

Palmitate-Induced Cardiac Lipotoxicity Is Relieved by the Redox-Active Motif of SELENOT through Improving Mitochondrial Function and Regulating Metabolic State
 , , , , , , , and
, , , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Peptides and Drugs
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Lactate Dehydrogenase (LDH) Assessment
2.5. Oil Red O Staining
2.6. Detection of Intracellular Reactive Oxygen Species (ROS) and Mitochondrial Superoxide Generation by MitoSOX
2.7. Immunofluorescence Analysis for CD36 Evaluation
2.8. Short Interfering RNA (siRNA) Transfection for SELENOT Silencing
2.9. Assessment of Mitochondrial Respiratory Function Using the Seahorse XF Analyzer
2.10. Western Blot
2.11. Attenuated Total Reflectance Fourier-Transform Infrared (ATR-FTIR) Spectroscopic Measurements
2.12. Transmission Electron Microscopy (TEM) Analysis
2.13. Statistical Analysis
3. Results
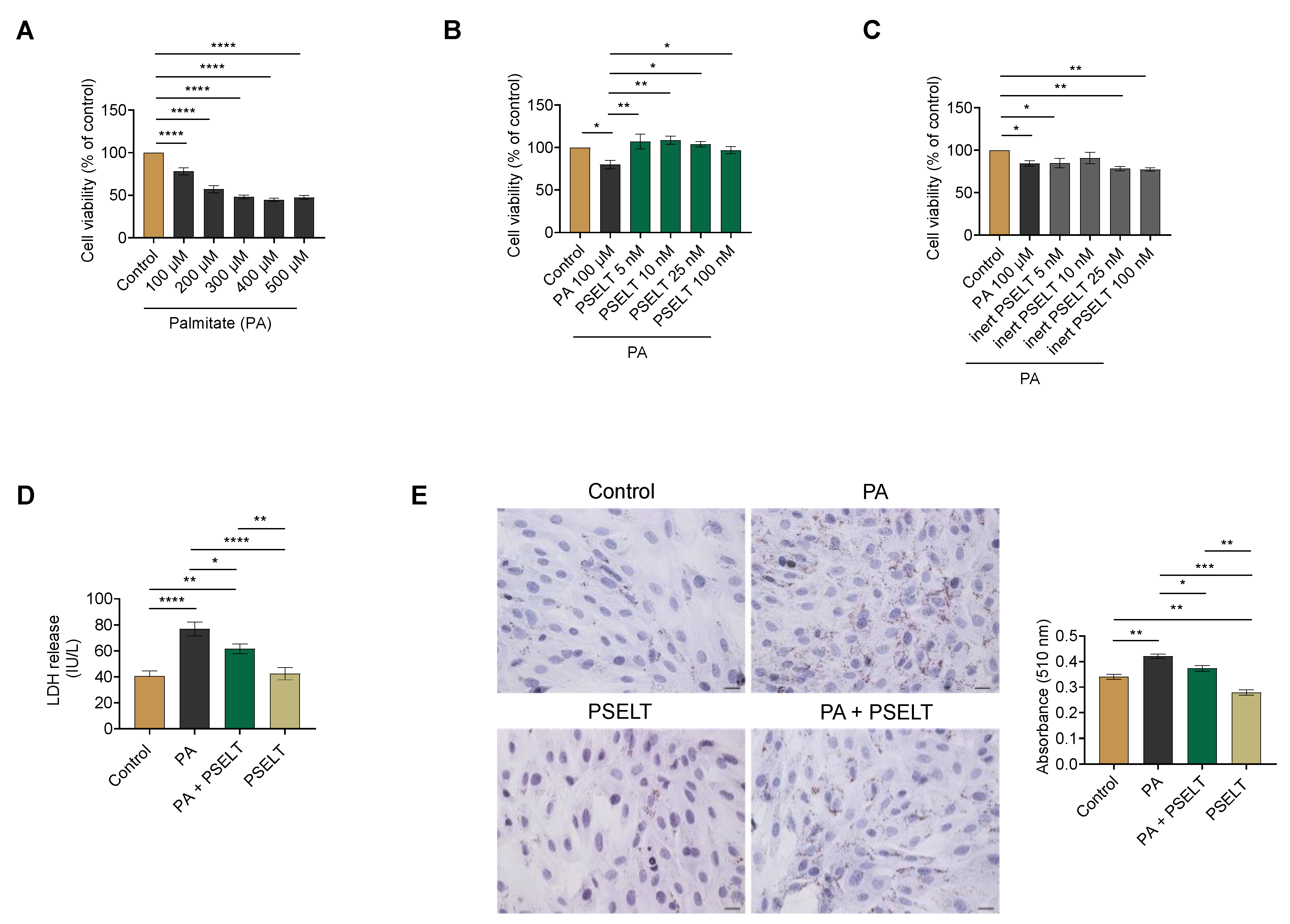
3.1. PSELT Mitigates PA-Induced Cytotoxicity and Lipid Accumulation in H9c2 Cardiomyocytes
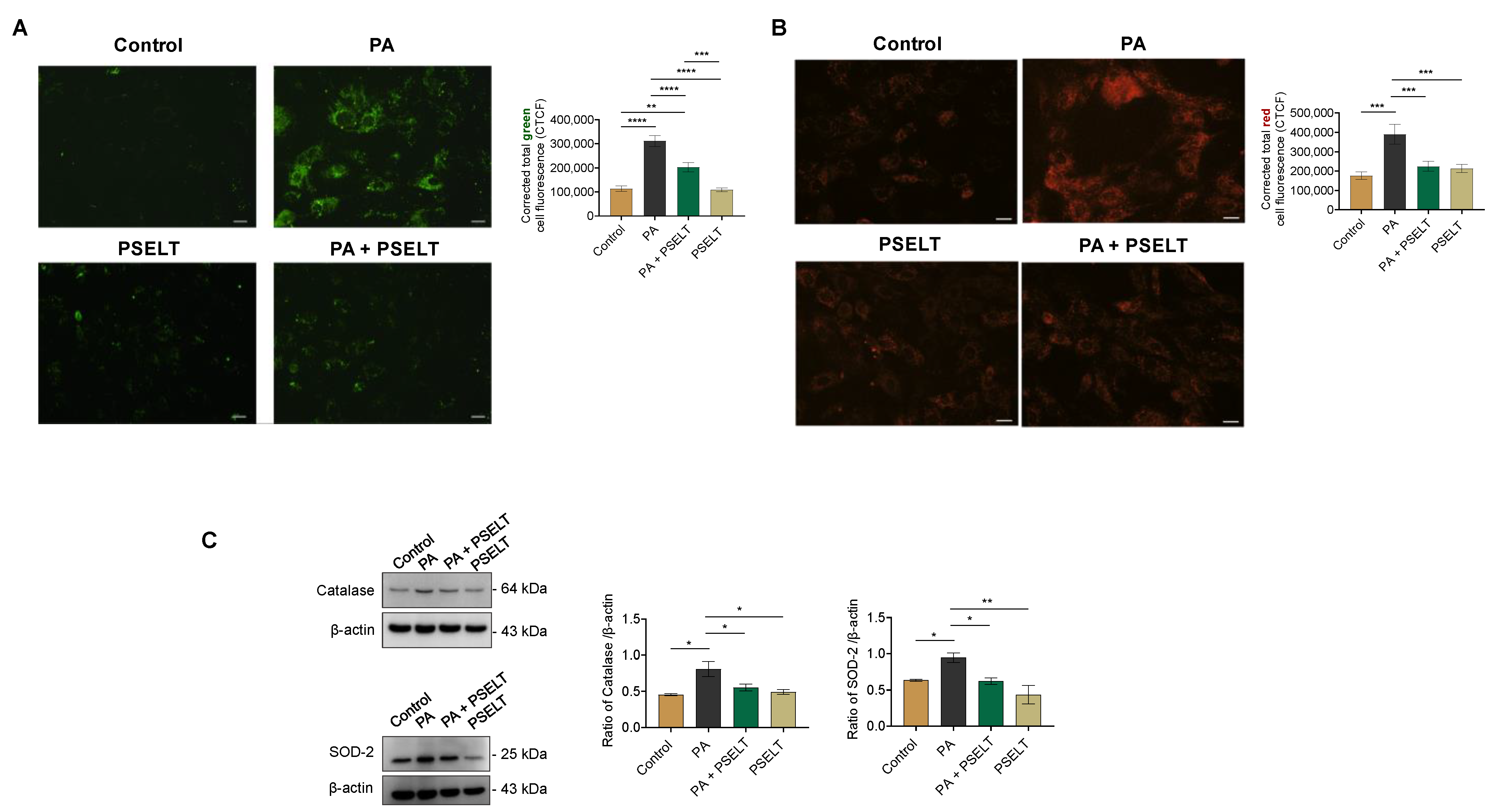
3.2. PSELT Protects H9c2 Cells against PA-Induced Oxidative Stress
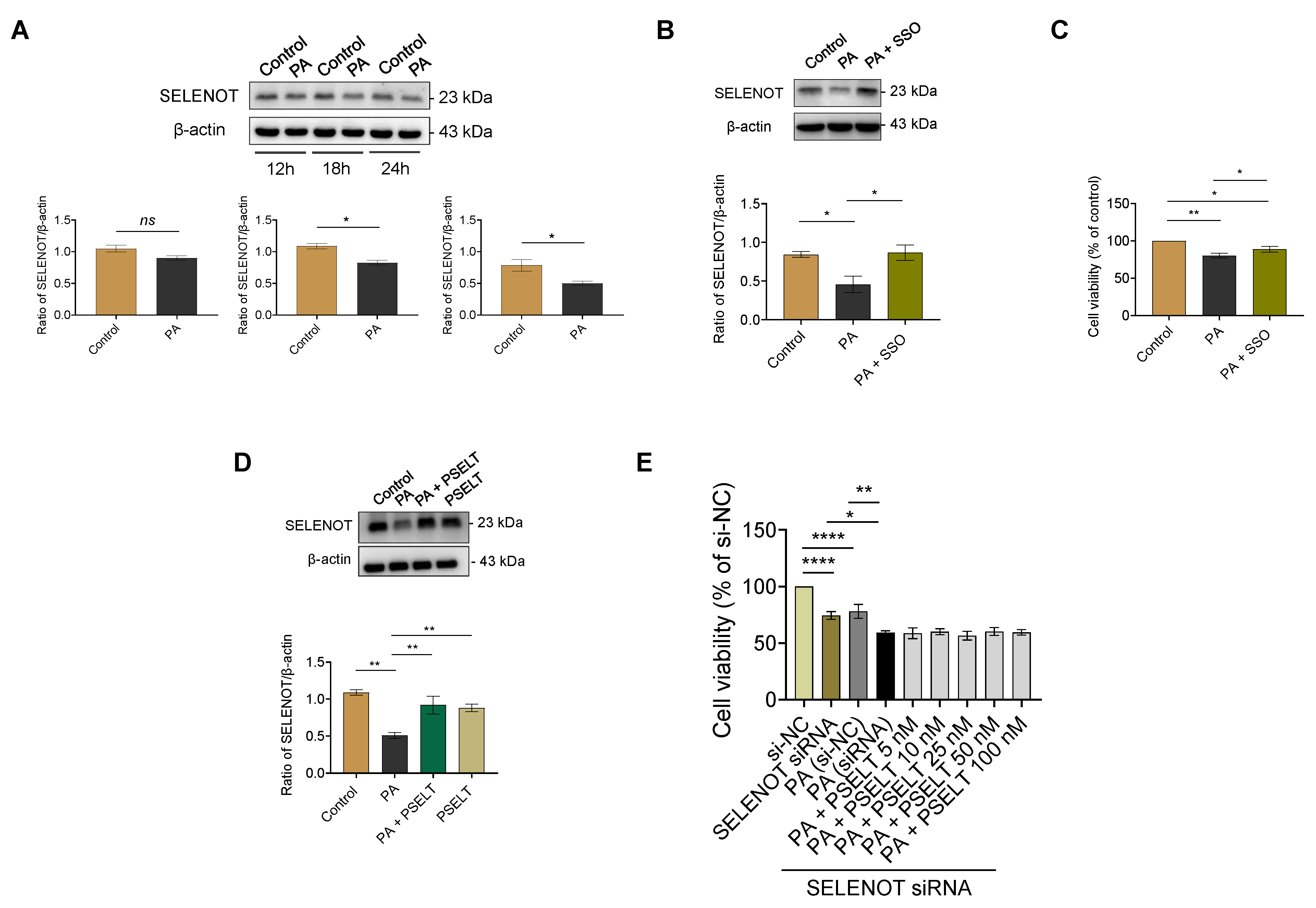
3.3. PSELT Rescues the PA-Induced Reduction of Endogenous SELENOT Expression in H9c2 Cells, and Endogenous SELENOT Is Fundamental for PSELT-Induced Cell Protection against the PA Effect
3.4. PSELT Reduces the PA-Dependent Upregulation of CD36 in H9c2 Cardiomyocytes
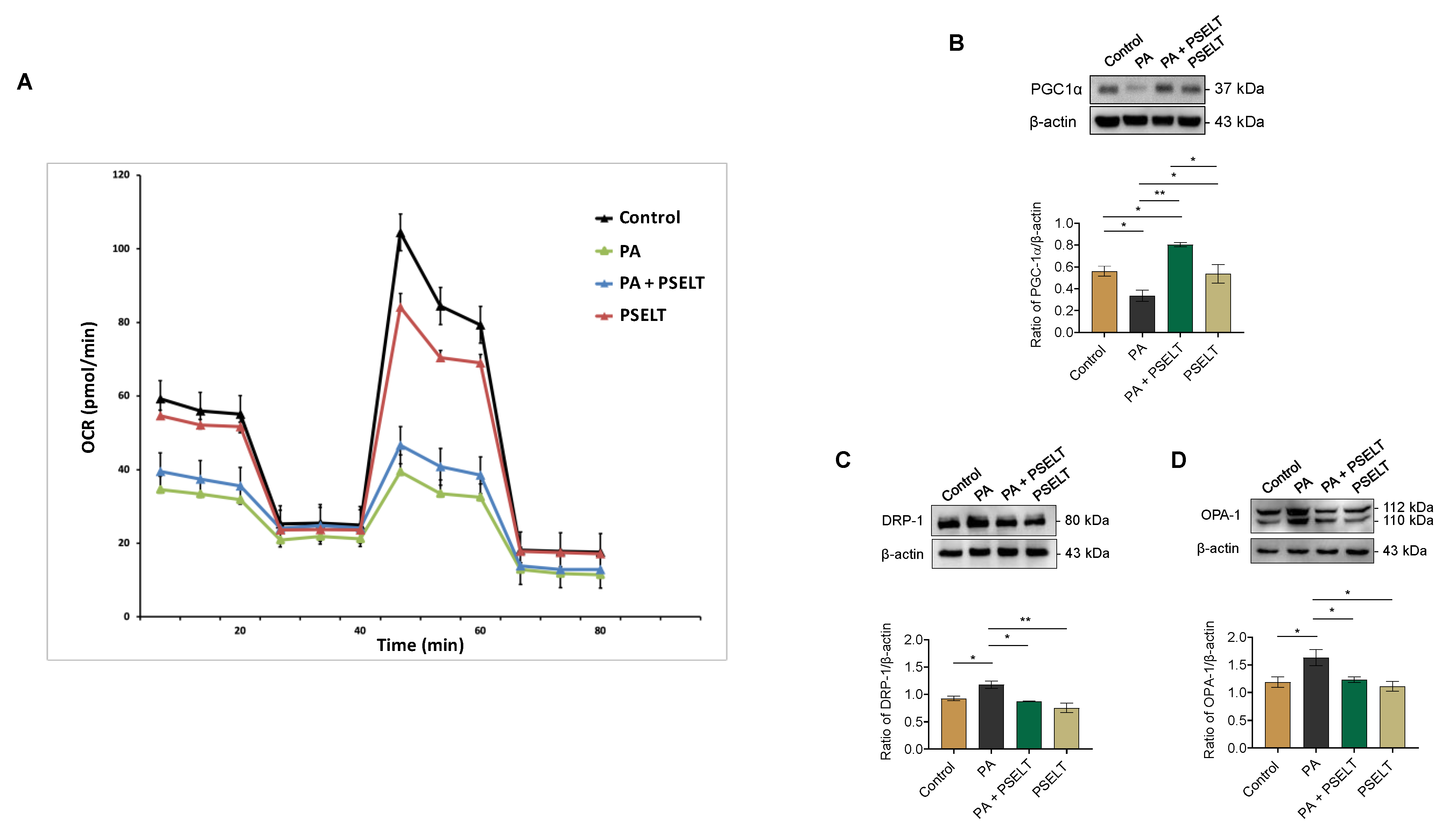
3.5. PSELT Mitigates the Detrimental Effects of PA on Mitochondrial Function, Biogenesis, and Dynamics
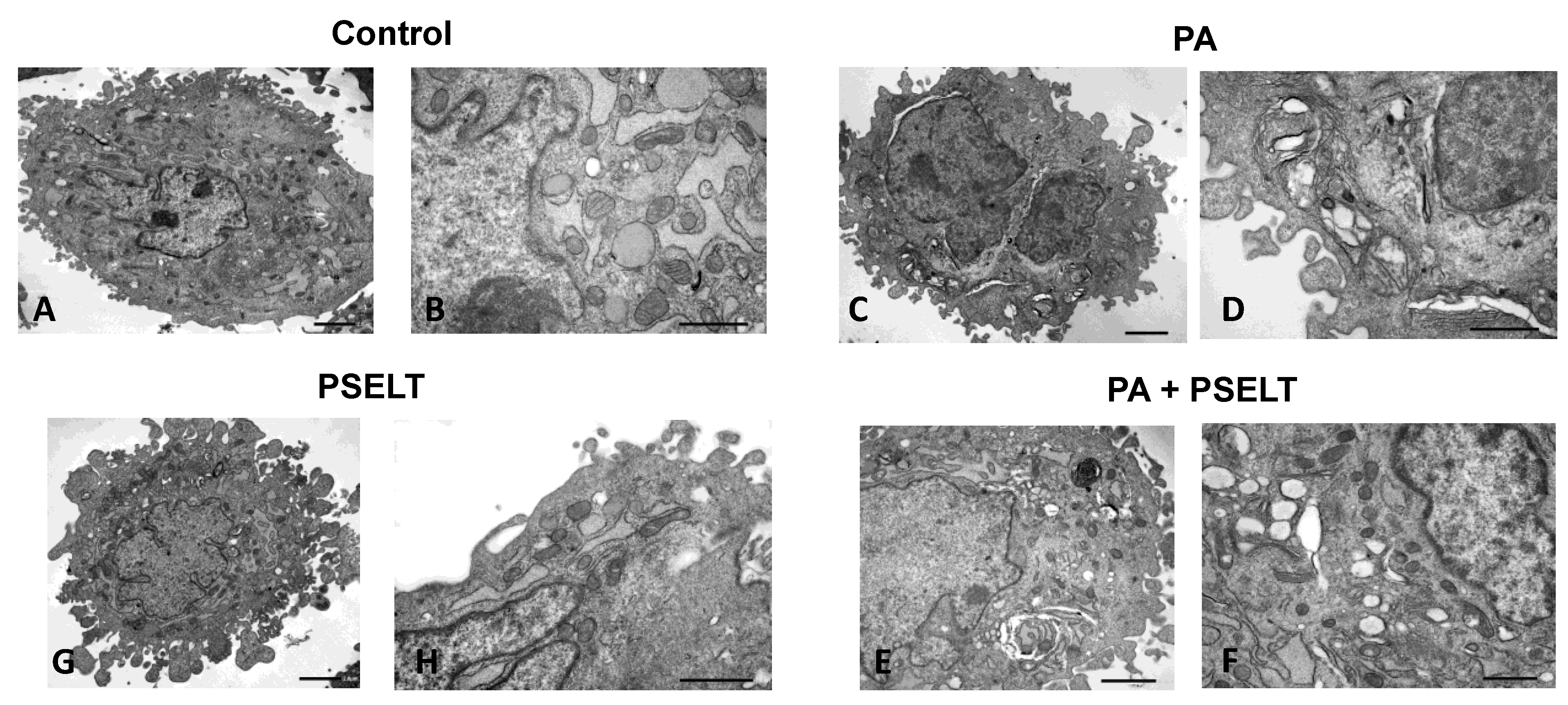
3.6. PSELT Mitigates PA-Dependent Ultrastructural Alterations in H9c2 Cardiomyocytes
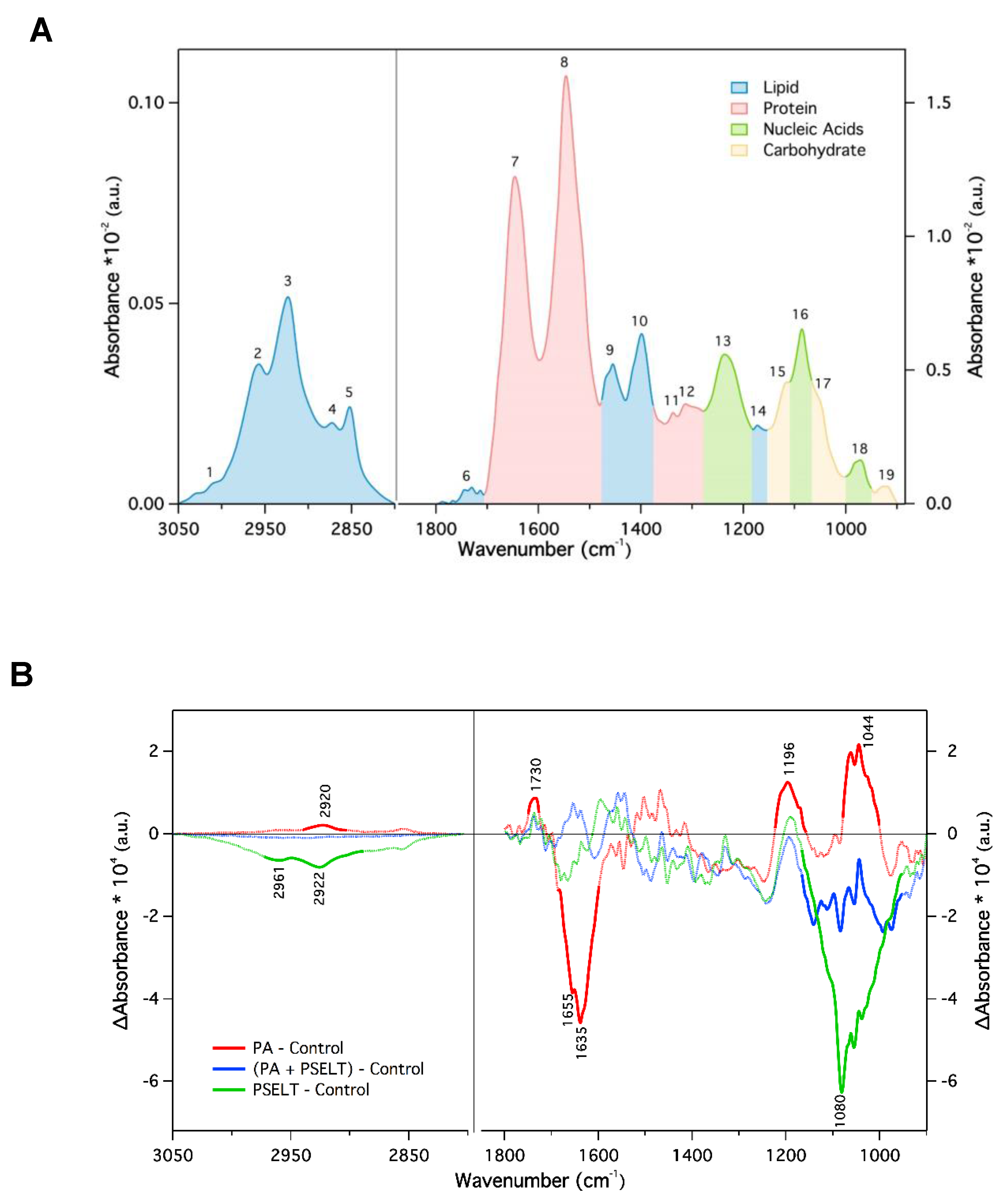
3.7. PSELT Mitigates Specific FTIR Spectral Alterations Induced by PA in H9c2 Cardiomyocytes
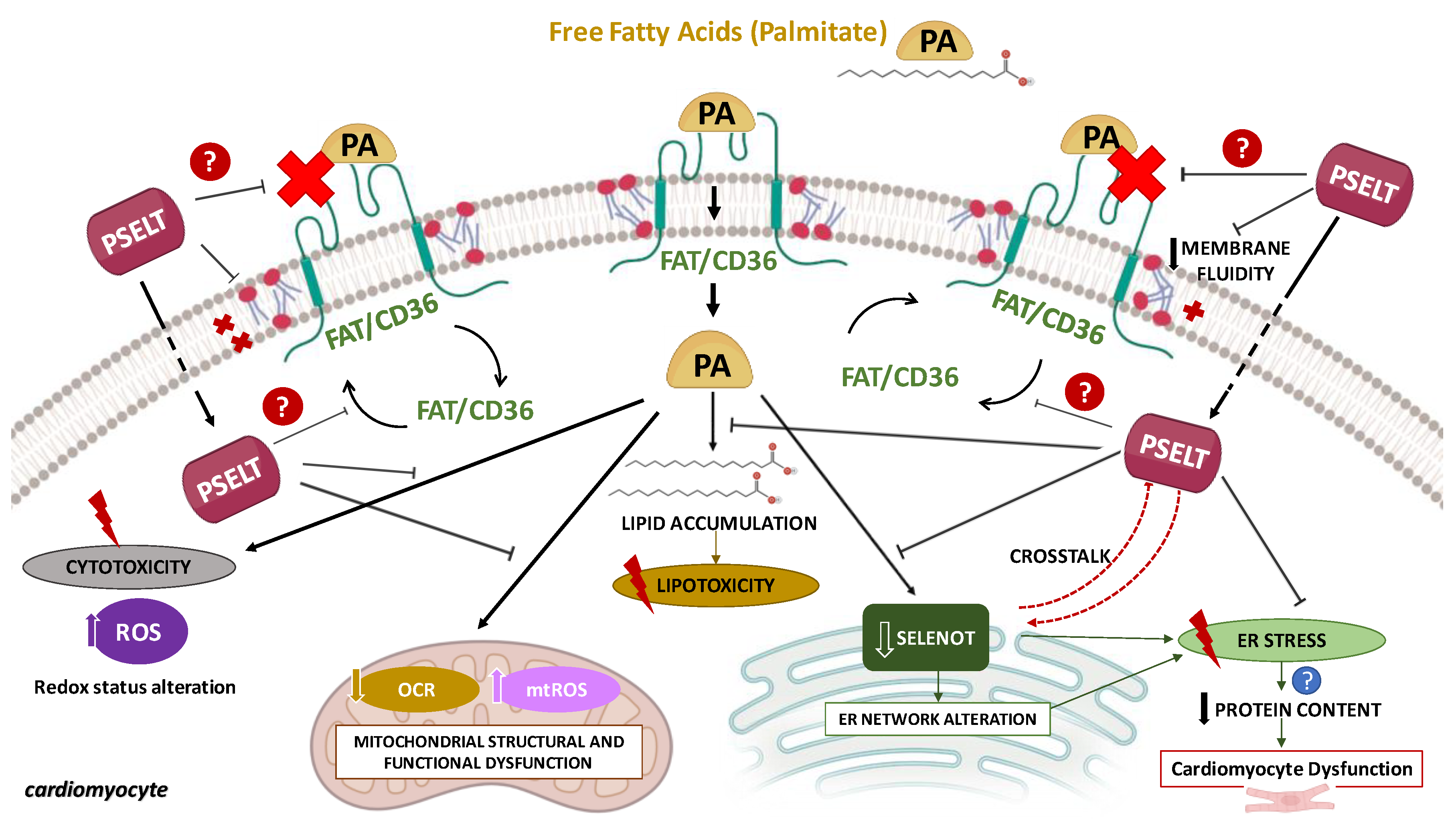
4. Discussion
4.1. PSELT Exerts Protective Effects against PA-Induced Cytotoxicity and Lipotoxicity through Its Redox Site Containing the Sec Residue
4.2. PSELT Counteracts PA-Induced Oxidative Stress and the Reduction of Endogenous SELENOT
4.3. PSELT Improves Mitochondrial Ultrastructure and Function in Terms of Respiration, Biogenesis, and Dynamics in PA-Treated Cardiomyocytes
4.4. PSELT Attenuates FTIR Spectral-Related Macromolecular Changes Induced by PA in H9c2 Cardiomyocytes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CAT | catalase |
| CM-H2DCFDA | (5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl ester) |
| CTCF | corrected total cell fluorescence |
| CVD | cardiovascular diseases |
| DRP-1 | dynamin-related protein 1 |
| ER | endoplasmic reticulum |
| FAs | fatty acids |
| FAT/CD36 | cluster of differentiation 36/fatty acid translocase |
| ATR-FTIR | Attenuated total Reflectance-Fourier-transform infrared spectroscopy |
| GSH | glutathione |
| GPX | glutathione peroxidase |
| HF | heart failure |
| I-PSELT | inert selenoprotein T-derived peptide 43–52 |
| LDH | lactate dehydrogenase |
