
Early Alterations of RNA Binding Protein (RBP) Homeostasis and ER Stress-Mediated Autophagy Contributes to Progressive Retinal Degeneration in the rd10 Mouse Model of Retinitis Pigmentosa (RP)
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. rd10 Mouse Model
2.3. Preparation of Retinal Sections and Homogenates
2.4. Diaminobenzidine (DAB) Immunohistochemistry
2.5. Immunofluorescence
2.6. Image Acquisition and Semi-Quantitative Analysis
2.7. Western Blot Analysis
2.8. Subcellular Fractionation
2.9. Transmission Electron Microscopy (TEM)
2.10. Statistics
3. Results
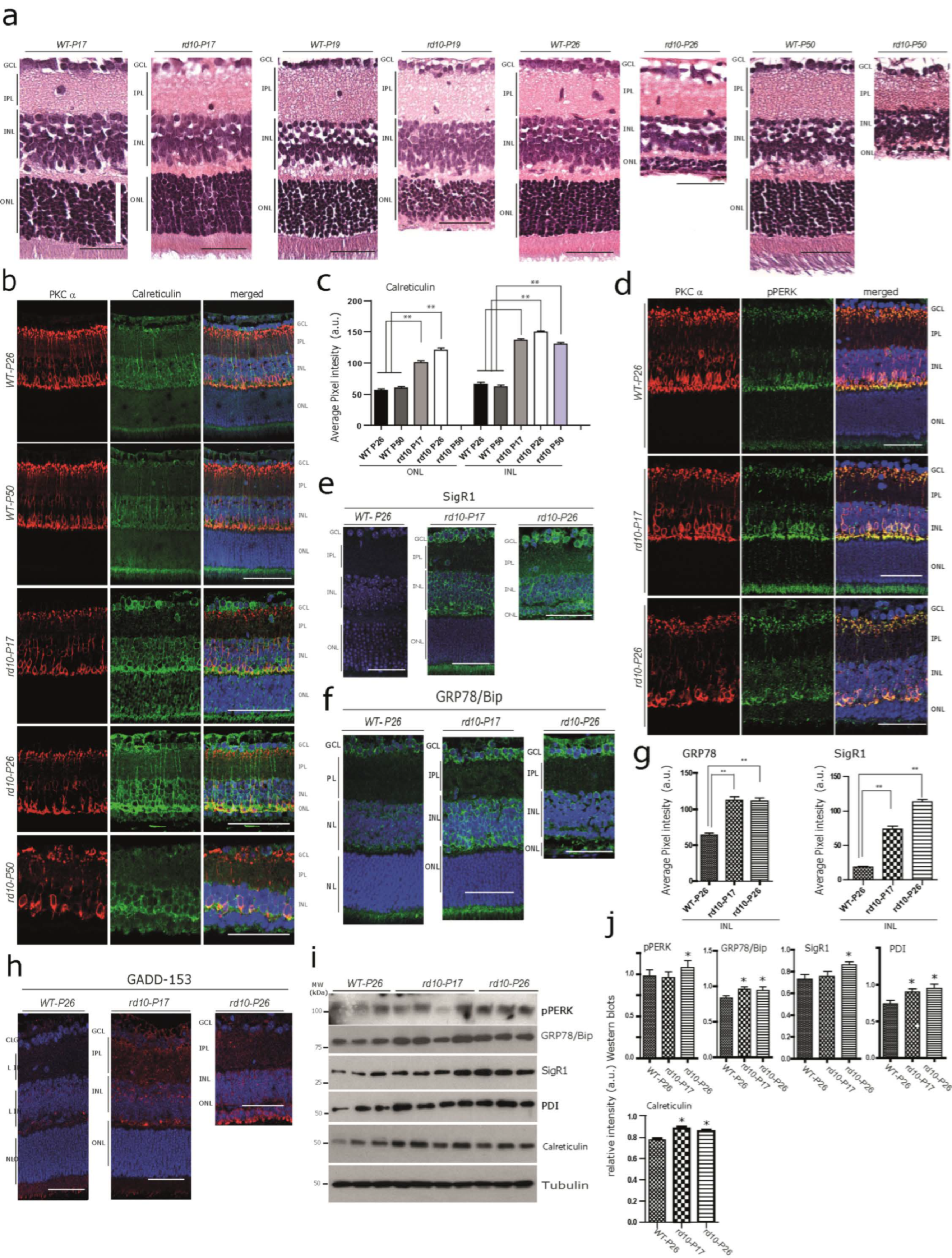
3.1. Increased Immunoreactivity and Protein Expression Levels of Calcium-Binding/Buffering Chaperones and ER Stress Markers in the rd10 Retina before any Noticeable Photoreceptor Loss at P17
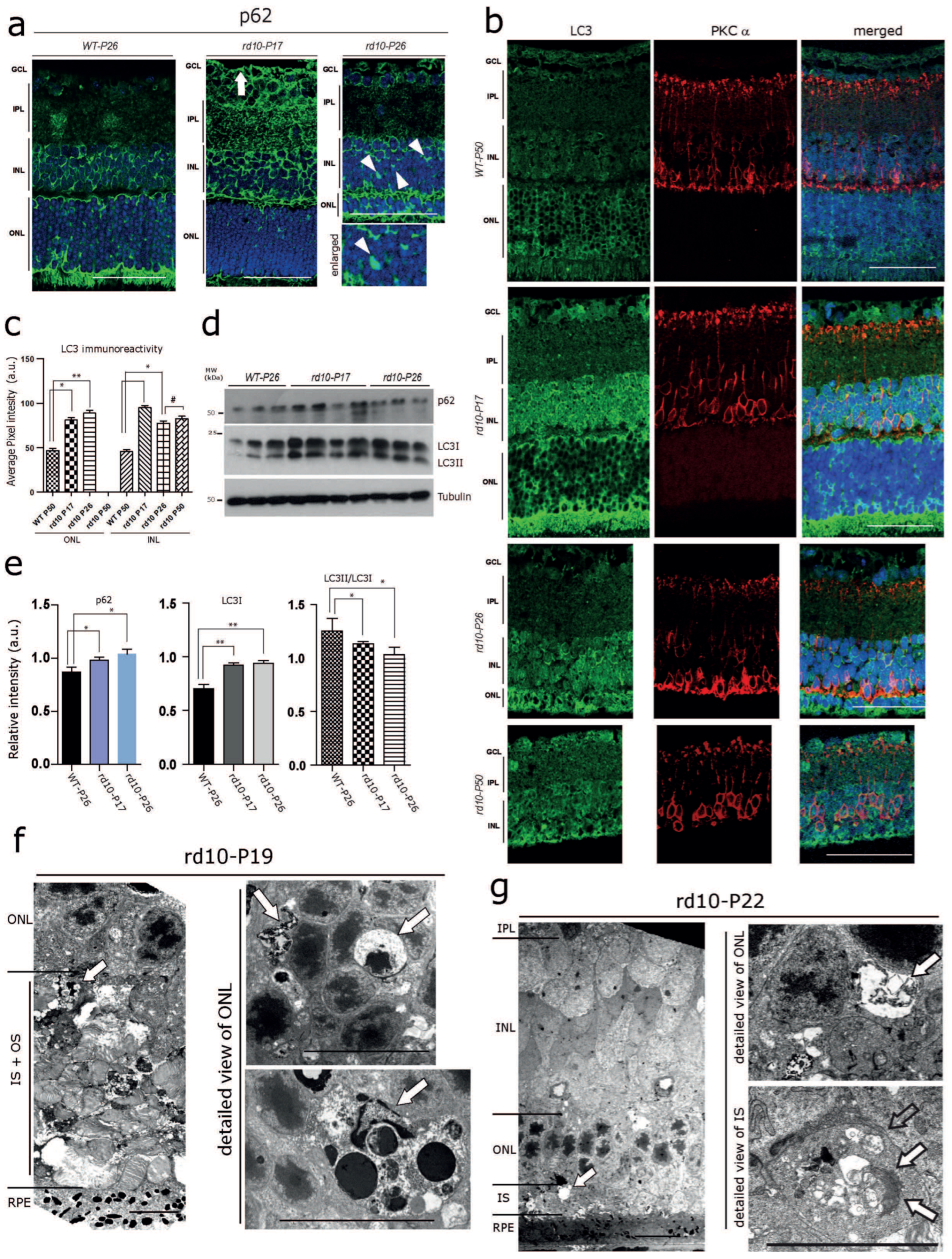
3.2. Early Autophagy Alterations in the rd10 Retina
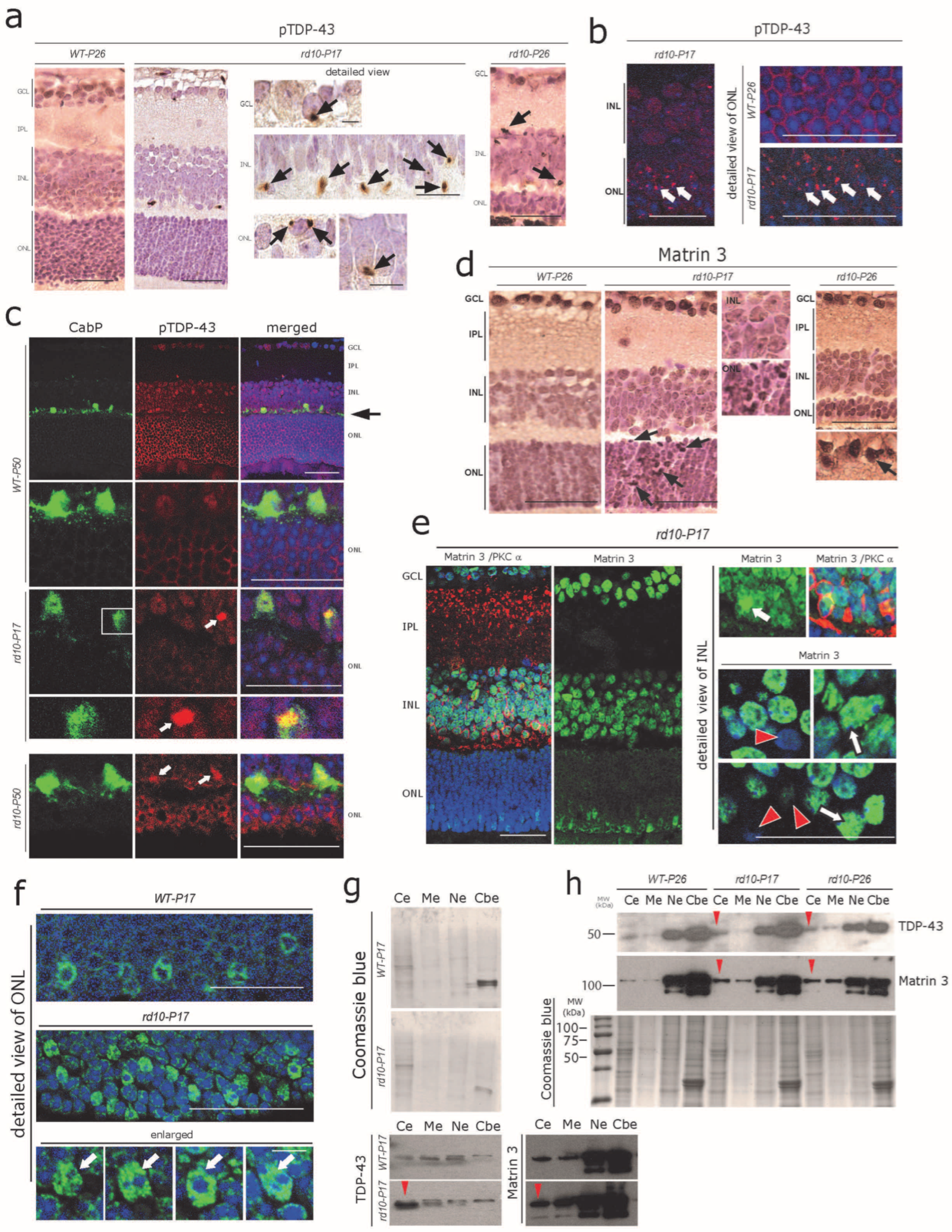
3.3. Abnormal Cytoplasmic Aggregation of pTDP-43 and Matrin 3 at Early Stages of rd10 Retina Degeneration
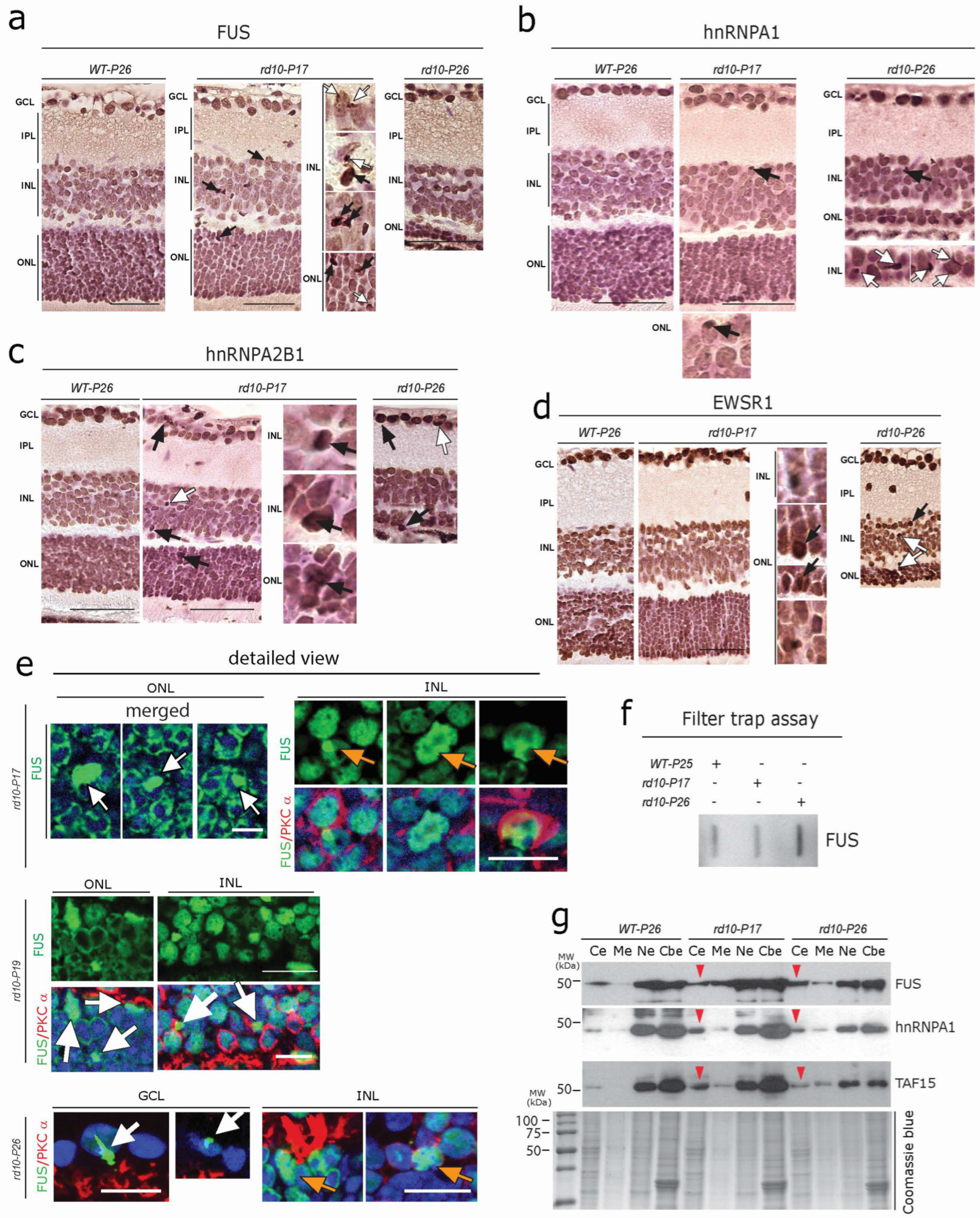
3.4. Aggregation of FET Family Proteins (FUS, EWRS1, and TAF15) in rd10 Retina
3.5. Formation of RNA Stress Granules (SGs) in rd10 Retina
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Sahel, J.A.; Boulanger-Scemama, E.; Pagot, C.; Arleo, A.; Galluppi, F.; Martel, J.N.; Esposti, S.D.; Delaux, A.; de Saint Aubert, J.B.; de Montleau, C.; et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 2021, 27, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Kennan, A.; Aherne, A.; Humphries, P. Light in retinitis pigmentosa. Trends. Genet. 2005, 21, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Pacione, L.R.; Szego, M.J.; Ikeda, S.; Nishina, P.M.; McInnes, R.R. Progress toward understanding the genetic and biochemical mechanisms of inherited photoreceptor degenerations. Annu. Rev. Neurosci. 2003, 26, 657–700. [Google Scholar] [CrossRef] [Green Version]
