pan, Q.; Smits, R. Blocking Wnt Secretion Reduces Growth of Hepatocellular Carcinoma Cell Lines Mostly Independent of β-Catenin Signaling. Neoplasia 2016, 18, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of Wnt/β-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014, 5, e1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, G.; Regel, I.; Lian, F.; Friedrich, T.; Hitkova, I.; Hofheinz, R.D.; Ströbel, P.; Langer, R.; Keller, G.; Röcken, C.; et al. WNT6 is a novel target gene of caveolin-1 promoting chemoresistance to epirubicin in human gastric cancer cells. Oncogene 2013, 32, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Oshima, H.; Matsunaga, A.; Fujimura, T.; Tsukamoto, T.; Taketo, M.M.; Oshima, M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology 2006, 131, 1086–1095. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.; Yang, H.; Li, N.; Ouyang, Y.; Xu, X.; Hong, J. Helicobacter pylori infection activates Wnt/β-catenin pathway to promote the occurrence of gastritis by upregulating ASCL1 and AQP5. Cell Death Discov. 2022, 8, 257. [Google Scholar] [CrossRef]
- Wu, C.; Zhuang, Y.; Jiang, S.; Liu, S.; Zhou, J.; Wu, J.; Teng, Y.; Xia, B.; Wang, R.; Zou, X. Interaction between Wnt/β-catenin pathway and microRNAs regulates epithelial-mesenchymal transition in gastric cancer (Review). Int. J. Oncol. 2016, 48, 2236–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachenmayer, A.; Alsinet, C.; Savic, R.; Cabellos, L.; Toffanin, S.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Newell, P.; Tsai, H.W.; et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin. Cancer Res. 2012, 18, 4997–5007. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Roberts, L.R.; Aderca, I.N.; Dong, X.; Qian, C.; Murphy, L.M.; Nagorney, D.M.; Burgart, L.J.; Roche, P.C.; Smith, D.I.; et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002, 21, 4863–4871. [Google Scholar] [CrossRef] [Green Version]
- Bengochea, A.; de Souza, M.M.; Lefrançois, L.; Le Roux, E.; Galy, O.; Chemin, I.; Kim, M.; Wands, J.R.; Trepo, C.; Hainaut, P.; et al. Common dysregulation of Wnt/Frizzled receptor elements in human hepatocellular carcinoma. Br. J. Cancer 2008, 99, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colnot, S.; Decaens, T.; Niwa-Kawakita, M.; Godard, C.; Hamard, G.; Kahn, A.; Giovannini, M.; Perret, C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc. Natl. Acad. Sci. USA 2004, 101, 17216–17221. [Google Scholar] [CrossRef] [Green Version]
- Satoh, S.; Daigo, Y.; Furukawa, Y.; Kato, T.; Miwa, N.; Nishiwaki, T.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Tokino, T.; et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat. Genet. 2000, 24, 245–250. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, H.X.; Growney, J.D.; Woolfenden, S.; Bottiglio, C.; Ng, N.; Lu, B.; Hsieh, M.H.; Bagdasarian, L.; Meyer, R.; et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12649–12654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammela, T.; Sanchez-Rivera, F.J.; Cetinbas, N.M.; Wu, K.; Joshi, N.S.; Helenius, K.; Park, Y.; Azimi, R.; Kerper, N.R.; Wesselhoeft, R.A.; et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Arai, F. Wnt signaling in the niche. Cell 2008, 132, 729–730. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kros, J.M.; Okamoto, Y.; Gaspert, A.; Huang, H.; Kurrer, M.O. APC mutations are infrequent but present in human lung cancer. Cancer Lett. 2004, 207, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Thomas de Montpréville, V.; Lacroix, L.; Rouleau, E.; Mamodaly, M.; Leclerc, J.; Tutuianu, L.; Planchard, D.; Boulate, D.; Mercier, O.; Besse, B.; et al. Non-small cell lung carcinomas with CTNNB1 (beta-catenin) mutations: A clinicopathological study of 26 cases. Ann. Diagn. Pathol. 2020, 46, 151522. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Wang, H.; Zhu, D. Wnt/β-catenin signaling pathway in lung cancer. Med. Drug Discov. 2022, 13, 100113. [Google Scholar] [CrossRef]
- Wagner, A.H.; Devarakonda, S.; Skidmore, Z.L.; Krysiak, K.; Ramu, A.; Trani, L.; Kunisaki, J.; Masood, A.; Waqar, S.N.; Spies, N.C.; et al. Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat. Commun. 2018, 9, 3787. [Google Scholar] [CrossRef]
- Stewart, D.J. Wnt signaling pathway in non-small cell lung cancer. J. Natl. Cancer Inst. 2014, 106, djt356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Morris, J.P.; Yan, W.; Schofield, H.K.; Gurney, A.; Simeone, D.M.; Millar, S.E.; Hoey, T.; Hebrok, M.; Pasca di Magliano, M. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013, 73, 4909–4922. [Google Scholar] [CrossRef] [Green Version]
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.M.; Yeung, V.P.; Cattaruzza, F.; Hussein, R.; Yen, W.C.; Murriel, C.; Evans, J.W.; O’Young, G.; Brunner, A.L.; Wang, M.; et al. RSPO3 antagonism inhibits growth and tumorigenicity in colorectal tumors harboring common Wnt pathway mutations. Sci. Rep. 2017, 7, 15270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Säfholm, A.; Tuomela, J.; Rosenkvist, J.; Dejmek, J.; Härkönen, P.; Andersson, T. The Wnt-5a-derived hexapeptide Foxy-5 inhibits breast cancer metastasis in vivo by targeting cell motility. Clin. Cancer Res. 2008, 14, 6556–6563. [Google Scholar] [CrossRef] [Green Version]
