
Olaparib-Resistant BRCA2MUT Ovarian Cancer Cells with Restored BRCA2 Abrogate Olaparib-Induced DNA Damage and G2/M Arrest Controlled by the ATR/CHK1 Pathway for Survival


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines
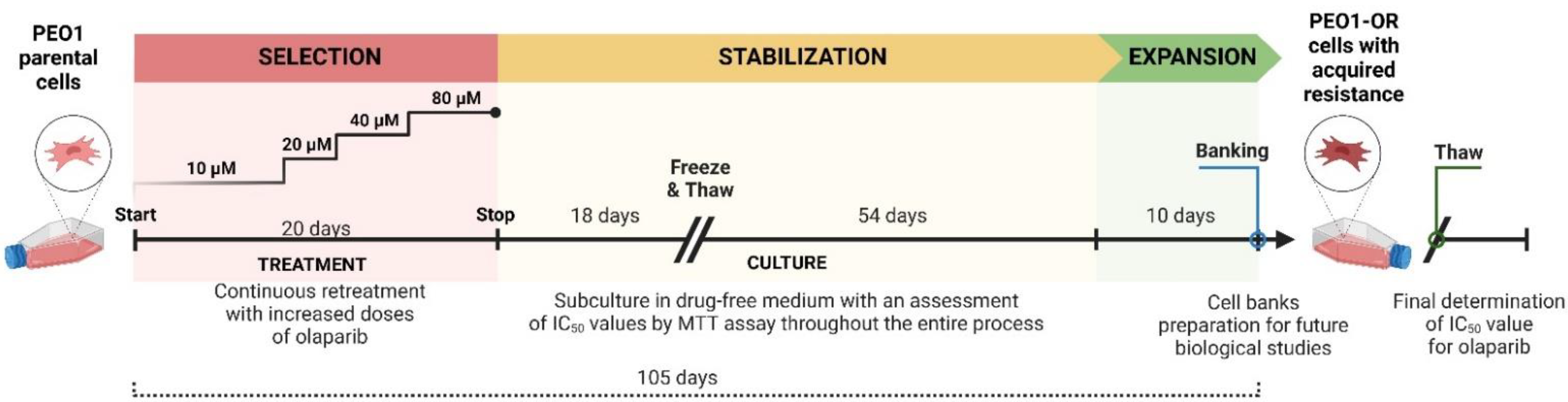
2.3. Development of Olaparib Resistance in HGSOC Cell Line In Vitro
2.4. Cell Proliferation Rate and Population Doubling Time
2.5. MTT Cell Viability Assay
2.6. Clonogenic Assay
2.7. Cellular Morphology Changes
2.8. Whole-Exome Sequencing and Variant Calling
2.9. Western Blotting
2.10. Immunofluorescence Analysis of γH2AX and RAD51 Foci Formation
2.11. Preparation of Metaphase Chromosomes for Structural Aberration Analysis
2.12. Neutral Comet Assay
2.13. Cell Cycle Analysis with Quantification of γH2AX by Flow Cytometry
2.14. Statistical Analysis
3. Results
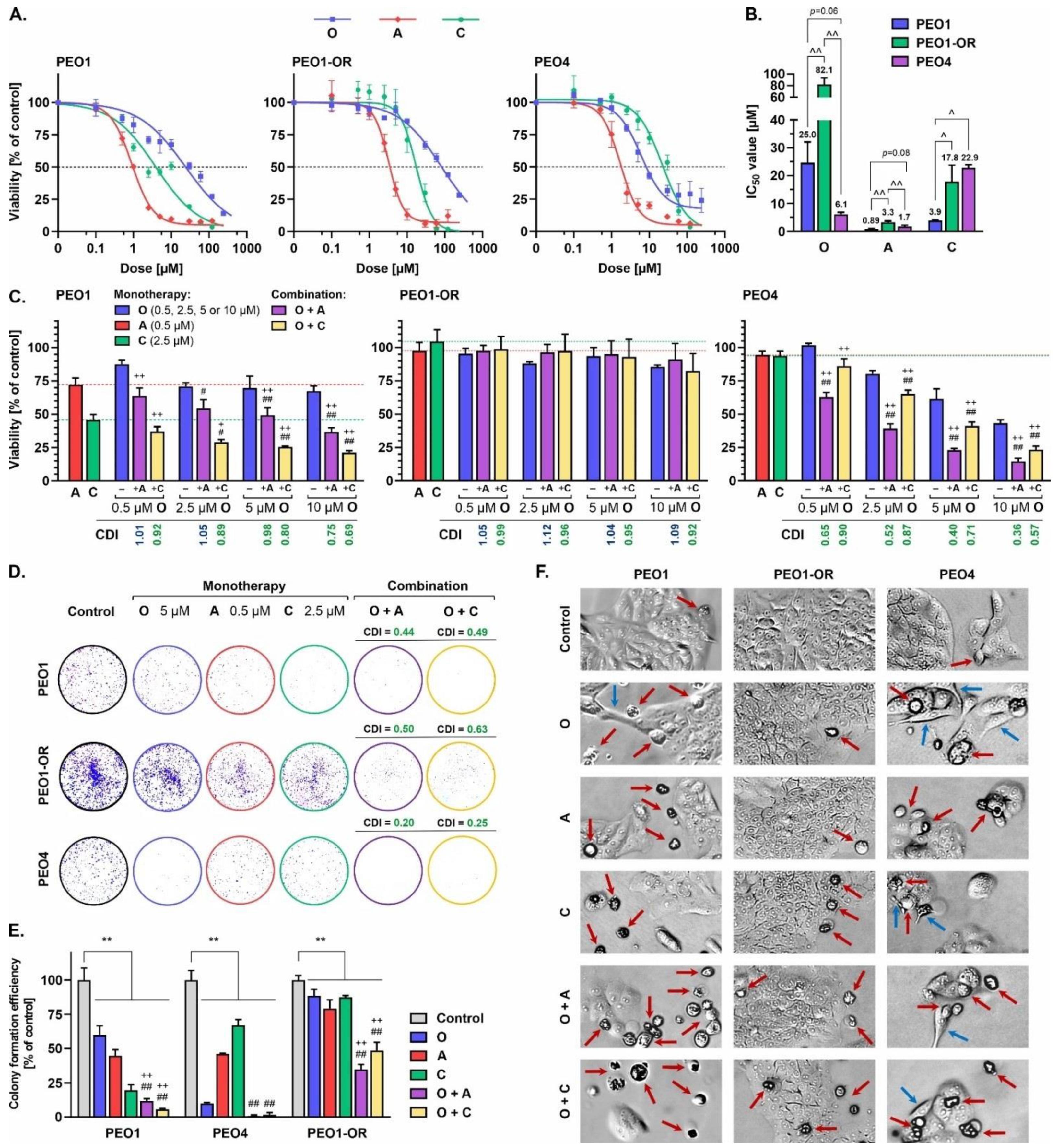
3.1. The PEO1-OR Cell Line with an Acquired Resistant Phenotype Displays Reduced Sensitivity to Olaparib and Increased Proliferation
3.2. The PEO1-OR Cell Line Displays Increased Resistance to Olaparib and Inhibitors of the ATR/CHK1 Pathway
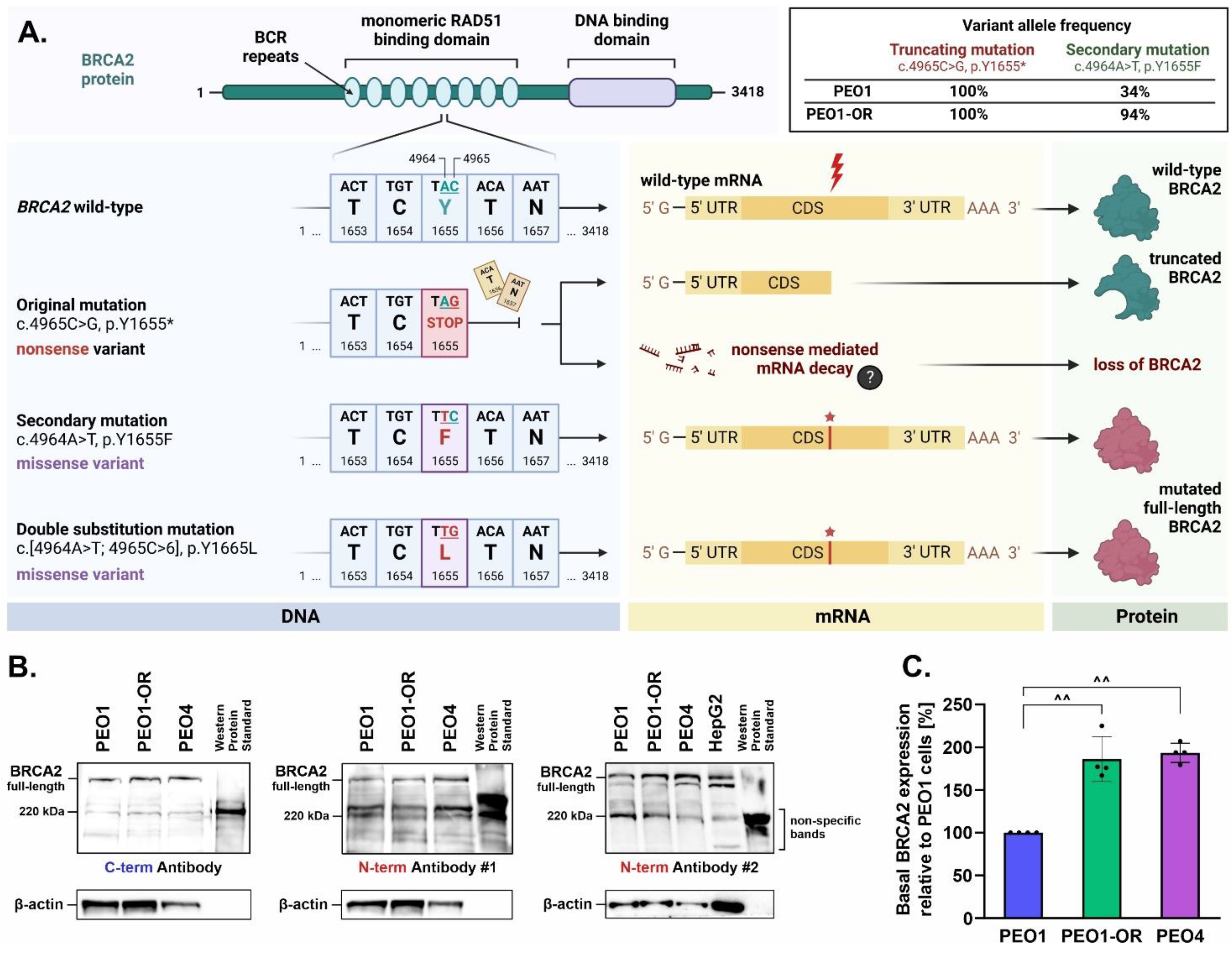
3.3. Relationship between Olaparib Resistance and Exonic Variants in Genes Involved in DNA Damage Repair, Cell Cycle Regulation, and Drug Efflux in OC Cells
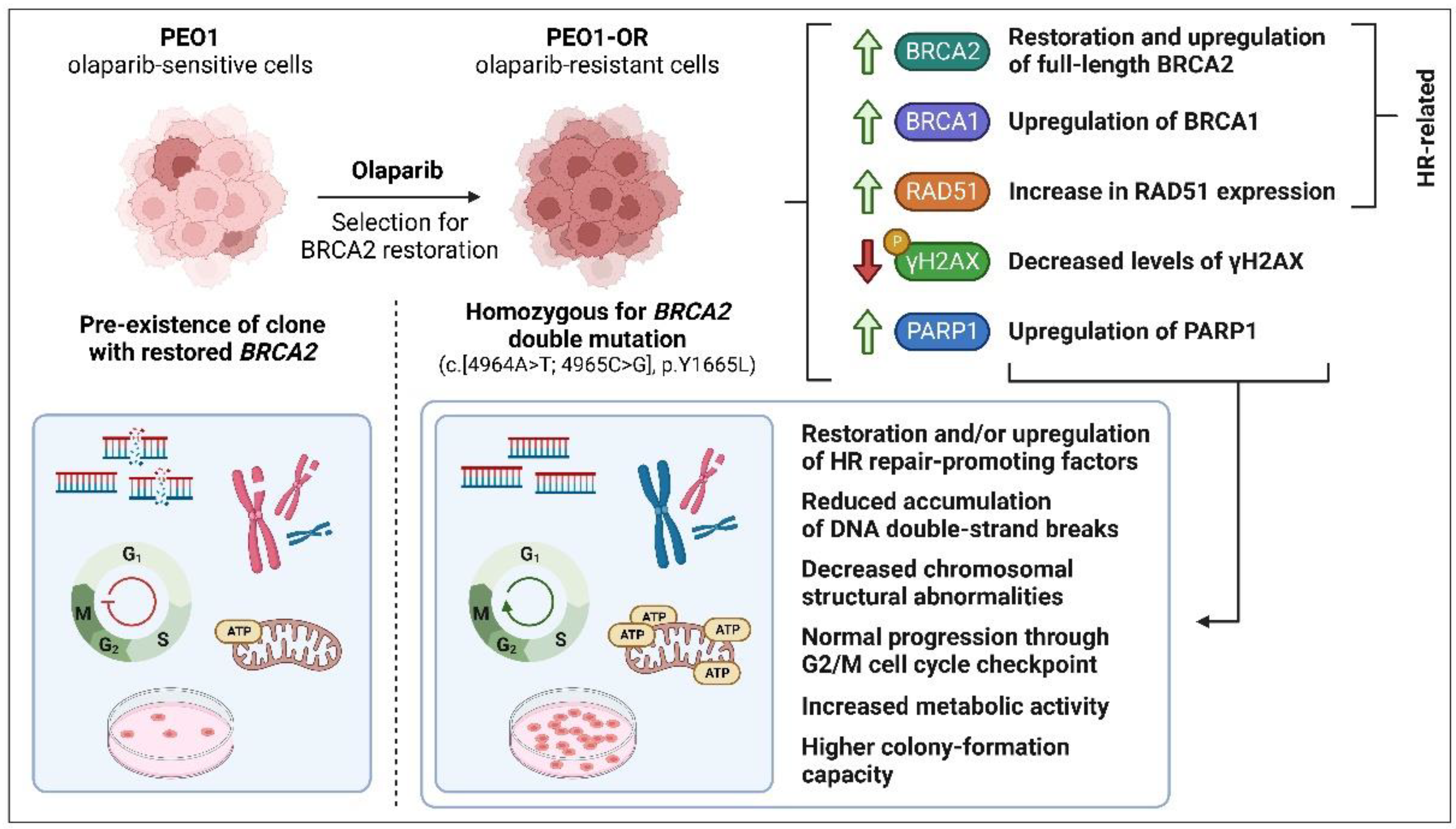
3.4. Upregulation of BRCA2 Is Correlated with a Secondary Reversion Mutation Accounting for Acquired Resistance to Olaparib in OC Cells
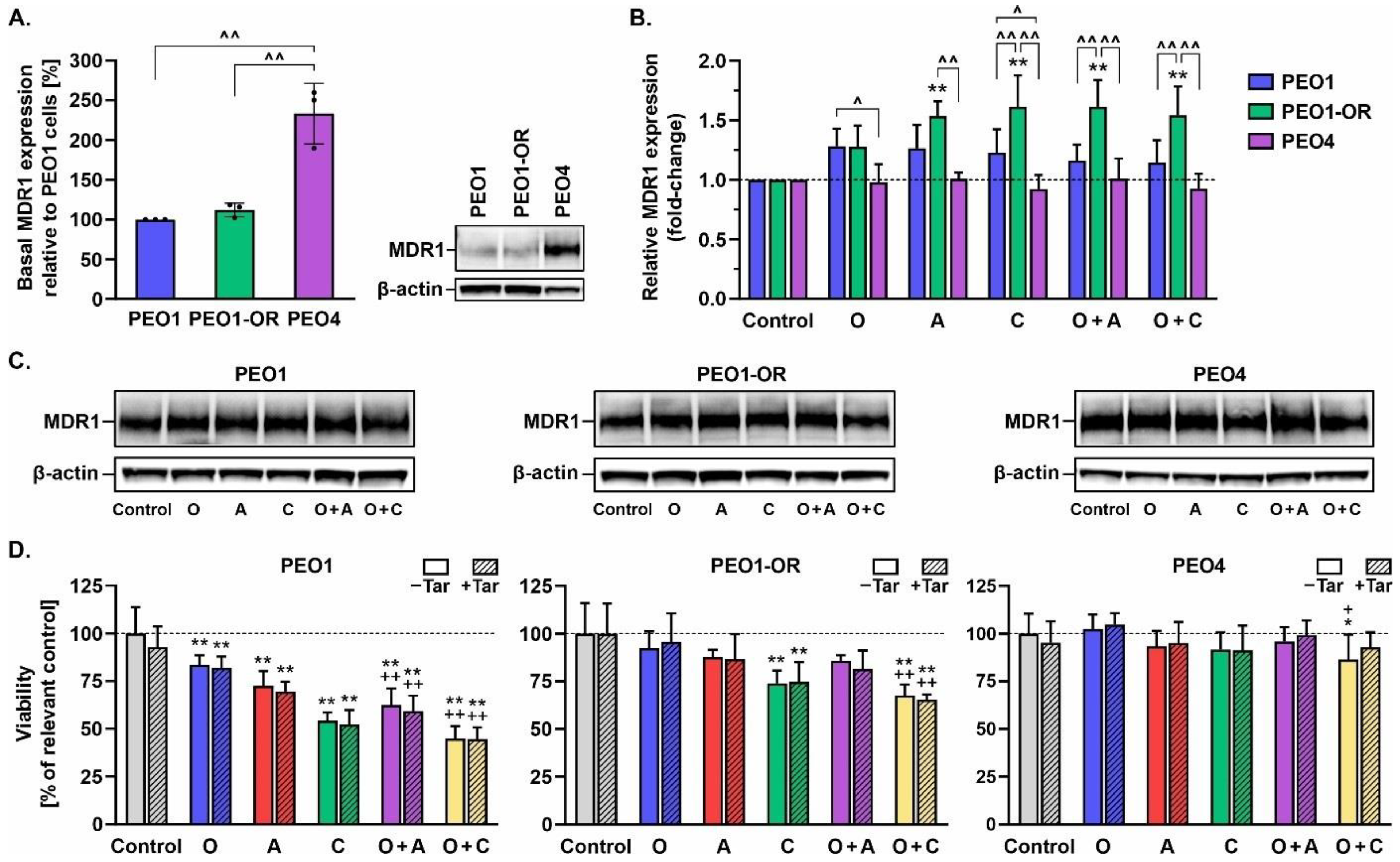
3.5. Inhibition of MDR1 Drug Efflux Pump Does Not Restore Sensitivity to Olaparib, ATRi, or CHK1i in Olaparib-Resistant Cells
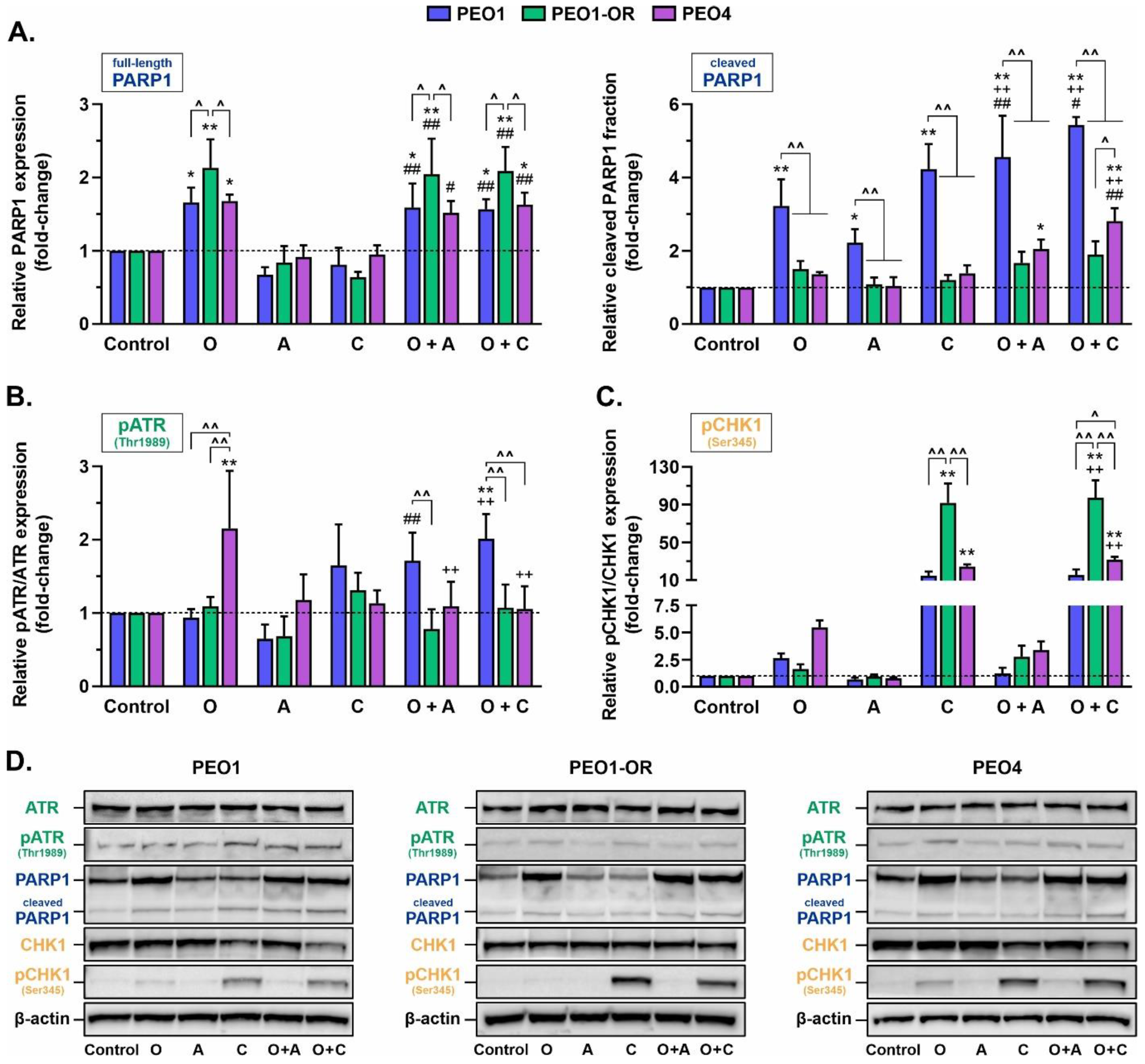