- Rattner, A.; Sun, H.; Nathans, J. Molecular genetics of human retinal disease. Annu. Rev. Genet. 1999, 33, 89–131. [Google Scholar] [CrossRef]
- McLaughlin, M.E.; Ehrhart, T.L.; Berson, E.L.; Dryja, T.P. Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 1995, 92, 3249–3253. [Google Scholar] [CrossRef] [Green Version]
- Lolley, R.N.; Farber, D.B.; Rayborn, M.E.; Hollyfield, J.G. Cyclic GMP accumulation causes degeneration of photoreceptor cells: Simulation of an inherited disease. Science 1977, 196, 664–666. [Google Scholar] [CrossRef]
- Nishiwaki, Y.; Komori, A.; Sagara, H.; Suzuki, E.; Manabe, T.; Hosoya, T.; Nojima, Y.; Wada, H.; Tanaka, H.; Okamoto, H.; et al. Mutation of cGMP phosphodiesterase 6alpha’-subunit gene causes progressive degeneration of cone photoreceptors in zebrafish. Mech. Dev. 2008, 125, 932–946. [Google Scholar] [CrossRef]
- Hong, D.H.; Pawlyk, B.S.; Shang, J.; Sandberg, M.A.; Berson, E.L.; Li, T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3). Proc. Natl. Acad. Sci. USA 2000, 97, 3649–3654. [Google Scholar] [CrossRef]
- Naash, M.I.; Hollyfield, J.G.; al-Ubaidi, M.R.; Baehr, W. Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc. Natl. Acad. Sci. USA 1993, 90, 5499–5503. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.; Hawes, N.L.; Pardue, M.T.; German, A.M.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Rengarajan, K.; Boyd, A.P.; Sidney, S.S.; et al. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. 2007, 47, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vision Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, S.L.; Wyse-Jackson, A.C.; Byrne, A.M.; Ruiz-Lopez, A.M.; Cotter, T.G. Alterations to retinal architecture prior to photoreceptor loss in a mouse model of retinitis pigmentosa. Int. J. Dev. Biol. 2016, 60, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Gargini, C.; Terzibasi, E.; Mazzoni, F.; Strettoi, E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: A morphological and ERG study. J. Comp. Neurol. 