- Rodon, J.; Argilés, G.; Connolly, R.M.; Vaishampayan, U.; de Jonge, M.; Garralda, E.; Giannakis, M.; Smith, D.C.; Dobson, J.R.; McLaughlin, M.E.; et al. Phase 1 study of single-agent WNT974, a first-in-class Porcupine inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2021, 125, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Bhamra, I.; Eagle, C.; Flanagan, E.; Armer, R.; Jones, C.D.; Bingham, M.; Calcraft, P.; Edmenson Cook, A.; Thompson, B. The Wnt Pathway Inhibitor RXC004 Blocks Tumor Growth and Reverses Immune Evasion in Wnt Ligand–dependent Cancer Models. Cancer Res. Commun. 2022, 2, 914–928. [Google Scholar] [CrossRef]
- Kelly, J.; Woodcock, S.; Bhamra, I.; Jones, C.; Armer, R.; Robertson, J.; Phillips, C. Pre-clinical efficacy of the Wnt pathway inhibitor RXC004 in combination with anti-cancer therapies. Cancer Res. 2022, 82, 2566. [Google Scholar] [CrossRef]
- Bhamra, I.; Adams, N.; Armer, R.; Bingham, M.; McKeever, H.; Phillips, C.; Thompson, B.; Woodcock, S. Novel porcupine (PORCN) inhibitor RXC004: Evaluation in models of RNF43 loss of function cancers. J. Clin. Oncol. 2017, 35, e14094. [Google Scholar] [CrossRef]
- Ng, M.; Tan, D.S.; Subbiah, V.; Weekes, C.D.; Teneggi, V.; Diermayr, V.; Ethirajulu, K.; Yeo, P.; Chen, D.; Blanchard, S. First-in-human phase 1 study of ETC-159 an oral PORCN inhbitor in patients with advanced solid tumours. J. Clin. Oncol. 2017, 35, 2584. [Google Scholar] [CrossRef]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [Green Version]
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.C.; Gordon, M.; Messersmith, W.; Chugh, R.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Brachmann, R.K. Abstract B79: A first-in-human Phase 1 study of anti-cancer stem cell (CSC) agent OMP-54F28 (FZD8-Fc) targeting the WNT pathway in patients with advanced solid tumors. Mol. Cancer Ther. 2013, 12, B79. [Google Scholar] [CrossRef]
- Yeung, P.; Beviglia, L.; Cancilla, B.; Dee-Hoskins, C.; Evans, J.W.; Fischer, M.M.; Yen, W.-C.; Gurney, A.; Lewicki, J.; Hoey, T. Wnt pathway antagonist OMP-54F28 (FZD8-Fc) inhibits tumor growth and reduces tumor-initiating cell frequency in patient-derived hepatocellular carcinoma and ovarian cancer xenograft models. Cancer Res. 2014, 74, 1907. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell. Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L.; et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef]
- Gurney, A.; Axelrod, F.; Bond, C.J.; Cain, J.; Chartier, C.; Donigan, L.; Fischer, M.; Chaudhari, A.; Ji, M.; Kapoun, A.M.; et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 11717–11722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraudet, A.L.; Cassier, P.A.; Iwao-Fukukawa, C.; Garin, G.; Badel, J.N.; Kryza, D.; Chabaud, S.; Gilles-Afchain, L.; Clapisson, G.; Desuzinges, C.; et al. A first-in-human study investigating biodistribution, safety and recommended dose of a new radiolabeled MAb targeting FZD10 in metastatic synovial sarcoma patients. BMC Cancer 2018, 18, 646. [Google Scholar] [CrossRef]
- Inglis, D.J.; Licari, J.; Georgiou, K.R.; Wittwer, N.L.; Hamilton, R.W.; Beaumont, D.M.; Scherer, M.A.; Lavranos, T.C. Characterization of BNC101 a human specific monoclonal antibody targeting the GPCR LGR5: First-in-human evidence of target engagement. Cancer Res. 2018, 78, 3910. [Google Scholar] [CrossRef]
- Edenfield, W.J.; Richards, D.A.; Vukelja, S.J.; Weiss, G.J.; Sirard, C.A.; Landau, S.B.; Ramanathan, R.K. A phase 1 study evaluating the safety and efficacy of DKN-01, an investigational monoclonal antibody (Mab) in patients (pts) with advanced non-small cell lung cancer. J. Clin. Oncol. 2014, 32, 8068. [Google Scholar] [CrossRef]
- Eads, J.R.; Goyal, L.; Stein, S.; El-Khoueiry, A.B.; Manji, G.A.; Abrams, T.A.; Landau, S.B.; Sirard, C.A. Phase I study of DKN-01, an anti-DKK1 antibody, in combination with gemcitabine (G) and cisplatin (C) in patients (pts) with advanced biliary cancer. J. Clin. Oncol. 2016, 34, e15603. [Google Scholar] [CrossRef]
- Goyal, L.; Sirard, C.; Schrag, M.; Kagey, M.H.; Eads, J.R.; Stein, S.; El-Khoueiry, A.B.; Manji, G.A.; Abrams, T.A.; Khorana, A.A.; et al. Phase I and Biomarker Study of the Wnt Pathway Modulator DKN-01 in Combination with Gemcitabine/Cisplatin in Advanced Biliary Tract Cancer. Clin. Cancer Res. 2020, 26, 6158–6167. [Google Scholar] [CrossRef]
- Arend, R.C.; Castro, C.M.; Matulonis, U.A.; Hamilton, E.; Gunderson, C.C.; Lybarger, K.S.S.; Goodman, H.M.; Duska, L.R.; Mahdi, H.; ElNaggar, A.C. Dkn-01 treated patients with recurrent epithelial endometrial (EEC) or ovarian (EOC) cancers which harbor Wnt activating mutations have longer progression-free survival and improved clinical benefit. Gynecol. Oncol. 2020, 159, 5–6. [Google Scholar] [CrossRef]
- Klempner, S.J.; Bendell, J.C.; Villaflor, V.M.; Tenner, L.L.; Stein, S.; Naik, G.S.; Sirard, C.A.; Kagey, M.; Chaney, M.F.; Strickler, J.H. DKN-01 in combination with pembrolizumab in patients with advanced gastroesophageal adenocarcinoma (GEA): Tumoral DKK1 expression as a predictor of response and survival. J. Clin. Oncol. 2020, 38, 357. [Google Scholar] [CrossRef]
- Lee, H.J.; Wang, N.X.; Shi, D.L.; Zheng, J.J. Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein Dishevelled. Angew. Chem. Int. Ed. Engl. 2009, 48, 6448–6452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef] [Green Version]
- McGonigle, S.; Chen, Z.; Wu, J.; Chang, P.; Kolber-Simonds, D.; Ackermann, K.; Twine, N.C.; Shie, J.L.; Miu, J.T.; Huang, K.C.; et al. E7449: A dual inhibitor of PARP1/2 and tankyrase1/2 inhibits growth of DNA repair deficient tumors and antagonizes Wnt signaling. Oncotarget 2015, 6, 41307–41323. [Google Scholar] [CrossRef]