3.6. Olaparib Synergistically Increases Cleavage of PARP1 in Olaparib-Sensitive OC Cells in Combination with ATRi or CHK1i
3.7. PEO1-OR Cells Bypass Olaparib-Induced Activation of the ATR/CHK1 Pathway
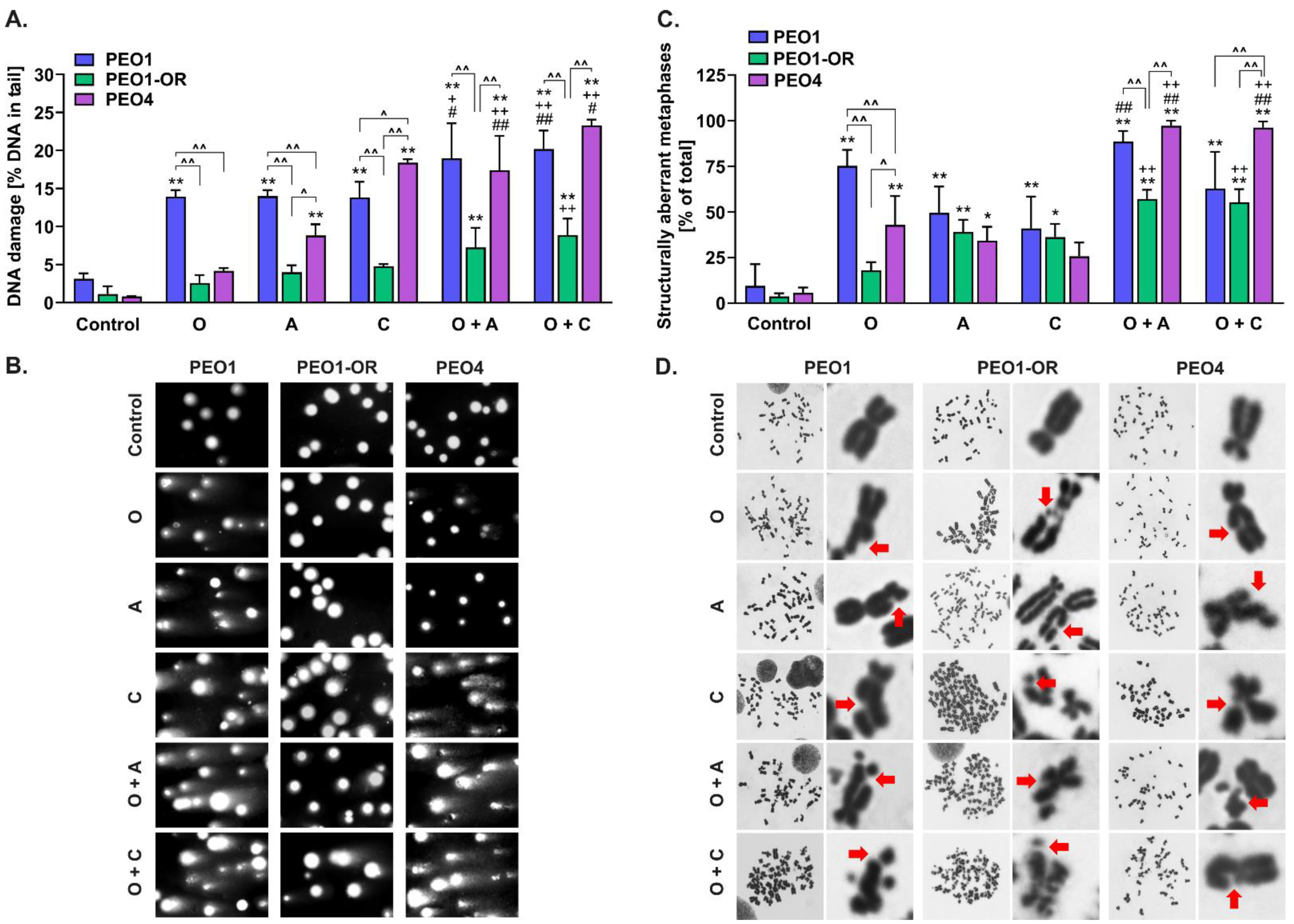
3.8. Olaparib Combined with Blockage of ATR/CHK1 Enhances DNA Double-Strand Breaks Irrespective of RAD51-Mediated HR Activity in BRCA2MUT Ovarian Cancer Cells
3.9. Olaparib-Resistant Cells Exhibit Decreased Accumulation of Chromosomal Aberrations and DNA Double-Strand Breaks Compared to Sensitive OC Cell Lines
3.10. PEO1-OR Cells Progress Normally through the Cell Cycle upon Inhibition of PARP1 and the ATR/CHK1 Axis
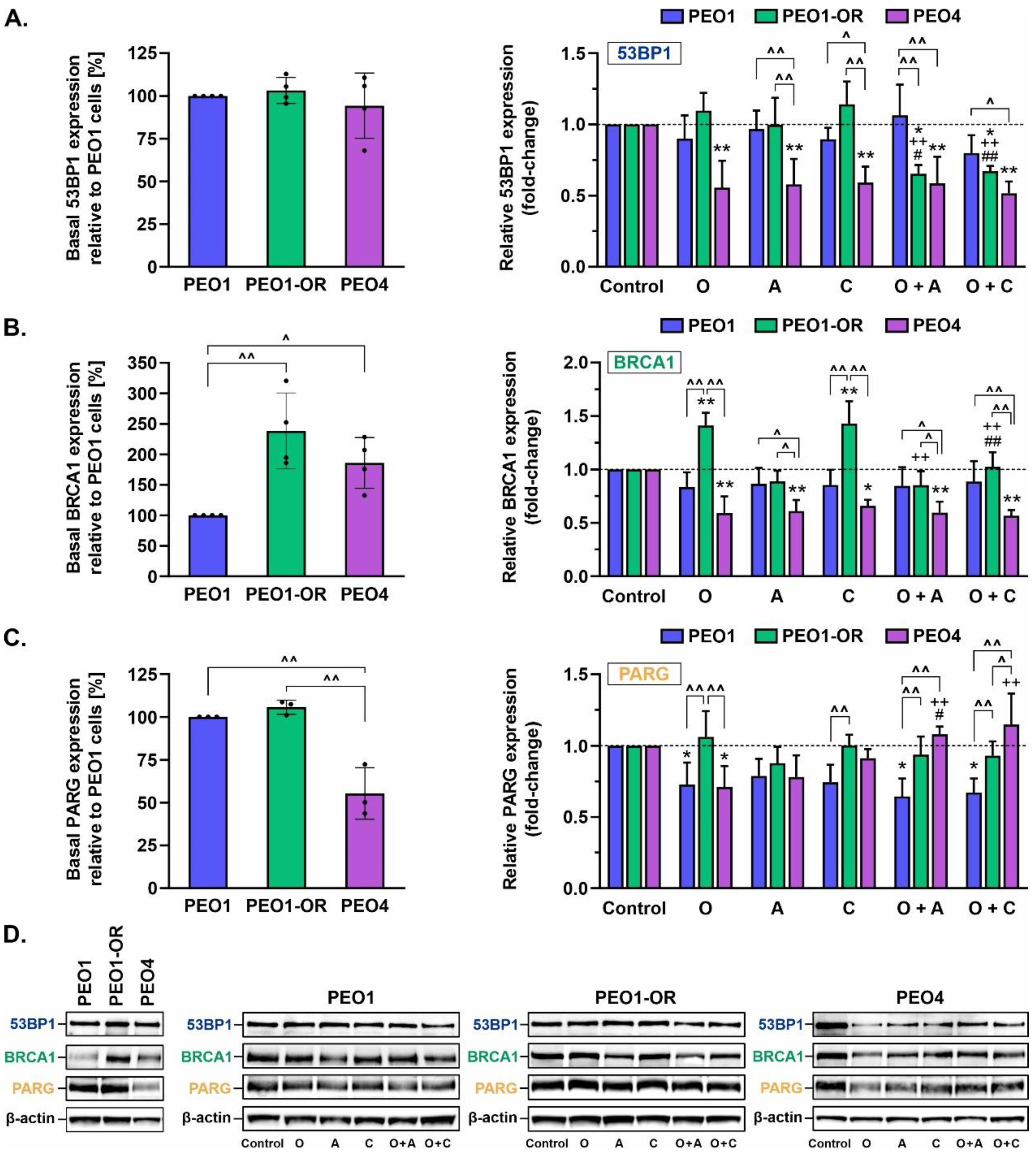
3.11. Crosstalk between BRCA1 and 53BP1 as an Indicator of Sensitivity to Olaparib and ATR/CHK1 Pathway Blockers
3.12. Olaparib Alone and Combined with Blockage of the ATR/CHK1 Pathway Suppresses PARG Expression in PEO1 Olaparib-Sensitive OC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A | ATR inhibitor (ceralasertib) |
| ABC | ATP-binding cassette |
| ATCC | American Type Culture Collection |
| ATR | ataxia telangiectasia and RAD3-related protein |
| ATRi | ATR inhibitor(s) |
| BRCA1 | breast cancer type 1 susceptibility protein |
| BRCA2 | breast cancer type 2 susceptibility protein |
| BrdU | bromodeoxyuridine |
| C | CHK1 inhibitor (MK-8776) |
| CDI | coefficient of drug interaction |
| CHK1 | checkpoint kinase 1 |
| CHK1i | CHK1 inhibitor(s) |
| DDR | DNA damage response |
| DSB | double-strand break |
| ECACC | The European Collection of Authenticated Cell Cultures |
| HGSOC | high-grade serous ovarian cancer |
| HI-FBS | heat-inactivated fetal bovine serum |
| IC50 | concentration of drug required to reduce cell viability to 50% |
| MDR | multidrug resistance |