2007, 500, 222–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Reingruber, J.; Woodruff, M.L.; Majumder, A.; Camarena, A.; Artemyev, N.O.; Fain, G.L.; Chen, J. The PDE6 mutation in the rd10 retinal degeneration mouse model causes protein mislocalization and instability and promotes cell death through increased ion influx. J. Biol. Chem. 2018, 293, 15332–15346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comitato, A.; Sanges, D.; Rossi, A.; Humphries, M.M.; Marigo, V. Activation of Bax in three models of retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3555–3562. [Google Scholar] [CrossRef] [Green Version]
- Arango-Gonzalez, B.; Trifunovic, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef]
- Rodriguez-Muela, N.; Hernandez-Pinto, A.M.; Serrano-Puebla, A.; Garcia-Ledo, L.; Latorre, S.H.; de la Rosa, E.J.; Boya, P. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ. 2015, 22, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, C.; Liu, Y.; You, J.; Su, G. Autophagy, lysosome dysfunction and mTOR inhibition in MNU-induced photoreceptor cell damage. Tissue Cell 2019, 61, 98–108. [Google Scholar] [CrossRef]
- De la Rosa, E.J.; Cotter, T.G. The Cellular Course of Retinal Degenerative Conditions : Therapies for Retinal Degeneration: Targeting Common Processes. R. Soc. Chem. 2018, 66, 1–30. [Google Scholar]
- Narayan, D.S.; Wood, J.P.; Chidlow, G.; Casson, R.J. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016, 94, 748–754. [Google Scholar] [CrossRef]