- Plummer, R.; Dua, D.; Cresti, N.; Drew, Y.; Stephens, P.; Foegh, M.; Knudsen, S.; Sachdev, P.; Mistry, B.M.; Dixit, V.; et al. First-in-human study of the PARP/tankyrase inhibitor E7449 in patients with advanced solid tumours and evaluation of a novel drug-response predictor. Br. J. Cancer 2020, 123, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Cranmer, L.D.; Abdul Razak, A.R.; Ratan, R.; Choy, E.; George, S.; Liebner, D.A.; Stenehjem, D.D.; Gounder, M.M. Results of a phase I dose escalation and expansion study of tegavivint (BC2059), a first-in-class TBL1 inhibitor for patients with progressive, unresectable desmoid tumor. J. Clin. Oncol. 2022, 40, 11523. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Saha, S.; Shah, B.; Devaraj, S.G.; Sun, B.; Horrigan, S.; Leveque, C.; Zu, Y.; Iyer, S.; et al. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia 2015, 29, 1267–1278. [Google Scholar] [CrossRef]
- Savvidou, I.; Khong, T.; Cuddihy, A.; McLean, C.; Horrigan, S.; Spencer, A. β-catenin inhibitor BC2059 is efficacious as monotherapy or in combination with proteasome inhibitor bortezomib in multiple myeloma. Mol. Cancer Ther. 2017, 16, 1765–1778. [Google Scholar] [CrossRef] [Green Version]
- Osawa, Y.; Oboki, K.; Imamura, J.; Kojika, E.; Hayashi, Y.; Hishima, T.; Saibara, T.; Shibasaki, F.; Kohara, M.; Kimura, K. Inhibition of Cyclic Adenosine Monophosphate (cAMP)-response Element-binding Protein (CREB)-binding Protein (CBP)/β-Catenin Reduces Liver Fibrosis in Mice. EBioMedicine 2015, 2, 1751–1758. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, R.R.; Ko, A.H.; Chiorean, E.G.; Kwak, E.L.; Lenz, H.-J.; Nadler, P.I.; Wood, D.L.; Fujimori, M.; Morita, K.; Inada, T. A phase Ib dose-escalation study of PRI-724, a CBP/beta-catenin modulator, plus gemcitabine (GEM) in patients with advanced pancreatic adenocarcinoma (APC) as second-line therapy after FOLFIRINOX or FOLFOX. J. Clin. Oncol. 2015, 33, e15270. [Google Scholar] [CrossRef]
- Scott, A.; Call, J.; Chandana, S.; Borazanci, E.; Falchook, G.; Bordoni, R.; Richey, S.; Starodub, A.; Chung, V.; Lakhani, N. 451O Preliminary evidence of clinical activity from phase I and Ib trials of the CLK/DYRK inhibitor cirtuvivint (CIRT) in subjects with advanced solid tumors. Ann. Oncol. 2022, 33, S742–S743. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Coelen, R.J.S.; Khurshed, M.; Roos, E.; Caan, M.W.A.; van Linde, M.E.; Kouwenhoven, M.; Bramer, J.A.M.; Bovée, J.V.M.G.; Mathôt, R.A.; et al. Study protocol of a phase IB/II clinical trial of metformin and chloroquine in patients with. BMJ Open 2017, 7, e014961. [Google Scholar] [CrossRef] [Green Version]
- Arora, S.P.; Tenner, L.; Sarantopoulos, J.; Morris, J.; Liu, Q.; Mendez, J.A.; Curiel, T.; Michalek, J.; Mahalingam, D. Modulation of autophagy: A Phase II study of vorinostat plus hydroxychloroquine versus regorafenib in chemotherapy-refractory metastatic colorectal cancer (mCRC). Br. J. Cancer 2022, 127, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef]
- Loaiza-Bonilla, A.; O’Hara, M.H.; Redlinger, M.; Damjanov, N.; Teitelbaum, U.R.; Vasilevskaya, I.; Rosen, M.A.; Heitjan, D.F.; Amaravadi, R.K.; O’Dwyer, P.J. Phase II trial of autophagy inhibition using hydroxychloroquine (HCQ) with FOLFOX/bevacizumab in the first-line treatment of advanced colorectal cancer. J. Clin. Oncol. 2015, 33, 3614. [Google Scholar] [CrossRef]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined autophagy and HDAC inhibition: A phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Dodge, M.E.; Tang, W.; Lu, J.; Ma, Z.; Fan, C.W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Moon, J.; Dodge, M.E.; Pan, X.; Zhang, L.; Hanson, J.M.; Tuladhar, R.; Ma, Z.; Shi, H.; Williams, N.S.; et al. The development of highly potent inhibitors for porcupine. J. Med. Chem. 2013, 56, 2700–2704. [Google Scholar] [CrossRef] [Green Version]
- Proffitt, K.D.; Madan, B.; Ke, Z.; Pendharkar, V.; Ding, L.; Lee, M.A.; Hannoush, R.N.; Virshup, D.M. Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT-driven mammary cancer. Cancer Res. 2013, 73, 502–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Liu, J.; Han, D.; Zhang, G.; Gao, W.; Hsieh, M.H.; Ng, N.; Kasibhatla, S.; Tompkins, C.; Li, J.; et al. Discovery of Pyridinyl Acetamide Derivatives as Potent, Selective, and Orally Bioavailable Porcupine Inhibitors. ACS Med. Chem. Lett. 2016, 7, 676–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mook, R.A.; Wang, J.; Ren, X.R.; Chen, M.; Spasojevic, I.; Barak, L.S.; Lyerly, H.K.; Chen, W. Structure-activity studies of Wnt/β-catenin inhibition in the Niclosamide chemotype: Identification of derivatives with improved drug exposure. Bioorg. Med. Chem. 2015, 23, 5829–5838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mook, R.A.; Ren, X.R.; Zhang, Q.; Jing, G.; Lu, M.; Spasojevic, I.; Lyerly, H.K.; Hsu, D.; Chen, W. Identification of DK419, a potent inhibitor of Wnt/β-catenin signaling and colorectal cancer growth. Bioorg. Med. Chem. 2018, 26, 5435–5442. [Google Scholar] [CrossRef] [PubMed]
- Nile, A.H.; de Sousa, E.; Melo, F.; Mukund, S.; Piskol, R.; Hansen, S.; Zhou, L.; Zhang, Y.; Fu, Y.; Gogol, E.B.; et al. A selective peptide inhibitor of Frizzled 7 receptors disrupts intestinal stem cells. Nat. Chem. Biol. 2018, 14, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, W.; Ananthan, S.; Suto, M.J.; Li, Y. Discovery of novel frizzled-7 inhibitors by targeting the receptor’s transmembrane domain. Oncotarget 2017, 8, 91459–91470. [Google Scholar] [CrossRef] [PubMed]