| MDR1 | multidrug resistance protein 1 |
| MDRi | multidrug resistance inhibitor(s) |
| MFI | geometric mean fluorescence intensity |
| NGS | next-generation sequencing |
| NHEJ | non-homologous end joining |
| O | olaparib |
| OC | ovarian cancer |
| ORF | open reading frame |
| PARG | poly(ADP-ribose) glycohydrolase |
| PARP1 | poly(ADP-ribose) polymerase 1 |
| PARPi | PARP inhibitor(s) |
| PDT | population doubling time |
| PEO1-OR | PEO1 olaparib-resistant cell line |
| QCI-IT | Qiagen Clinical Insight Interpret Translational |
| SF | surviving fraction |
| Tar | tariquidar |
| WES | whole-exome sequencing |
References
- Ledermann, J.A.; Pujade-Lauraine, E. Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919849753. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.G.; Yao, M.; Chambers, L.M.; Mahdi, H.; DeBernardo, R.; Michener, C.M.; AlHilli, M.; Ricci, S.; Vargas, R. PARP inhibitors decrease response to subsequent platinum-based chemotherapy in patients with BRCA mutated ovarian cancer. Anticancer Drugs 2021, 32, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Moubarak, M.; Harter, P.; Ataseven, B.; Traut, A.; Welz, J.; Baert, T.; Heitz, F. Re-treatment with PARPi in patients with recurrent epithelial ovarian cancer: A single institutional experience. Gynecol. Oncol. Rep. 2022, 40, 100939. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, R.M.; Wahner Hendrickson, A.E.; Visscher, D.W.; Ansell, P.; Harrell, M.I.; Wagner, J.M.; Negron, V.; Goergen, K.M.; Maurer, M.J.; Oberg, A.L.; et al. 53BP1 as a potential predictor of response in PARP inhibitor-treated homologous recombination-deficient ovarian cancer. Gynecol. Oncol. 2019, 153, 127–134. [Google Scholar] [CrossRef]