- Wang, A.L.; Knight, D.K.; Vu, T.T.; Mehta, M.C. Retinitis Pigmentosa: Review of Current Treatment. Int. Ophthalmol. Clin. 2019, 59, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; LaFerla, F.M.; Chan, S.L.; Leissring, M.A.; Shepel, P.N.; Geiger, J.D. Calcium signaling in the ER: Its role in neuronal plasticity and neurodegenerative disorders. Trends. Neurosci. 2000, 23, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium. 2018, 70, 87–94. [Google Scholar] [CrossRef]
- Jesse, C.M.; Bushuven, E.; Tripathi, P.; Chandrasekar, A.; Simon, C.M.; Drepper, C.; Yamoah, A.; Dreser, A.; Katona, I.; Johann, S.; et al. ALS-Associated Endoplasmic Reticulum Proteins in Denervated Skeletal Muscle: Implications for Motor Neuron Disease Pathology. Brain. Pathl. 2017, 27, 781–794. [Google Scholar] [CrossRef]
- Prause, J.; Goswami, A.; Katona, I.; Roos, A.; Schnizler, M.; Bushuven, E.; Dreier, A.; Buchkremer, S.; Johann, S.; Beyer, C.; et al. Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 1581–1600. [Google Scholar] [CrossRef]
- Yamoah, A.; Tripathi, P.; Sechi, A.; Kohler, C.; Guo, H.; Chandrasekar, A.; Nolte, K.W.; Wruck, C.J.; Katona, I.; Anink, J.; et al. Aggregates of RNA Binding Proteins and ER Chaperones Linked to Exosomes in Granulovacuolar Degeneration of the Alzheimer’s Disease Brain. J. Alzheimers Dis. 2020, 75, 139–156. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Haselier, C.; Mataruga, A.; Thumann, G.; Walter, P.; Muller, F. Pharmacological analysis of intrinsic neuronal oscillations in rd10 retina. PLoS ONE 2014, 9, e99075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, P.; Guo, H.; Dreser, A.; Yamoah, A.; Sechi, A.; Jesse, C.M.; Katona, I.; Doukas, P.; Nikolin, S.; Ernst, S.; et al. Pathomechanisms of ALS8: Altered autophagy and defective RNA binding protein (RBP) homeostasis due to the VAPB P56S mutation. Cell Death. Dis. 2021, 12, 466. [Google Scholar] [CrossRef] [PubMed]