| MI/R | myocardial ischemia/reperfusion |
| OCR | oxygen consumption rate |
| OPA-1 | optic Atrophy 1 |
| OST | oligosaccharyl transferase |
| PA | palmitate |
| PGC1-α | peroxisome proliferator-activated receptor-gamma coactivator1-α |
| PSELT | selenoprotein T-derived peptide 43–52 |
| ROS | reactive oxygen species |
| Sec, U | selenocysteine |
| SELENOT | selenoprotein T |
| siRNA | short interfering RNA |
| SOD | superoxide dismutase |
| SSO | sulfo-N-succinimidyl oleate |
| UPR | unfolded protein response |
References
- Powell-Wiley, T.M.; Poirier, P.; Burke, L.E.; Després, J.P.; Gordon-Larsen, P.; Lavie, C.J.; Lear, S.A.; Ndumele, C.E.; Neeland, I.J.; Sanders, P.; et al. American Heart Association Council on Lifestyle and Cardiometabolic Health; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; Council on Epidemiology and Prevention; and Stroke Council. Obesity and Cardiovascular Disease: A Scientific Statement from the American Heart Association. Circulation 2021, 143, e984–e1010. [Google Scholar] [PubMed]
- Szczepaniak, L.S.; Dobbins, R.L.; Metzger, G.J.; Sartoni-D’Ambrosia, G.; Arbique, D.; Vongpatanasin, W.; Unger, R.; Victor, R.G. Myocardial triglycerides and systolic function in humans: In vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn. Reson. Med. 2003, 49, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: Molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebong, I.A.; Goff, D.C., Jr.; Rodriguez, C.J.; Chen, H.; Bertoni, A.G. Mechanisms of heart failure in obesity. Obes. Res. Clin. Pract. 2014, 8, e540–e548. [Google Scholar] [CrossRef] [Green Version]
- Pasqua, T.; Rocca, C.; Giglio, A.; Angelone, T. Cardiometabolism as an Interlocking Puzzle between the Healthy and Diseased Heart: New Frontiers in Therapeutic Applications. J. Clin. Med. 2021, 10, 721. [Google Scholar] [CrossRef]
- Sletten, A.C.; Peterson, L.R.; Schaffer, J.E. Manifestations and mechanisms of myocardial lipotoxicity in obesity. J. Intern. Med. 2018, 284, 478–491. [Google Scholar] [CrossRef] [Green Version]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V., Jr.; Ory, D.S.; Schaffer, J.E. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [Green Version]
- Rocca, C.; Pasqua, T.; Boukhzar, L.; Anouar, Y.; Angelone, T. Progress in the emerging role of selenoproteins in cardiovascular disease: Focus on endoplasmic reticulum-resident selenoproteins. Cell. Mol. Life Sci. 2019, 76, 3969–3985. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Anouar, Y.; Lihrmann, I.; Falluel-Morel, A.; Boukhzar, L. Selenoprotein T is a key player in ER proteostasis, endocrine homeostasis and neuroprotection. Free Radic. Biol. Med. 2018, 127, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Pothion, H.; Jehan, C.; Tostivint, H.; Cartier, D.; Bucharles, C.; Falluel-Morel, A.; Boukhzar, L.; Anouar, Y.; Lihrmann, I. Selenoprotein T: An Essential Oxidoreductase Serving as a Guardian of Endoplasmic Reticulum Homeostasis. Antioxid. Redox Signal. 2020, 33, 1257–1275. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, A.; Cartier, D.; Abid, H.; Calas, A.; Burel, C.; Bucharles, C.; Jehan, C.; Grumolato, L.; Landry, M.; Lerouge, P.; et al. Selenoprotein T is a novel OST subunit that regulates UPR signaling and hormone secretion. EMBO Rep. 2017, 18, 1935–1946. [Google Scholar] [CrossRef]