- Nambotin, S.B.; Lefrancois, L.; Sainsily, X.; Berthillon, P.; Kim, M.; Wands, J.R.; Chevallier, M.; Jalinot, P.; Scoazec, J.Y.; Trepo, C.; et al. Pharmacological inhibition of Frizzled-7 displays anti-tumor properties in hepatocellular carcinoma. J. Hepatol. 2011, 54, 288–299. [Google Scholar] [CrossRef]
- Lee, H.J.; Bao, J.; Miller, A.; Zhang, C.; Wu, J.; Baday, Y.C.; Guibao, C.; Li, L.; Wu, D.; Zheng, J.J. Structure-based Discovery of Novel Small Molecule Wnt Signaling Inhibitors by Targeting the Cysteine-rich Domain of Frizzled. J. Biol. Chem. 2015, 290, 30596–30606. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Ma, S.; Kim, H.Y.; Yun, J.H.; Heo, J.N.; Lee, W.; Choi, K.Y.; No, K.T. Identification of small-molecule compounds targeting the dishevelled PDZ domain by virtual screening and binding studies. Bioorg. Med. Chem. 2016, 24, 3259–3266. [Google Scholar] [CrossRef]
- Grandy, D.; Shan, J.; Zhang, X.; Rao, S.; Akunuru, S.; Li, H.; Zhang, Y.; Alpatov, I.; Zhang, X.A.; Lang, R.A.; et al. Discovery and characterization of a small molecule inhibitor of the PDZ domain of dishevelled. J. Biol. Chem. 2009, 284, 16256–16263. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Zhang, X.; Bao, J.; Cassell, R.; Zheng, J.J. Synthesis of potent dishevelled PDZ domain inhibitors guided by virtual screening and NMR studies. Chem. Biol. Drug Des. 2012, 79, 376–383. [Google Scholar] [CrossRef]
- Fujii, N.; You, L.; Xu, Z.; Uematsu, K.; Shan, J.; He, B.; Mikami, I.; Edmondson, L.R.; Neale, G.; Zheng, J.; et al. An antagonist of dishevelled protein-protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res. 2007, 67, 573–579. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Choi, S.; Yoon, J.H.; Lim, H.J.; Lee, H.; Choi, J.; Ro, E.J.; Heo, J.N.; Lee, W.; No, K.T.; et al. Small molecule inhibitors of the Dishevelled-CXXC5 interaction are new drug candidates for bone anabolic osteoporosis therapy. EMBO Mol. Med. 2016, 8, 375–387. [Google Scholar] [CrossRef]
- Shan, J.; Zheng, J.J. Optimizing Dvl PDZ domain inhibitor by exploring chemical space. J. Comput. Aided Mol. Des. 2009, 23, 37–47. [Google Scholar] [CrossRef]
- Zhang, Y.; Appleton, B.A.; Wiesmann, C.; Lau, T.; Costa, M.; Hannoush, R.N.; Sidhu, S.S. Inhibition of Wnt signaling by Dishevelled PDZ peptides. Nat. Chem. Biol. 2009, 5, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Orton, D.; Neitzel, L.R.; Astudillo, L.; Shen, C.; Long, J.; Chen, X.; Kirkbride, K.C.; Doundoulakis, T.; Guerra, M.L.; et al. Differential abundance of CK1α provides selectivity for pharmacological CK1α activators to target WNT-dependent tumors. Sci. Signal. 2017, 10, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewan, K.; Pajak, B.; Stubbs, M.; Todd, H.; Barbeau, O.; Quevedo, C.; Botfield, H.; Young, R.; Ruddle, R.; Samuel, L.; et al. A useful approach to identify novel small-molecule inhibitors of Wnt-dependent transcription. Cancer Res. 2010, 70, 5963–5973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos, J.A.; Godoy, J.A.; Inestrosa, N.C. Wnt3a ligand facilitates autophagy in hippocampal neurons by modulating a novel GSK-3β-AMPK axis. Cell Commun. Signal. 2018, 16, 15. [Google Scholar] [CrossRef]
- Scarborough, H.A.; Helfrich, B.A.; Casás-Selves, M.; Schuller, A.G.; Grosskurth, S.E.; Kim, J.; Tan, A.C.; Chan, D.C.; Zhang, Z.; Zaberezhnyy, V.; et al. AZ1366: An Inhibitor of Tankyrase and the Canonical Wnt Pathway that Limits the Persistence of Non-Small Cell Lung Cancer Cells Following EGFR Inhibition. Clin. Cancer Res. 2017, 23, 1531–1541. [Google Scholar] [CrossRef] [Green Version]
- Lau, T.; Chan, E.; Callow, M.; Waaler, J.; Boggs, J.; Blake, R.A.; Magnuson, S.; Sambrone, A.; Schutten, M.; Firestein, R.; et al. A novel tankyrase small-molecule inhibitor suppresses APC mutation-driven colorectal tumor growth. Cancer Res. 2013, 73, 3132–3144. [Google Scholar] [CrossRef] [Green Version]
- Okada-Iwasaki, R.; Takahashi, Y.; Watanabe, Y.; Ishida, H.; Saito, J.; Nakai, R.; Asai, A. The Discovery and Characterization of K-756, a Novel Wnt/β-Catenin Pathway Inhibitor Targeting Tankyrase. Mol. Cancer Ther. 2016, 15, 1525–1534. [Google Scholar] [CrossRef] [Green Version]
- Shultz, M.D.; Cheung, A.K.; Kirby, C.A.; Firestone, B.; Fan, J.; Chen, C.H.; Chen, Z.; Chin, D.N.; Dipietro, L.; Fazal, A.; et al. Identification of NVP-TNKS656: The use of structure-efficiency relationships to generate a highly potent, selective, and orally active tankyrase inhibitor. J. Med. Chem. 2013, 56, 6495–6511. [Google Scholar] [CrossRef]
- Narwal, M.; Koivunen, J.; Haikarainen, T.; Obaji, E.; Legala, O.E.; Venkannagari, H.; Joensuu, P.; Pihlajaniemi, T.; Lehtiö, L. Discovery of tankyrase inhibiting flavones with increased potency and isoenzyme selectivity. J. Med. Chem. 2013, 56, 7880–7889. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, A.; Yashiroda, Y.; Muramatsu, Y.; Yoshida, H.; Chikada, T.; Tsumura, T.; Okue, M.; Shirai, F.; Fukami, T.; Yoshida, M.; et al. RK-287107, a potent and specific tankyrase inhibitor, blocks colorectal cancer cell growth in a preclinical model. Cancer Sci. 2018, 109, 4003–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, R.G.; Davidson, K.C.; Bosch, K.A.; Biechele, T.L.; Robin, N.C.; Taylor, R.J.; Major, M.B.; Camp, N.D.; Fowler, K.; Martins, T.J.; et al. WIKI4, a novel inhibitor of tankyrase and Wnt/ß-catenin signaling. PLoS ONE 2012, 7, e50457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, M.; Elliott, R.; Bowers, L.; Balan, N.; Rafiq, R.; Costa-Cabral, S.; Munkonge, F.; Trinidade, I.; Porter, R.; Campbell, A.D.; et al. A novel tankyrase inhibitor, MSC2504877, enhances the effects of clinical CDK4/6 inhibitors. Sci. Rep. 2019, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Voronkov, A.; Holsworth, D.D.; Waaler, J.; Wilson, S.R.; Ekblad, B.; Perdreau-Dahl, H.; Dinh, H.; Drewes, G.; Hopf, C.; Morth, J.P.; et al. Structural basis and SAR for G007-LK, a lead stage 1,2,4-triazole based specific tankyrase 1/2 inhibitor. J. Med. Chem. 2013, 56, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Waaler, J.; Machon, O.; Tumova, L.; Dinh, H.; Korinek, V.; Wilson, S.R.; Paulsen, J.E.; Pedersen, N.M.; Eide, T.J.; Machonova, O.; et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012, 72, 2822–2832. [Google Scholar] [CrossRef] [Green Version]
- Waaler, J.; Machon, O.; von Kries, J.P.; Wilson, S.R.; Lundenes, E.; Wedlich, D.; Gradl, D.; Paulsen, J.E.; Machonova, O.; Dembinski, J.L.; et al. Novel synthetic antagonists of canonical Wnt signaling inhibit colorectal cancer cell growth. Cancer Res. 2011, 71, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Shetti, D.; Zhang, B.; Fan, C.; Mo, C.; Lee, B.H.; Wei, K. Low Dose of Paclitaxel Combined with XAV939 Attenuates Metastasis, Angiogenesis and Growth in Breast Cancer by Suppressing Wnt Signaling. Cells 2019, 8, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, P.H.; Cho, Y.H.; Lee, S.K.; Lee, J.; Jeong, W.J.; Moon, B.S.; Yun, J.H.; Yang, J.S.; Choi, S.; Yoon, J.; et al. Small-molecule binding of the axin RGS domain promotes β-catenin and Ras degradation. Nat. Chem. Biol. 2016, 12, 593–600. [Google Scholar] [CrossRef]
- Trosset, J.Y.; Dalvit, C.; Knapp, S.; Fasolini, M.; Veronesi, M.; Mantegani, S.; Gianellini, L.M.; Catana, C.; Sundström, M.; Stouten, P.F.; et al. Inhibition of protein-protein interactions: The discovery of druglike beta-catenin inhibitors by combining virtual and biophysical screening. Proteins 2006, 64, 60–67. [Google Scholar] [CrossRef]
- Yao, H.; Ashihara, E.; Strovel, J.W.; Nakagawa, Y.; Kuroda, J.; Nagao, R.; Tanaka, R.; Yokota, A.; Takeuchi, M.; Hayashi, Y.; et al. AV-65, a novel Wnt/β-catenin signal inhibitor, successfully suppresses progression of multiple myeloma in a mouse model. Blood Cancer J. 2011, 1, e43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Ao, A.; Zhou, L.; Murphy, C.K.; Frist, A.Y.; Keel, J.J.; Thorne, C.A.; Kim, K.; Lee, E.; Hong, C.C. Selective small molecule targeting β-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 2013, 4, 898–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordonaro, M.; Lazarova, D.L. Determination of the Role of CBP- and p300-Mediated Wnt Signaling on Colonic Cells. JMIR Res. Protoc. 2016, 5, e66. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Zhu, D.; Bird, G.H.; Sukhdeo, K.; Zhao, J.J.; Mani, M.; Lemieux, M.; Carrasco, D.E.; Ryan, J.; Horst, D.; et al. Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 2012, 4, 148ra117. [Google Scholar] [CrossRef] [Green Version]
- De la Roche, M.; Rutherford, T.J.; Gupta, D.; Veprintsev, D.B.; Saxty, B.; Freund, S.M.; Bienz, M. An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 2012, 3, 680. [Google Scholar] [PubMed] [Green Version]
- Zhang, H.; Liu, C.; Zhu, D.; Zhang, Q.; Li, J. Medicinal Chemistry Strategies for the Development of Inhibitors Disrupting β-Catenin’s Interactions with Its Nuclear Partners. J. Med. Chem. 2023, 66, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Lavergne, E.; Hendaoui, I.; Coulouarn, C.; Ribault, C.; Leseur, J.; Eliat, P.A.; Mebarki, S.; Corlu, A.; Clément, B.; Musso, O. Blocking Wnt signaling by SFRP-like molecules inhibits in vivo cell proliferation and tumor growth in cells carrying active β-catenin. Oncogene 2011, 30, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Bovolenta, P.; Esteve, P.; Ruiz, J.M.; Cisneros, E.; Lopez-Rios, J. Beyond Wnt inhibition: New functions of secreted Frizzled-related proteins in development and disease. J. Cell Sci. 2008, 121, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Glinka, A.; Wu, W.; Delius, H.; Monaghan, A.P.; Blumenstock, C.; Niehrs, C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998, 391, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Ribe, E.M.; Al-Shawi, R.; Malik, B.; Hooper, C.; Fernandes, C.; Dobson, R.; Nolan, P.M.; Lourdusamy, A.; Furney, S.; et al. Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt-PCP-JNK pathway. Mol. Psychiatry 2014, 19, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Krause, U.; Ryan, D.M.; Clough, B.H.; Gregory, C.A. An unexpected role for a Wnt-inhibitor: Dickkopf-1 triggers a novel cancer survival mechanism through modulation of aldehyde-dehydrogenase-1 activity. Cell Death Dis. 2014, 5, e1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, Y.; Beauchamp, E.; Woods, D.; Taylor, W.G.; Toretsky, J.A.; Uren, A.; Rubin, J.S. Wnt-3a and Dickkopf-1 stimulate neurite outgrowth in Ewing tumor cells via a Frizzled3- and c-Jun N-terminal kinase-dependent mechanism. Mol. Cell. Biol. 2008, 28, 2368–2379. [Google Scholar] [CrossRef] [Green Version]