- Kristeleit, R.; Lisyanskaya, A.; Fedenko, A.; Dvorkin, M.; de Melo, A.C.; Shparyk, Y.; Rakhmatullina, I.; Bondarenko, I.; Colombo, N.; Svintsitskiy, V. 1Rucaparib versus chemotherapy in patients with advanced, relapsed ovarian cancer and a deleterious BRCA mutation: Efficacy and safety from ARIEL4, a randomized phase III study. Gynecol. Oncol. 2021, 162, S3–S4. [Google Scholar] [CrossRef]
- Creeden, J.F.; Nanavaty, N.S.; Einloth, K.R.; Gillman, C.E.; Stanbery, L.; Hamouda, D.M.; Dworkin, L.; Nemunaitis, J. Homologous recombination proficiency in ovarian and breast cancer patients. BMC Cancer 2021, 21, 1154. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Tan, D.S.P. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: Do we need it? ESMO Open 2021, 6, 100144. [Google Scholar] [CrossRef]
- van Wijk, L.M.; Nilas, A.B.; Vrieling, H.; Vreeswijk, M.P.G. RAD51 as a functional biomarker for homologous recombination deficiency in cancer: A promising addition to the HRD toolbox? Expert Rev. Mol. Diagn. 2022, 22, 185–199. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Biegala, L.; Gajek, A.; Marczak, A.; Rogalska, A. PARP inhibitor resistance in ovarian cancer: Underlying mechanisms and therapeutic approaches targeting the ATR/CHK1 pathway. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188633. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.K.; Illuzzi, G.; Colomer, C.; Churchman, M.; Hollis, R.L.; O’Connor, M.J.; Gourley, C.; Leo, E.; Melton, D.W. Identifying and Overcoming Mechanisms of PARP Inhibitor Resistance in Homologous Recombination Repair-Deficient and Repair-Proficient High Grade Serous Ovarian Cancer Cells. Cancers 2020, 12, 1503. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Camacho, C.V.; Kraus, W.L. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp. Mol. Med. 2021, 53, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Wang, J.; Jia, Y.; Duan, Z.; Shi, H. ATR and p-ATR are emerging prognostic biomarkers and DNA damage response targets in ovarian cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920982853. [Google Scholar] [CrossRef]
- Gupta, N.; Huang, T.T.; Horibata, S.; Lee, J.M. Cell cycle checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor-resistant cancer. Pharmacol. Res. 2022, 178, 106162. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Mikula, M.; Ostrowski, J.; Sliwinska, A.; Rogalska, A. PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality-An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. Int. J. Mol. Sci. 2020, 21, 9715. [Google Scholar] [CrossRef] [PubMed]
- Burgess, B.T.; Anderson, A.M.; McCorkle, J.R.; Wu, J.; Ueland, F.R.; Kolesar, J.M. Olaparib Combined with an ATR or Chk1 Inhibitor as a Treatment Strategy for Acquired Olaparib-Resistant BRCA1 Mutant Ovarian Cells. Diagnostics 2020, 10, 121. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef]
- Mahdi, H.; Hafez, N.; Doroshow, D.; Sohal, D.; Keedy, V.; Do, K.T.; LoRusso, P.; Jurgensmeier, J.; Avedissian, M.; Sklar, J.; et al. Ceralasertib-Mediated ATR Inhibition Combined with Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis. Oncol. 2021, 5, 1432–1442. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef]
- Wethington, S.L.; Shah, P.D.; Martin, L.P.; Tanyi, J.L.; Latif, N.A.; Morgan, M.A.; Torigian, D.A.; Pagan, C.; Rodriguez, D.; Domchek, S.M.; et al. Combination of PARP and ATR inhibitors (olaparib and ceralasertib) shows clinical activity in acquired PARP inhibitor-resistant recurrent ovarian cancer. J. Clin. Oncol. 2021, 39, 5516. [Google Scholar] [CrossRef]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2020, 11, 629266. [Google Scholar] [CrossRef]