- Prevost, G.; Arabo, A.; Jian, L.; Quelennec, E.; Cartier, D.; Hassan, S.; Falluel-Morel, A.; Tanguy, Y.; Gargani, S.; Lihrmann, I.; et al. The PACAP-regulated gene selenoprotein T is abundantly expressed in mouse and human β-cells and its targeted inactivation impairs glucose tolerance. Endocrinology 2013, 154, 3796–3806. [Google Scholar] [CrossRef] [Green Version]
- Rocca, C.; Boukhzar, L.; Granieri, M.C.; Alsharif, I.; Mazza, R.; Lefranc, B.; Tota, B.; Leprince, J.; Cerra, M.C.; Anouar, Y.; et al. A selenoprotein T-derived peptide protects the heart against ischaemia/reperfusion injury through inhibition of apoptosis and oxidative stress. Acta Physiol. 2018, 223, e13067. [Google Scholar] [CrossRef] [PubMed]
- Rocca, C.; De Bartolo, A.; Granieri, M.C.; Rago, V.; Amelio, D.; Falbo, F.; Malivindi, R.; Mazza, R.; Cerra, M.C.; Boukhzar, L.; et al. The Antioxidant Selenoprotein T Mimetic, PSELT, Induces Preconditioning-like Myocardial Protection by Relieving Endoplasmic-Reticulum Stress. Antioxidants 2022, 11, 571. [Google Scholar] [CrossRef]
- Alsharif, I.; Boukhzar, L.; Lefranc, B.; Godefroy, D.; Aury-Landas, J.; Rego, J.D.; Rego, J.D.; Naudet, F.; Arabo, A.; Chagraoui, A.; et al. Cell-penetrating, antioxidant SELENOT mimetic protects dopaminergic neurons and ameliorates motor dysfunction in Parkinson’s disease animal models. Redox Biol. 2021, 40, 101839. [Google Scholar] [CrossRef]
- Rocca, C.; De Bartolo, A.; Grande, F.; Rizzuti, B.; Pasqua, T.; Giordano, F.; Granieri, M.C.; Occhiuzzi, M.A.; Garofalo, A.; Amodio, N.; et al. Cateslytin abrogates lipopolysaccharide-induced cardiomyocyte injury by reducing inflammation and oxidative stress through toll like receptor 4 interaction. Int. Immunopharmacol. 2021, 94, 107487. [Google Scholar] [CrossRef]
- Grande, F.; De Bartolo, A.; Occhiuzzi, M.A.; Caruso, A.; Rocca, C.; Pasqua, T.; Carocci, A.; Rago, V.; Angelone, T.; Sinicropi, M.S. Carbazole and Simplified Derivatives: Novel Tools toward β-Adrenergic Receptors Targeting. Appl. Sci. 2021, 11, 5486. [Google Scholar] [CrossRef]
- Rocca, C.; Grande, F.; Granieri, M.C.; Colombo, B.; De Bartolo, A.; Giordano, F.; Rago, V.; Amodio, N.; Tota, B.; Cerra, M.C.; et al. The chromogranin A1-373 fragment reveals how a single change in the protein sequence exerts strong cardioregulatory effects by engaging neuropilin-1. Acta Physiol. 2021, 231, e13570. [Google Scholar] [CrossRef]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar] [CrossRef]
- McQueen, M.J. Optimal Assay of LDH and α-HBD at 37 °C. Ann. Clin. Biochem. 1972, 9, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Rocca, C.; Scavello, F.; Colombo, B.; Gasparri, A.M.; Dallatomasina, A.; Granieri, M.C.; Amelio, D.; Pasqua, T.; Cerra, M.C.; Tota, B.; et al. Physiological levels of chromogranin A prevent doxorubicin-induced cardiotoxicity without impairing its anticancer activity. FASEB J. 2019, 33, 7734–7747. [Google Scholar] [CrossRef]