- Caneparo, L.; Huang, Y.L.; Staudt, N.; Tada, M.; Ahrendt, R.; Kazanskaya, O.; Niehrs, C.; Houart, C. Dickkopf-1 regulates gastrulation movements by coordinated modulation of Wnt/beta catenin and Wnt/PCP activities, through interaction with the Dally-like homolog Knypek. Genes Dev. 2007, 21, 465–480. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Song, J.; Chen, W.; Yuan, D.; Wang, W.; Chen, X.; Liu, H.; Su, H.; Zhu, J. Expression and Role of Dickkopf-1 (Dkk1) in Tumors: From the Cells to the Patients. Cancer Manag. Res. 2021, 13, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Hinoda, Y.; Nakajima, K.; Kawamoto, K.; Kikuno, N.; Ueno, K.; Yamamura, S.; Zaman, M.S.; Khatri, G.; Chen, Y.; et al. Wnt antagonist DKK1 acts as a tumor suppressor gene that induces apoptosis and inhibits proliferation in human renal cell carcinoma. Int. J. Cancer 2011, 128, 1793–1803. [Google Scholar] [CrossRef]
- Mikheev, A.M.; Mikheeva, S.A.; Maxwell, J.P.; Rivo, J.V.; Rostomily, R.; Swisshelm, K.; Zarbl, H. Dickkopf-1 mediated tumor suppression in human breast carcinoma cells. Breast Cancer Res. Treat. 2008, 112, 263–273. [Google Scholar] [CrossRef]
- Cowling, V.H.; D’Cruz, C.M.; Chodosh, L.A.; Cole, M.D. c-Myc transforms human mammary epithelial cells through repression of the Wnt inhibitors DKK1 and SFRP1. Mol. Cell. Biol. 2007, 27, 5135–5146. [Google Scholar] [CrossRef] [Green Version]
- Kuphal, S.; Lodermeyer, S.; Bataille, F.; Schuierer, M.; Hoang, B.H.; Bosserhoff, A.K. Expression of Dickkopf genes is strongly reduced in malignant melanoma. Oncogene 2006, 25, 5027–5036. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar] [PubMed]
- Kimelman, D.; Xu, W. beta-catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.A.; Johnson, P.E. Pyrvinium pamoate in the treatment of pinworm infection (enterobiasis) in the home. J. Pediatr. 1962, 60, 243–251. [Google Scholar] [CrossRef]
- Li, B.; Flaveny, C.A.; Giambelli, C.; Fei, D.L.; Han, L.; Hang, B.I.; Bai, F.; Pei, X.H.; Nose, V.; Burlingame, O.; et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS ONE 2014, 9, e101969. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, M.; Resnick, L.; Gamliel, E.; Kesaraju, S.; Weissbach, H.; Binninger, D. Sulindac enhances the killing of cancer cells exposed to oxidative stress. PLoS ONE 2009, 4, e5804. [Google Scholar] [CrossRef] [Green Version]
- Riffell, J.L.; Lord, C.J.; Ashworth, A. Tankyrase-targeted therapeutics: Expanding opportunities in the PARP family. Nat. Rev. Drug Discov. 2012, 11, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Cruciat, C.M.; Ohkawara, B.; Acebron, S.P.; Karaulanov, E.; Reinhard, C.; Ingelfinger, D.; Boutros, M.; Niehrs, C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science 2010, 327, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Jun, S.; Kim, M.J.; Lee, S.H.; Suh, H.N.; Lien, E.M.; Jung, H.Y.; Lee, S.; Zhang, J.; Yang, J.I.; et al. TMEM9 promotes intestinal tumorigenesis through vacuolar-ATPase-activated Wnt/β-catenin signalling. Nat. Cell Biol. 2018, 20, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Pálmer, H.G.; González-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D3 promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef] [Green Version]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, S.; Beilschmidt, M.; To, M.; Lin, K.; Lui, F.; Jmeian, Y.; Ng, M.; Fernandez, M.; Fu, Y.; Mascall, K.; et al. Structure-guided design fine-tunes pharmacokinetics, tolerability, and antitumor profile of multispecific frizzled antibodies. Proc. Natl. Acad. Sci. USA 2019, 116, 6812–6817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.; Li, X.; Zhao, L.; Wang, Y.; Wang, X.; Wu, Y.; Zhou, X.; Fu, W.; Liu, L.; Hu, H.G.; et al. A PROTAC peptide induces durable β-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020, 6, 35. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetta, K.R.; Taygerly, J.; Boyle, K.; Basham, S.E.; Padovani, C.; Lou, Y.; Cummins, T.J.; Yung, S.L.; von Soly, S.K.; Kayser, F.; et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat. Commun. 2019, 10, 1402. [Google Scholar] [CrossRef] [Green Version]
- Do, M.; Wu, C.C.N.; Sonavane, P.R.; Juarez, E.F.; Adams, S.R.; Ross, J.; Rodriguez, Y.; Baena, A.; Patel, C.; Mesirov, J.P.; et al. A FZD7-specific Antibody-Drug Conjugate Induces Ovarian Tumor Regression in Preclinical Models. Mol. Cancer Ther. 2022, 21, 113–124. [Google Scholar] [CrossRef]
- Katoh, M. Antibody-drug conjugate targeting protein tyrosine kinase 7, a receptor tyrosine kinase-like molecule involved in WNT and vascular endothelial growth factor signaling: Effects on cancer stem cells, tumor microenvironment and whole-body homeostasis. Ann. Transl. Med. 2017, 5, 462. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; Mao, W.; Wang, X.; Wang, B.E.; Pham, T.; Flygare, J.; Yu, S.F.; Yee, S.; Goldenberg, D.; Fields, C.; et al. Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci. Transl. Med. 2015, 7, 314ra186. [Google Scholar] [CrossRef]
- Li, M.; Ding, X.; Zhang, Y.; Li, X.; Zhou, H.; Yang, L.; Li, Y.; Yang, P.; Zhang, X.; Hu, J.; et al. Antisense oligonucleotides targeting lncRNA AC104041.1 induces antitumor activity through Wnt2B/β-catenin pathway in head and neck squamous cell carcinomas. Cell Death Dis. 2020, 11, 672. [Google Scholar] [CrossRef] [PubMed]