- Molinaro, C.; Martoriati, A.; Cailliau, K. Proteins from the DNA Damage Response: Regulation, Dysfunction, and Anticancer Strategies. Cancers 2021, 13, 3819. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.A.; Zhao, R.; Glick, G.G.; Bansbach, C.E.; Friedman, D.B.; Cortez, D. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J. Biol. Chem. 2011, 286, 28707–28714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; McCorvie, T.J.; Yates, L.A.; Zhang, X. Structural basis of homologous recombination. Cell. Mol. Life Sci. 2020, 77, 3–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Jian Ma, C.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Acevedo, M.; Grace, L.; Teoh, D.; Whitaker, R.; Adams, D.J.; Jia, J.; Nixon, A.B.; Secord, A.A. Dasatinib (BMS-35482) potentiates the activity of gemcitabine and docetaxel in uterine leiomyosarcoma cell lines. Gynecol. Oncol. Res. Pract. 2014, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yao, Y.; He, H.; Shen, J. Clinical Interpretation of Sequence Variants. Curr. Protoc. Hum. Genet. 2020, 106, e98. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, B.; Umrigar, A.; Tsien, F. Chromosome preparation from cultured cells. J. Vis. Exp. 2014, 83, e50203. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.; Eustace, A.; Busschots, S.; Breen, L.; Clynes, M.; O’Donovan, N.; Stordal, B. In vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Cai, S.; Han, C.; Banerjee, A.; Wu, D.; Cui, T.; Xie, G.; Zhang, J.; Zhang, X.; McLaughlin, E.; et al. ALDH1A1 Contributes to PARP Inhibitor Resistance via Enhancing