- Ivan, A.; Herman, H.; Balta, C.; Hadaruga, D.I.; Mihali, C.V.; Ardelean, A.; Hermenean, A. Berberis vulgaris extract/β-cyclodextrin complex increases protection of hepatic cells via suppression of apoptosis and lipogenesis pathways. Exp. Ther. Med. 2017, 13, 2143–2150. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Kong, B.; Shuai, W.; Liu, Y.; Wang, G.J.; Xu, M.; Zhao, J.J.; Fang, J.; Fu, H.; Jiang, X.B.; et al. Myeloid differentiation protein 1 protected myocardial function against high-fat stimulation induced pathological remodelling. J. Cell. Mol. Med. 2019, 23, 5303–5316. [Google Scholar]
- Nasci, V.L.; Chuppa, S.; Griswold, L.; Goodreau, K.A.; Dash, R.K.; Kriegel, A.J. miR-21-5p regulates mitochondrial respiration and lipid content in H9C2 cells. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H710–H721. [Google Scholar] [CrossRef] [PubMed]
- Güçlü, O.; Doğanlar, O.; Yüksel, V.; Doğanlar, Z.B. FOLFIRI-Mediated Toxicity in Human Aortic Smooth Muscle Cells and Possible Amelioration with Curcumin and Quercetin. Cardiovasc. Toxicol. 2020, 20, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, M.; Carresi, C.; Nucera, S.; Ruga, S.; Maiuolo, J.; Macrì, R.; Scarano, F.; Bosco, F.; Mollace, R.; Cardamone, A.; et al. Icariin Protects H9c2 Rat Cardiomyoblasts from Doxorubicin-Induced Cardiotoxicity: Role of Caveolin-1 Upregulation and Enhanced Autophagic Response. Nutrients 2021, 13, 4070. [Google Scholar] [CrossRef]
- Benoist, L.; Chadet, S.; Genet, T.; Lefort, C.; Heraud, A.; Danila, M.D.; Muntean, D.M.; Baron, C.; Angoulvant, D.; Babuty, D.; et al. Stimulation of P2Y11 receptor protects human cardiomyocytes against Hypoxia/Reoxygenation injury and involves PKCε signaling pathway. Sci. Rep. 2019, 9, 11613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Francesco, E.M.; Ózsvári, B.; Sotgia, F.; Lisanti, M.P. Dodecyl-TPP Targets Mitochondria and Potently Eradicates Cancer Stem Cells (CSCs): Synergy With FDA-Approved Drugs and Natural Compounds (Vitamin C and Berberine). Front. Oncol. 2019, 9, 615. [Google Scholar] [CrossRef] [Green Version]
- Urso, C.J.; Zhou, H. Role of CD36 in Palmitic Acid Lipotoxicity in Neuro-2a Neuroblastoma Cells. Biomolecules 2021, 11, 1567. [Google Scholar] [CrossRef]
- Rehman, I.U.; Movasaghi, Z.; Rehman, S. Vibrational Spectroscopy for Tissue Analysis, 1st ed.; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Güler, G.; Guven, U.; Oktem, G. Characterization of CD133+/CD44+ human prostate cancer stem cells with ATR-FTIR spectroscopy. Analyst 2019, 144, 2138–2149. [Google Scholar] [CrossRef]
- Tamm, L.K.; Tatulian, S.A. Infrared spectroscopy of proteins and peptides in lipid bilayers. Q. Rev. Biophys. 1997, 30, 365–429. [Google Scholar] [CrossRef] [Green Version]