- Dreser, A.; Vollrath, J.T.; Sechi, A.; Johann, S.; Roos, A.; Yamoah, A.; Katona, I.; Bohlega, S.; Wiemuth, D.; Tian, Y.; et al. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017, 24, 1655–1671. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Otteson, D.C.; Sherry, D.M. Progression of neuronal and synaptic remodeling in the rd10 mouse model of retinitis pigmentosa. J. Comp. Neurol. 2010, 518, 2071–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puthussery, T.; Taylor, W.R. Functional changes in inner retinal neurons in animal models of photoreceptor degeneration. Adv Exp. Med. Biol. 2010, 664, 525–532. [Google Scholar] [CrossRef]
- Engedal, N.; Torgersen, M.L.; Guldvik, I.J.; Barfeld, S.J.; Bakula, D.; Saetre, F.; Hagen, L.K.; Patterson, J.B.; Proikas-Cezanne, T.; Seglen, P.O.; et al. Modulation of intracellular calcium homeostasis blocks autophagosome formation. Autophagy 2013, 9, 1475–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotemeier, A.; Alers, S.; Pfisterer, S.G.; Paasch, F.; Daubrawa, M.; Dieterle, A.; Viollet, B.; Wesselborg, S.; Proikas-Cezanne, T.; Stork, B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal. 2010, 22, 914–925. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Cuervo, A.M.; Seglen, P.O. Methods for monitoring autophagy from yeast to human. Autophagy 2007, 3, 181–206. [Google Scholar] [CrossRef] [Green Version]
- Aggad, D.; Veriepe, J.; Tauffenberger, A.; Parker, J.A. TDP-43 toxicity proceeds via calcium dysregulation and necrosis in aging Caenorhabditis elegans motor neurons. J. Neurosci. 2014, 34, 12093–12103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Fan, H.; Ying, Z.; Li, B.; Wang, H.; Wang, G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci. Lett. 2010, 469, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.H.; Jun, M.H.; Min, K.J.; Jang, D.J.; Lee, Y.S.; Kim, H.K.; Lee, J.A. Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol. Aging 2014, 35, 2822–2831. [Google Scholar] [CrossRef]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef]