DNA Repair in BRCA2(-/-) Ovarian Cancer Cells. Mol. Cancer Ther. 2020, 19, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Stukova, M.; Hall, M.D.; Tsotsoros, S.D.; Madigan, J.P.; Farrell, N.P.; Gottesman, M.M. Reduced accumulation of platinum drugs is not observed in drug-resistant ovarian cancer cell lines derived from cisplatin-treated patients. J. Inorg. Biochem. 2015, 149, 45–48. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.K.; Cooke, S.L.; Howe, K.; Newman, S.; Xian, J.; Temple, J.; Batty, E.M.; Pole, J.C.; Langdon, S.P.; Edwards, P.A.; et al. The role of tandem duplicator phenotype in tumour evolution in high-grade serous ovarian cancer. J. Pathol. 2012, 226, 703–712. [Google Scholar] [CrossRef]
- Langdon, S.P.; Lawrie, S.S.; Hay, F.G.; Hawkes, M.M.; McDonald, A.; Hayward, I.P.; Schol, D.J.; Hilgers, J.; Leonard, R.C.; Smyth, J.F. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988, 48, 6166–6172. [Google Scholar]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef] [Green Version]
- Dickson, K.A.; Xie, T.; Evenhuis, C.; Ma, Y.; Marsh, D.J. PARP Inhibitors Display Differential Efficacy in Models of BRCA Mutant High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 8506. [Google Scholar] [CrossRef]
- Chiappa, M.; Guffanti, F.; Anselmi, M.; Lupi, M.; Panini, N.; Wiesmuller, L.; Damia, G. Combinations of ATR, Chk1 and Wee1 Inhibitors with Olaparib Are Active in Olaparib Resistant Brca1 Proficient and Deficient Murine Ovarian Cells. Cancers 2022, 14, 1807. [Google Scholar] [CrossRef] [PubMed]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.N.; Zethoven, M.; McInerny, S.; Morgan, J.A.; Rowley, S.M.; Lee, J.E.A.; Li, N.; Gorringe, K.L.; James, P.A.; Campbell, I.G. Exome sequencing of familial high-grade serous ovarian carcinoma reveals heterogeneity for rare candidate susceptibility genes. Nat. Commun. 