- Ozek, N.S.; Tuna, S.; Erson-Bensan, A.E.; Severcan, F. Characterization of microRNA-125b expression in MCF7 breast cancer cells by ATR-FTIR spectroscopy. Analyst 2010, 135, 3094–3102. [Google Scholar] [CrossRef]
- Cheon, H.G.; Cho, Y.S. Protection of palmitic acid-mediated lipotoxicity by arachidonic acid via channeling of palmitic acid into triglycerides in C2C12. J. Biomed. Sci. 2014, 21, 13. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Kim, C.H.; Kim, K.Y.; Cheon, H.G. Protective effects of arachidonic acid against palmitic acid-mediated lipotoxicity in HIT-T15 cells. Mol. Cell. Biochem. 2012, 364, 19–28. [Google Scholar] [CrossRef]
- Estadella, D.; da Penha Oller do Nascimento, C.M.; Oyama, L.M.; Ribeiro, E.B.; Dâmaso, A.R.; de Piano, A. Lipotoxicity: Effects of dietary saturated and transfatty acids. Mediat. Inflamm. 2013, 2013, 137579. [Google Scholar] [CrossRef] [Green Version]
- Stanley, W.C.; Dabkowski, E.R.; Ribeiro, R.F., Jr.; O’Connell, K.A. Dietary fat and heart failure: Moving from lipotoxicity to lipoprotection. Circ. Res. 2012, 110, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.Y.; Velmurugan, B.K.; Day, C.H.; Shen, C.Y.; Chun, L.C.; Tsai, Y.C.; Lin, Y.M.; Chen, R.J.; Kuo, C.H.; Huang, C.Y. High density lipoprotein (HDL) reverses palmitic acid induced energy metabolism imbalance by switching CD36 and GLUT4 signaling pathways in cardiomyocyte. J. Cell. Physiol. 2017, 232, 3020–3029. [Google Scholar] [CrossRef]
- Hescheler, J.; Meyer, R.; Plant, S.; Krautwurst, D.; Rosenthal, W.; Schultz, G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 1991, 69, 1476–1486. [Google Scholar] [CrossRef] [Green Version]
- Branco, A.F.; Pereira, S.P.; Gonzalez, S.; Gusev, O.; Rizvanov, A.A.; Oliveira, P.J. Gene Expression Profiling of H9c2 Myoblast Differentiation towards a Cardiac-Like Phenotype. PLoS ONE 2015, 10, e0129303. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.Y.; Rabkin, S.W. Palmitate-induced apoptosis in cardiomyocytes is mediated through alterations in mitochondria: Prevention by cyclosporin A. Biochim. Biophys. Acta 2000, 1485, 45–55. [Google Scholar] [CrossRef]
- Leroy, C.; Tricot, S.; Lacour, B.; Grynberg, A. Protective effect of eicosapentaenoic acid on palmitate-induced apoptosis in neonatal cardiomyocytes. Biochim. Biophys. Acta 2008, 1781, 685–693. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Li, X.; Wu, N.; Jia, P.; Liu, C.; Jia, D. Palmitate induces myocardial lipotoxic injury via the endoplasmic reticulum stress-mediated apoptosis pathway. Mol. Med. Rep. 2017, 16, 6934–6939. [Google Scholar] [CrossRef] [Green Version]
- De Geest, B.; Mishra, M. Role of Oxidative Stress in Diabetic Cardiomyopathy. Antioxidants 2022, 11, 784. [Google Scholar] [CrossRef]
- Remigante, A.; Morabito, R.; Spinelli, S.; Trichilo, V.; Loddo, S.; Sarikas, A.; Dossena, S.; Marino, A. d-Galactose Decreases Anion Exchange Capability through Band 3 Protein in Human Erythrocytes. Antioxidants 2020, 9, 689. [Google Scholar] [CrossRef]