- Ito, D.; Hatano, M.; Suzuki, N. RNA binding proteins and the pathological cascade in ALS/FTD neurodegeneration. Sci. Transl. Med. 2017, 9, 5436. [Google Scholar] [CrossRef] [Green Version]
- Salton, M.; Elkon, R.; Borodina, T.; Davydov, A.; Yaspo, M.L.; Halperin, E.; Shiloh, Y. Matrin 3 binds and stabilizes mRNA. PLoS ONE 2011, 6, e23882. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [Green Version]
- Ramaswami, M.; Taylor, J.P.; Parker, R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 2013, 154, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 40–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchan, J.R. mRNP granules. Assembly, function, and connections with disease. RNA Biol. 2014, 11, 1019–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, S.; Mateju, D.; Mediani, L.; Carra, S. Granulostasis: Protein Quality Control of RNP Granules. Front. Mol. Neurosci. 2017, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Wolozin, B. Regulated protein aggregation: Stress granules and neurodegeneration. Mol. Neurodegener. 2012, 7, 56. [Google Scholar] [CrossRef] [Green Version]
- Stasheff, S.F. Emergence of sustained spontaneous hyperactivity and temporary preservation of OFF responses in ganglion cells of the retinal degeneration (rd1) mouse. J. Neurophysiol. 2008, 99, 1408–1421. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, J.; Cui, X.; Mysona, B.A.; Navneet, S.; Saul, A.; Ahuja, M.; Lambert, N.; Gazaryan, I.G.; Thomas, B.; et al. The molecular chaperone sigma 1 receptor mediates rescue of retinal cone photoreceptor cells via modulation of NRF2. Free Radic. Biol. Med. 2019, 134, 604–616. [Google Scholar] [CrossRef]