- Popov, V.B.; Jornayvaz, F.R.; Akgul, E.O.; Kanda, S.; Jurczak, M.J.; Zhang, D.; Abudukadier, A.; Majumdar, S.K.; Guigni, B.; Petersen, K.F.; et al. Second-generation antisense oligonucleotides against β-catenin protect mice against diet-induced hepatic steatosis and hepatic and peripheral insulin resistance. FASEB J. 2016, 30, 1207–1217. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-X. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Ji, C.H.; Kim, H.Y.; Lee, M.J.; Heo, A.J.; Park, D.Y.; Lim, S.; Shin, S.; Ganipisetti, S.; Yang, W.S.; Jung, C.A.; et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 2022, 13, 904. [Google Scholar] [CrossRef] [PubMed]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Marei, H.; Tsai, W.K.; Kee, Y.S.; Ruiz, K.; He, J.; Cox, C.; Sun, T.; Penikalapati, S.; Dwivedi, P.; Choi, M.; et al. Antibody targeting of E3 ubiquitin ligases for receptor degradation. Nature 2022, 610, 182–189. [Google Scholar] [CrossRef]
- Trail, P.A. Antibody drug conjugates as cancer therapeutics. Antibodies 2013, 2, 113–129. [Google Scholar]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- de Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Schepers, A.G.; Snippert, H.J.; Stange, D.E.; van den Born, M.; van Es, J.H.; van de Wetering, M.; Clevers, H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 2012, 337, 730–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, X.; Azhdarinia, A.; Ghosh, S.C.; Xiong, W.; An, Z.; Liu, Q.; Carmon, K.S. LGR5-Targeted Antibody-Drug Conjugate Eradicates Gastrointestinal Tumors and Prevents Recurrence. Mol. Cancer Ther. 2016, 15, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonci, D.; Coppola, V.; Musumeci, M.; Addario, A.; Giuffrida, R.; Memeo, L.; D’Urso, L.; Pagliuca, A.; Biffoni, M.; Labbaye, C.; et al. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat. Med. 2008, 14, 1271–1277. [Google Scholar] [CrossRef]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Rennoll, S.; Yochum, G. Regulation of MYC gene expression by aberrant Wnt/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290–300. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Martinez, D.; Wood, D.L.; Fielman, B.; Sharma, M.; Janisch, L.A.; Brown, B.D. Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 11006. [Google Scholar] [CrossRef]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther.-Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense oligonucleotides: An emerging area in drug discovery and development. J. Clin. Med. 2020, 9, 2004. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Heo, Y.A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Aartsma-Rus, A. FDA Approval of Nusinersen for Spinal Muscular Atrophy Makes 2016 the Year of Splice Modulating Oligonucleotides. Nucleic Acid Ther. 2017, 27, 67–69. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Corey, D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Velasco, M.A.; Kura, Y.; Sakai, K.; Hatanaka, Y.; Davies, B.R.; Campbell, H.; Klein, S.; Kim, Y.; MacLeod, A.R.; Sugimoto, K.; et al. Targeting castration-resistant prostate cancer with androgen receptor antisense oligonucleotide therapy. JCI Insight 2019, 4, e122688. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Sliwkowski, M.X.; Lofgren, J.A.; Lewis, G.D.; Hotaling, T.E.; Fendly, B.M.; Fox, J.A. Nonclinical studies addressing the mechanism of action of trastuzumab (Herceptin). Semin. Oncol. 1999, 26, 60–70. [Google Scholar]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.; Henner, D.; Wong, W.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Wnt signaling and its alteration in various cancers. Signal transduction of β-catenin-dependent and independent Wnt signaling. The frequencies of genetic alteration of individual Wnt signaling modules in indicated cancers were summarized.
Figure 1.
Wnt signaling and its alteration in various cancers. Signal transduction of β-catenin-dependent and independent Wnt signaling. The frequencies of genetic alteration of individual Wnt signaling modules in indicated cancers were summarized.

Figure 2.
Wnt signaling modulating agents in clinical trials for cancer treatment. Promising clinical agents targeting the Wnt signaling module were displayed along with their targets.
Figure 2.
Wnt signaling modulating agents in clinical trials for cancer treatment. Promising clinical agents targeting the Wnt signaling module were displayed along with their targets.

Figure 3.
Targeting Wnt signaling pathway by the latest technologies. β-catenin targeted PROTACs (xStAx-VHL) recruits E3 ligase to β-catenin, inducing the degradation of β-catenin using the ubiquitin–proteasome system. Additionally, the molecular glue of β-catenin and its E3 ligase β-TrCP, NRX-252114, enhances the β-catenin degradation. ADCs (LGR5-mc-vc-PAB-MMAE, LRG5-NMs818, and PF-06647020) targeting LGR5- or PTK7-expressing Wnt responder cells bind to the target cells and release a cytotoxic drug to induce the cell death. ASO (LNA-modified ASO, β-catenin targeting ASO) binds to the target region of RNA and suppresses expression.
Figure 3.
Targeting Wnt signaling pathway by the latest technologies. β-catenin targeted PROTACs (xStAx-VHL) recruits E3 ligase to β-catenin, inducing the degradation of β-catenin using the ubiquitin–proteasome system. Additionally, the molecular glue of β-catenin and its E3 ligase β-TrCP, NRX-252114, enhances the β-catenin degradation. ADCs (LGR5-mc-vc-PAB-MMAE, LRG5-NMs818, and PF-06647020) targeting LGR5- or PTK7-expressing Wnt responder cells bind to the target cells and release a cytotoxic drug to induce the cell death. ASO (LNA-modified ASO, β-catenin targeting ASO) binds to the target region of RNA and suppresses expression.


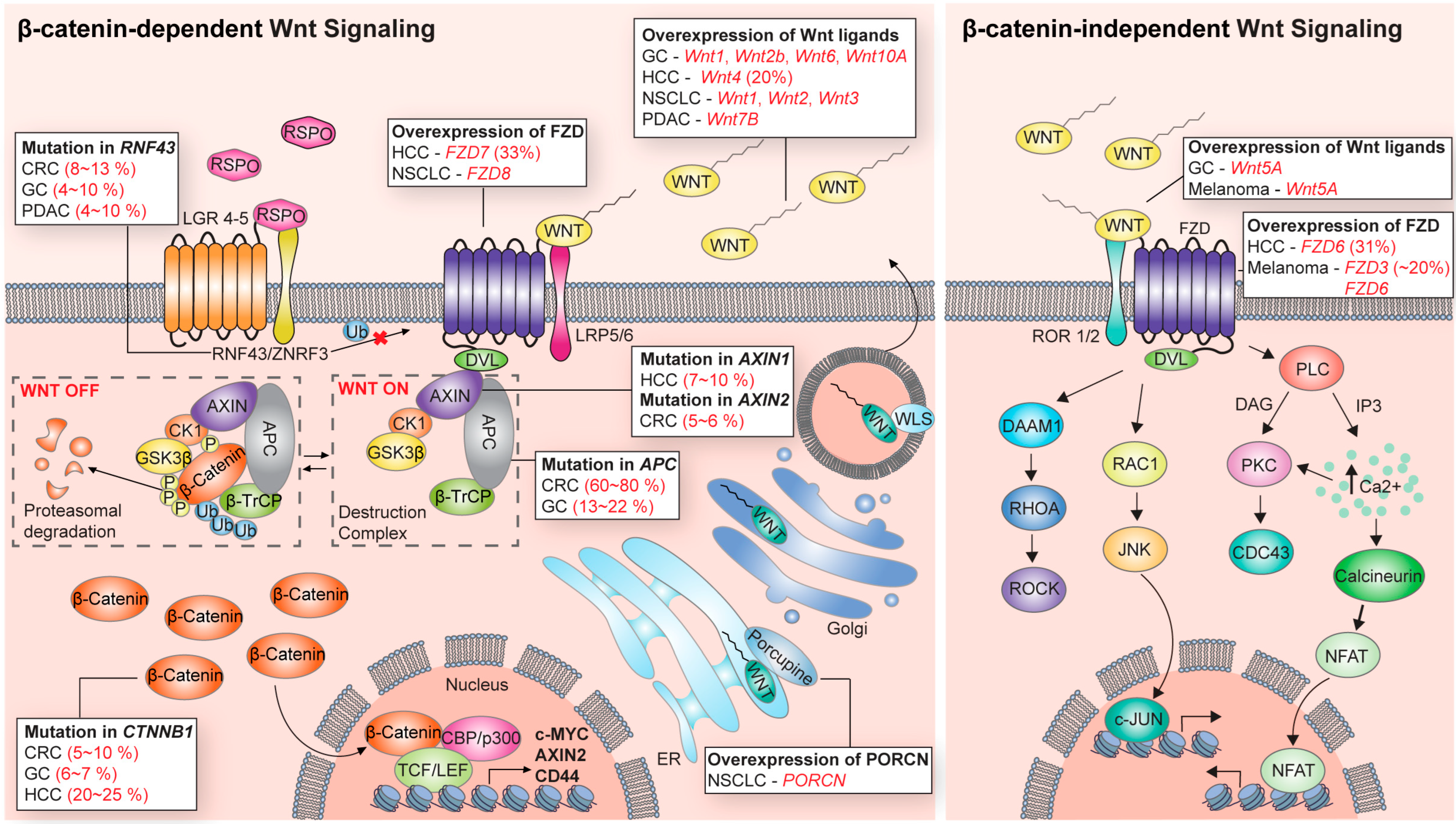{kind=link}
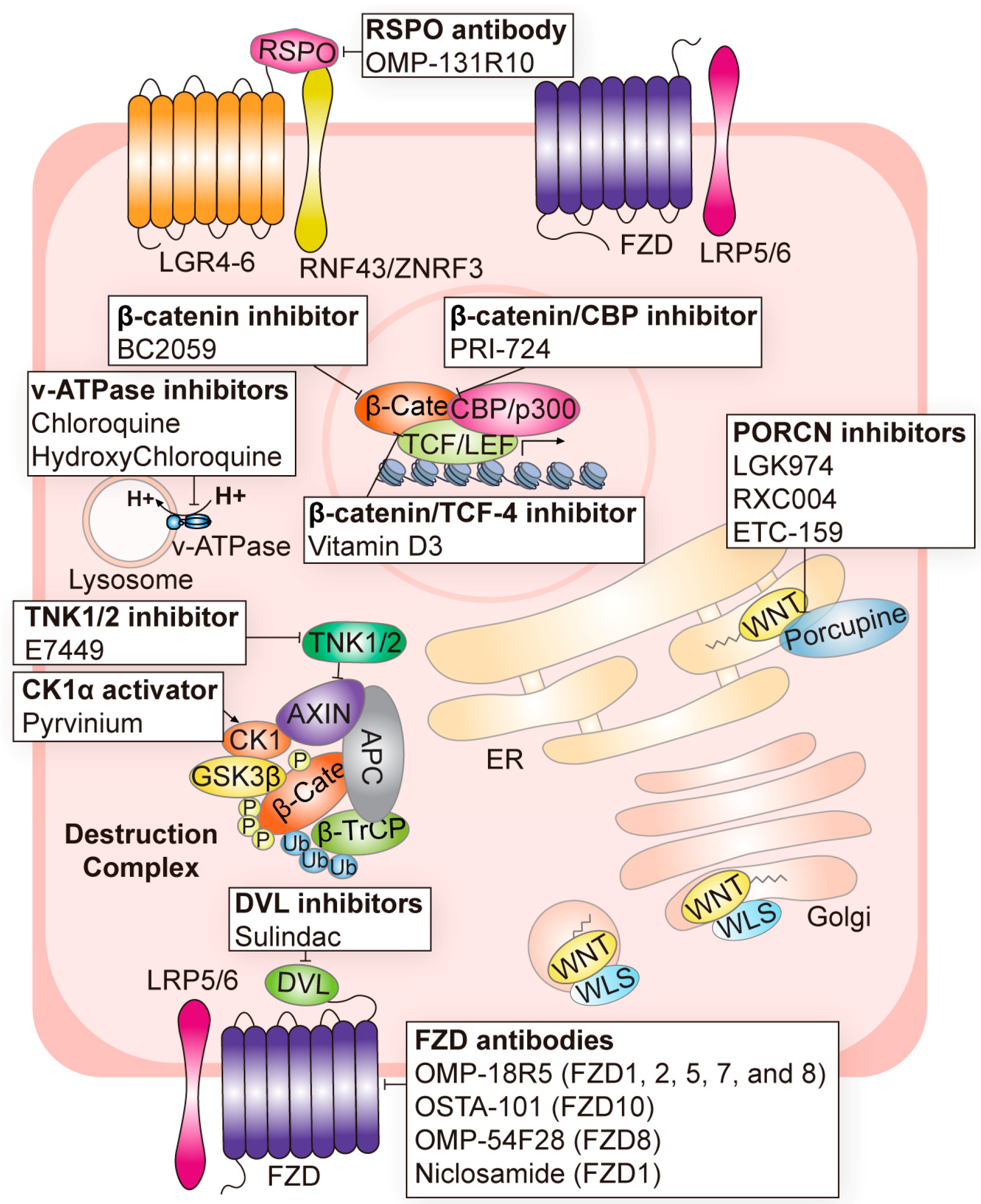{kind=link}
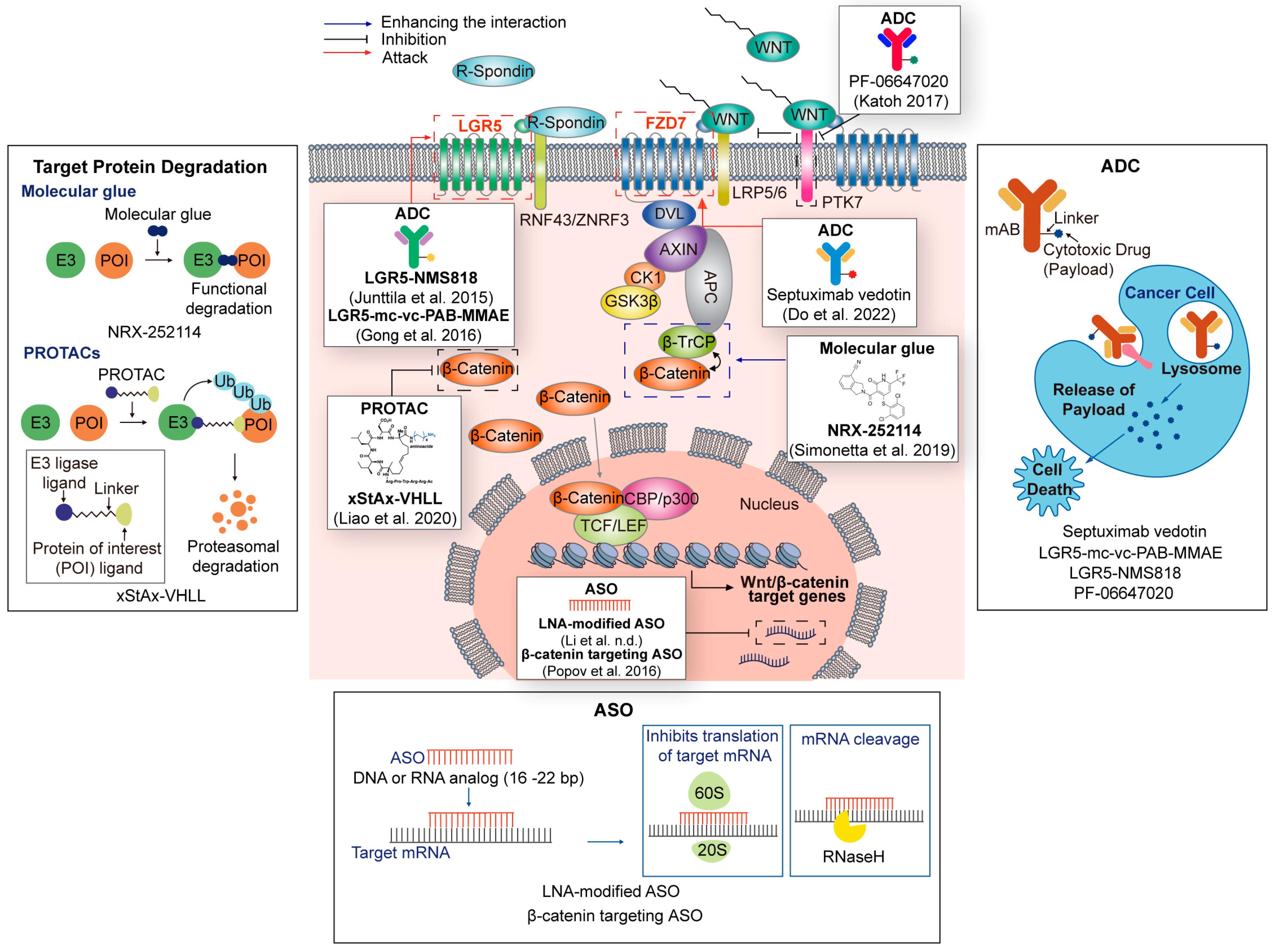{kind=link}
Table 1.