- Crupi, R.; Morabito, R.; Remigante, A.; Gugliandolo, E.; Britti, D.; Cuzzocrea, S.; Marino, A. Susceptibility of erythrocytes from different sources to xenobiotics-induced lysis. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2019, 221, 68–72. [Google Scholar] [CrossRef]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Mangali, S.; Bhat, A.; Udumula, M.P.; Dhar, I.; Sriram, D.; Dhar, A. Inhibition of protein kinase R protects against palmitic acid-induced inflammation, oxidative stress, and apoptosis through the JNK/NF-kB/NLRP3 pathway in cultured H9C2 cardiomyocytes. J. Cell. Biochem. 2019, 120, 3651–3663. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Alnahdi, A.; John, A.; Raza, H. N-acetyl cysteine attenuates oxidative stress and glutathione-dependent redox imbalance caused by high glucose/high palmitic acid treatment in pancreatic Rin-5F cells. PLoS ONE 2019, 14, e0226696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. [Google Scholar] [CrossRef]
- Tiwary, S.; Nandwani, A.; Khan, R.; Datta, M. GRP75 mediates endoplasmic reticulum-mitochondria coupling during palmitate-induced pancreatic β-cell apoptosis. J. Biol. Chem. 2021, 297, 101368. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Chen, Z.; Chen, R.; Lin, L.; Ren, L.; Zhang, M.; Liu, L. Increased fatty acid metabolism attenuates cardiac resistance to β-adrenoceptor activation via mitochondrial reactive oxygen species: A potential mechanism of hypoglycemia-induced myocardial injury in diabetes. Redox Biol. 2022, 52, 102320. [Google Scholar] [CrossRef]
- Grumolato, L.; Ghzili, H.; Montero-Hadjadje, M.; Gasman, S.; Lesage, J.; Tanguy, Y.; Galas, L.; Ait-Ali, D.; Leprince, J.; Guérineau, N.C.; et al. Selenoprotein T is a PACAP-regulated gene involved in intracellular Ca2+ mobilization and neuroendocrine secretion. FASEB J. 2008, 22, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2022, 118, 115–129. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Glatz, J.F.C.; Luiken, J.J.F.P.; Nabben, M. CD36 (SR-B2) as a Target to Treat Lipid Overload-Induced Cardiac Dysfunction. J. Lipid Atheroscler. 2020, 9, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Boukhzar, L.; Hamieh, A.; Cartier, D.; Tanguy, Y.; Alsharif, I.; Castex, M.; Arabo, A.; El Hajji, S.; Bonnet, J.J.; Errami, M.; et al. Selenoprotein T Exerts an Essential Oxidoreductase Activity That Protects Dopaminergic Neurons in Mouse Models of Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Santesmasses, D.; Mariotti, M.; Gladyshev, V.N. Tolerance to Selenoprotein Loss Differs between Human and Mouse. Mol. Biol. Evol. 2020, 37, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, Å.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Kakimoto, P.A.; Kowaltowski, A.J. Effects of high fat diets on rodent liver bioenergetics and oxidative imbalance. Redox Biol. 2016, 8, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial Dynamics is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell. Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sousa, I.F.; Migliaccio, V.; Lepretti, M.; Paolella, G.; Di Gregorio, I.; Caputo, I.; Ribeiro, E.B.; Lionetti, L. Dose- and Time-Dependent Effects of Oleate on Mitochondrial Fusion/Fission Proteins and Cell Viability in HepG2 Cells: Comparison with Palmitate Effects. Int. J. Mol. Sci. 2021, 22, 9812. [Google Scholar] [CrossRef]