- Wang, J.; Saul, A.; Roon, P.; Smith, S.B. Activation of the molecular chaperone, sigma 1 receptor, preserves cone function in a murine model of inherited retinal degeneration. Proc. Natl. Acad. Sci. USA 2016, 113, E3764–E3772. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Saul, A.; Cui, X.; Roon, P.; Smith, S.B. Absence of Sigma 1 Receptor Accelerates Photoreceptor Cell Death in a Murine Model of Retinitis Pigmentosa. InvestIG. Ophthalmol. Vis. Sci. 2017, 58, 4545–4558. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Fu, Y.; Liu, X.; Shahi, P.K.; Mavlyutov, T.A.; Li, J.; Yao, A.; Guo, S.Z.; Pattnaik, B.R.; Guo, L.W. Role of the sigma-1 receptor chaperone in rod and cone photoreceptor degenerations in a mouse model of retinitis pigmentosa. Mol. Neurodegener. 2017, 12, 68. [Google Scholar] [CrossRef]
- Ilieva, E.V.; Ayala, V.; Jove, M.; Dalfo, E.; Cacabelos, D.; Povedano, M.; Bellmunt, M.J.; Ferrer, I.; Pamplona, R.; Portero-Otin, M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain 2007, 130, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P. Amyotrophic lateral sclerosis. unfolding the toxicity of the misfolded. Cell 2001, 104, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Lohr, H.R.; Kuntchithapautham, K.; Sharma, A.K.; Rohrer, B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp. Eye Re.s 2006, 83, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Mammadova, N.; Baron, T.; Verchere, J.; Greenlee, J.J.; Greenlee, M.H.W. Retina as a Model to Study In Vivo Transmission of alpha-Synuclein in the A53T Mouse Model of Parkinson’s Disease. Methods Mol. Biol. 2021, 2224, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.L.; Marc, R.E.; Jones, B.W. Persistent remodeling and neurodegeneration in late-stage retinal degeneration. Prog. Retin. Eye Res. 2020, 74, 100771. [Google Scholar] [CrossRef]