Wnt signaling targeting agents in clinical trials for cancer treatment.
| Components Name | Target | Cancer | Clinical Phase | Inhibition of Canonical or Non-Canonical Wnt Signaling | Refs. |
|---|---|---|---|---|---|
| OMP-131R10 | RSPO |
| Phase 1 (NCT02482441) | Canonical | [52] |
| Foxy-5 | WNT5A mimic |
| Phase 2 (NCT03883802) Phase 1 (NCT02020291) Phase 1 (NCT02655952) | Non-canonical | [53] |
| LGK974 | PORCN |
| Phase 1/2 (NCT02278133) Phase 2 (NCT02649530) Phase 1 (NCT01351103) | Canonical/Non-canonical | [3,43,54] |
| RXC004 | PORCN |
| Phase 2 (NCT04907539) Phase 1 (NCT03447470) Phase 2 (NCT04907851) | Canonical/Non-canonical | [55,56,57] |
| ETC-159 | PORCN |
| Phase 1 (NCT02521844) | Canonical/Non-canonical | [58,59] |
| OMP-54F28 | FZD8 |
| Phase 1 (NCT02069145) Phase 1 (NCT02092363) Phase 1 (NCT02050178) Phase 1 (NCT01608867) | Canonical | [60,61,62] |
| Niclosamide | FZD1 |
| Phase 1 (NCT02687009) Phase 1 (NCT03123978) Phase 1 (NCT02532114) Phase 2 (NCT02807805) Phase 1 (NCT05188170) FDA-approved antihelminth | Canonical | [63] |
| OMP-18R5 | FZD1/2/5/7/8 |
| Phase 1 (NCT01345201) Phase 1 (NCT01957007) Phase 1 (NCT02005315) Phase 1 (NCT01973309) | Canonical | [64,65] |
| OTSA-101 | FZD10 |
| Phase 1 (NCT01469975) | Canonical | [66] |
| BNC101 | LGR5 |
| Phase 1 (NCT02726334) | Canonical/Non-canonical | [67] |
| DKN-01 | DKK1 |
| Phase 1 (NCT01457417) Phase 1 (NCT01711671) Phase 1 (NCT02013154) Phase 1 (NCT02375880) Phase 2 (NCT03395080) | Canonical/Non-canonical | [68,69,70,71,72] |
| Sulindac | DVL |
| Phase 1 (NCT00245024) Phase 2 (NCT04542135) Phase 2 (NCT01856322) Phase 2 (NCT00062023) Phase 2 (NCT00368927) FDA-approved nonsteroidal anti-inflammatory drug | Canonical | [73] |
| Pyrvinium | CK1 |
| Phase 1 (NCT05055323) FDA-approved antihelminth | Canonical | [74] |
| E7449 | TNK1/2 |
| Phase 2 (NCT03878849) Phase 1 (NCT01618136) | Canonical | [75,76] |
| BC2059 | β-catenin |
| Phase 1 (NCT03459469) Phase 1 (NCT04780568) Phase 1 (NCT04874480) Phase 1/2 (NCT04851119) | Canonical | [77,78,79] |
| PRI-724 | β-catenin/CBP |
| Phase 1 (NCT01302405) Phase 1 (NCT01764477) Phase 1/2 (NCT01606579) | Canonical | [80,81] |
| SM08502 | CLK |
| Phase 1 (NCT05084859) Phase 1 (NCT03355066) | Canonical | [82] |
| Chloroquine | v-ATPase inhibitor |
| Phase 1 (NCT01777477) Phase 1 (NCT02071537) Phase 1/2 (NCT01023477) Phase 1/2 (NCT02496741) Phase 3 (NCT00224978) | Canonical | [83] |
| Hydroxy chloroquine | v-ATPase inhibitor |
| Phase 2 (NCT01006369), etc. (total of 30 trials completed) | Canonical | [84,85,86,87,88] |
Table 2.
Pre-clinical agents targeting Wnt signaling.
| Target | Pre-Clinical Agents | Refs. |
|---|---|---|
| PORCN | IWP1, IWP2, IWP3, IWP4, IWP12, IWP L6, C59, GNF-6231, GNF-1331 | [31,89,90,91,92] |
| FZD1 | DK-520, DK-419 | [93,94] |
| FZD5 | IgG-2919 | [51] |
| FZD7 | Fz7-21, RHPD-P1, SRI37892 | [95,96,97] |
| FZD8 | 1094-0205, 2124-0331, 3235-0367, NSC36784, NSC654259, IgG-2919 | [98] |
| WNT/FZD/LRP complex | Salinomycin | [32] |
| DVL | BMD4702, 3289-8625, J01-017a, FJ9, KY-02061, KY-02327, NSC668036, Peptide Pen-N3 | [99,100,101,102,103,104,105] |
| CK1 | SSTC3, CCT031374 | [106,107] |
| GSK3β mimic | TCS 183 | [108] |
| TNKS | XAV939, AZ1366, G007-LK, MSC2504877, G244-LM, IWR-1, JW74, JW55, K-756, NVP-TNKS656, MN-64, RK-287107, WIKI4 | [109,110,111,112,113,114,115,116,117,118,119,120] |
| β-catenin | KY1220, KYA1797K, MSAB, | [78,121] |
| β-catenin/TCF | PKF115-584, CGP049090, AV-65, PNU-74654 | [122,123] |
| β-catenin/EP300 | Windorphen, IQ-1 | [124,125] |
| β-catenin/BCL9 | PNPB-29, ZW4864, SAH-BCL9, Carnosic acid | [126,127,128] |
Table 3.
Wnt signaling targeting agents using the latest technologies (PROTACs, ADC, and ASO).
| Technologies | Name | Target | Refs. |
|---|---|---|---|
| PROTACs | xStAx-VHL | β-catenin | [153] |
| Molecular glue | NRX-252114 | β-catenin/ β-TrCP | [156] |
| ADC | Septuximab vedotin | FZD7 | [157] |
| PF-06647020 | PTK7 | [158] | |
| LGR5-mc-vc-PAB-MMAE | LGR5 | [159] | |
| LGR5-NMS818 | LGR5 | ||
| ASO | LNA-modified ASO | AC104041.1 (lncRNA) | [160] |
| β-catenin targeting ASO | β-catenin | [161] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Park, W.-J.; Kim, M.J. A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities. Cells 2023, 12, 1110. https://doi.org/10.3390/cells12081110
AMA Style
Park W-J, Kim MJ. A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities. Cells. 2023; 12(8):1110. https://doi.org/10.3390/cells12081110
Chicago/Turabian StylePark, Woo-Jung, and Moon Jong Kim. 2023. "A New Wave of Targeting ‘Undruggable’ Wnt Signaling for Cancer Therapy: Challenges and Opportunities" Cells 12, no. 8: 1110. https://doi.org/10.3390/cells12081110
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.