- Coll, T.; Jové, M.; Rodríguez-Calvo, R.; Eyre, E.; Palomer, X.; Sánchez, R.M.; Merlos, M.; Laguna, J.C.; Vázquez-Carrera, M. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes 2006, 55, 2779–2787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.H.; Gerhart-Hines, Z.; Dominy, J.E.; Lee, Y.; Kim, S.; Tabata, M.; Xiang, Y.K.; Puigserver, P. Oleic acid stimulates complete oxidation of fatty acids through protein kinase A-dependent activation of SIRT1-PGC1α complex. J. Biol. Chem. 2013, 288, 7117–7126. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adaniya, S.M.; O-Uchi, J.; Cypress, M.W.; Kusakari, Y.; Jhun, B.S. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am. J. Physiol. Cell. Physiol. 2019, 316, C583–C604. [Google Scholar] [CrossRef] [PubMed]
- Hoppins, S.; Nunnari, J. Cell Biology. Mitochondrial dynamics and apoptosis--the ER connection. Science 2012, 337, 1052–1054. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guan, G.; Lei, L.; Liu, J.; Cao, L.; Wang, X. Oxidative and endoplasmic reticulum stresses are involved in palmitic acid-induced H9c2 cell apoptosis. Biosci. Rep. 2019, 39, BSR20190225. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zhou, L.; Fan, Z.; Liu, S.; Fang, W. Palmitic acid, but not high-glucose, induced myocardial apoptosis is alleviated by N-acetylcysteine due to attenuated mitochondrial-derived ROS accumulation-induced endoplasmic reticulum stress. Cell. Death Dis. 2018, 9, 568. [Google Scholar] [CrossRef]
- Xue, R.Q.; Zhao, M.; Wu, Q.; Yang, S.; Cui, Y.L.; Yu, X.J.; Liu, J.; Zang, W.J. Regulation of mitochondrial cristae remodelling by acetylcholine alleviates palmitate-induced cardiomyocyte hypertrophy. Free Radic. Biol. Med. 2019, 145, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Li, Y.; Zhang, S.; Zhang, N.; Sun, Y.; Liu, F.; Chen, Q.; Gai, Z. Docosahexaenoic acid protects against palmitate-induced mitochondrial dysfunction in diabetic cardiomyopathy. Biomed. Pharmacother. 2020, 128, 110306. [Google Scholar] [CrossRef]
- Wu, K.M.; Hsu, Y.M.; Ying, M.C.; Tsai, F.J.; Tsai, C.H.; Chung, J.G.; Yang, J.S.; Tang, C.H.; Cheng, L.Y.; Su, P.H.; et al. High-density lipoprotein ameliorates palmitic acid-induced lipotoxicity and oxidative dysfunction in H9c2 cardiomyoblast cells via ROS suppression. Nutr. Metab. 2019, 16, 36. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Zhang, H.; Gutiérrez Cortés, N.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef]
- Perry, B.D.; Rahnert, J.A.; Xie, Y.; Zheng, B.; Woodworth-Hobbs, M.E.; Price, S.R. Palmitate-induced ER stress and inhibition of protein synthesis in cultured myotubes does not require Toll-like receptor 4. PLoS ONE 2018, 13, e0191313. [Google Scholar] [CrossRef] [Green Version]