- Goswami, A.; Jesse, C.M.; Chandrasekar, A.; Bushuven, E.; Vollrath, J.T.; Dreser, A.; Katona, I.; Beyer, C.; Johann, S.; Feller, A.C.; et al. Accumulation of STIM1 is asso-ciated with the degenerative muscle fibre phenotype in ALS and other neurogenic atrophies. Neuropathol. Appl. Neurobiol. 2015, 41, 304–318. [Google Scholar] [CrossRef]
- Vollrath, J.T.; Sechi, A.; Dreser, A.; Katona, I.; Wiemuth, D.; Vervoorts, J.; Dohmen, M.; Chandrasekar, A.; Prause, J.; Brauers, E.; et al. Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death. Dis. 2014, 12, 243. [Google Scholar] [CrossRef] [Green Version]
- Filezac de L’Etang, A.; Maharjan, N.; Cordeiro Brana, M.; Ruegsegger, C.; Rehmann, R.; Goswami, A.; Roos, A.; Troost, D.; Schneider, B.L.; Weis, J.; et al. Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nat. Neurosci. 2015, 18, 227–238. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamoah, A.; Tripathi, P.; Guo, H.; Scheve, L.; Walter, P.; Johnen, S.; Müller, F.; Weis, J.; Goswami, A. Early Alterations of RNA Binding Protein (RBP) Homeostasis and ER Stress-Mediated Autophagy Contributes to Progressive Retinal Degeneration in the rd10 Mouse Model of Retinitis Pigmentosa (RP). Cells 2023, 12, 1094. https://doi.org/10.3390/cells12071094
Yamoah A, Tripathi P, Guo H, Scheve L, Walter P, Johnen S, Müller F, Weis J, Goswami A. Early Alterations of RNA Binding Protein (RBP) Homeostasis and ER Stress-Mediated Autophagy Contributes to Progressive Retinal Degeneration in the rd10 Mouse Model of Retinitis Pigmentosa (RP). Cells. 2023; 12(7):1094. https://doi.org/10.3390/cells12071094
Chicago/Turabian StyleYamoah, Alfred, Priyanka Tripathi, Haihong Guo, Leonie Scheve, Peter Walter, Sandra Johnen, Frank Müller, Joachim Weis, and Anand Goswami. 2023. "Early Alterations of RNA Binding Protein (RBP) Homeostasis and ER Stress-Mediated Autophagy Contributes to Progressive Retinal Degeneration in the rd10 Mouse Model of Retinitis Pigmentosa (RP)" Cells 12, no. 7: 1094. https://doi.org/10.3390/cells12071094