- Gudbjarnason, S.; De Schryver, C.; Chiba, C.; Yamanaka, J.; Bing, R. Protein and Nucleic Acid Synthesis During the Reparative Processes Following Myocardial Infarction. Circ. Res. 1964, 15, 320–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, M.; Panagia, V.; Paolillo, G.; Majumder, S.; Ou, C.; Dhalla, N.S. Inhibiton of cardiac phosphatidylethanolamine N-methylation by oxygen free radicals. Biochim. Biophys. Acta 1990, 1021, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Kumar, S.; Choi, E.H.; Chaudhary, S.; Kim, M.H. Molecular dynamic simulations of oxidized skin lipid bilayer and permeability of reactive oxygen species. Sci. Rep. 2019, 14, 4496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak Number | Wavenumber (cm−1) | Assignment of Functional Groups |
|---|---|---|
| 1 | 3010 | Olefinic = CH stretching vibration (unsaturated lipids) |
| 2 | 2957 | CH3 asymmetric stretching (lipids) |
| 3 | 2923 | CH2 asymmetric stretching (lipids) |
| 4 | 2872 | CH3 symmetric stretching (proteins and lipids) |
| 5 | 2852 | CH2 symmetric stretching (mainly lipids) |
| 6 | 1750–1715 | C=O stretching (ester functional groups in lipids) |
| 7 | 1646 | Amide I (protein C=O stretching) |
| 8 | 1546 | Amide II (protein NH bending, CN stretching) |
| 9 | 1456 | CH2 bending (mainly lipids) |
| 10 | 1399 | COO– symmetric stretching (fatty acids) |
| 11 | 1337 | CH3 symmetric bending (lipids) |
| 12 | 1313 | CH2 wagging (lipids) |
| 13 | 1236 | PO2– asymmetric stretching, fully hydrogen-bonded (mainly nucleic acids) |
| 14 | 1173 | CO–O–C asymmetric stretching (ester bonds in cholesteryl esters) |
| 15 | 1116 | Ribose ring vibrations (RNA) |
| 16 | 1086 | PO2– symmetric stretching (nucleic acids and phospholipids) |
| 17 | 1050 | C–O stretching (polysaccharides, glycogen) |
| 18 | 971 | C–N±–C stretching (nucleic acids) |
| 19 | 924 | Ribose ring vibrations (RNA) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocca, C.; De Bartolo, A.; Guzzi, R.; Crocco, M.C.; Rago, V.; Romeo, N.; Perrotta, I.; De Francesco, E.M.; Muoio, M.G.; Granieri, M.C.; et al. Palmitate-Induced Cardiac Lipotoxicity Is Relieved by the Redox-Active Motif of SELENOT through Improving Mitochondrial Function and Regulating Metabolic State. Cells 2023, 12, 1042. https://doi.org/10.3390/cells12071042
Rocca C, De Bartolo A, Guzzi R, Crocco MC, Rago V, Romeo N, Perrotta I, De Francesco EM, Muoio MG, Granieri MC, et al. Palmitate-Induced Cardiac Lipotoxicity Is Relieved by the Redox-Active Motif of SELENOT through Improving Mitochondrial Function and Regulating Metabolic State. Cells. 2023; 12(7):1042. https://doi.org/10.3390/cells12071042
Chicago/Turabian StyleRocca, Carmine, Anna De Bartolo, Rita Guzzi, Maria Caterina Crocco, Vittoria Rago, Naomi Romeo, Ida Perrotta, Ernestina Marianna De Francesco, Maria Grazia Muoio, Maria Concetta Granieri, and et al. 2023. "Palmitate-Induced Cardiac Lipotoxicity Is Relieved by the Redox-Active Motif of SELENOT through Improving Mitochondrial Function and Regulating Metabolic State" Cells 12, no. 7: 1042. https://doi.org/10.3390/cells12071042