2020, 11, 1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alenezi, W.M.; Fierheller, C.T.; Revil, T.; Serruya, C.; Mes-Masson, A.M.; Foulkes, W.D.; Provencher, D.; El Haffaf, Z.; Ragoussis, J.; Tonin, P.N. Case Review: Whole-Exome Sequencing Analyses Identify Carriers of a Known Likely Pathogenic Intronic BRCA1 Variant in Ovarian Cancer Cases Clinically Negative for Pathogenic BRCA1 and BRCA2 Variants. Genes 2022, 13, 697. [Google Scholar] [CrossRef]
- Kong, S.W.; Lee, I.H.; Liu, X.; Hirschhorn, J.N.; Mandl, K.D. Measuring coverage and accuracy of whole-exome sequencing in clinical context. Genet. Med. 2018, 20, 1617–1626. [Google Scholar] [CrossRef]
- Garziera, M.; Roncato, R.; Montico, M.; De Mattia, E.; Gagno, S.; Poletto, E.; Scalone, S.; Canzonieri, V.; Giorda, G.; Sorio, R.; et al. New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach. Cells 2019, 8, 584. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Dong, X.; Xie, S.; Zhang, L.; Zeng, P.; Zhang, L. Cellular Mechanism of Gene Mutations and Potential Therapeutic Targets in Ovarian Cancer. Cancer Manag. Res. 2021, 13, 3081–3100. [Google Scholar] [CrossRef]
- Carvalho, M.A.; Marsillac, S.M.; Karchin, R.; Manoukian, S.; Grist, S.; Swaby, R.F.; Urmenyi, T.P.; Rondinelli, E.; Silva, R.; Gayol, L.; et al. Determination of cancer risk associated with germ line BRCA1 missense variants by functional analysis. Cancer Res. 2007, 67, 1494–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, V.C.; Golubeva, V.A.; Di Pietro, G.; Shields, C.; Amankwah, K.; Nepomuceno, T.C.; de Gregoriis, G.; Abreu, R.B.V.; Harro, C.; Gomes, T.T.; et al. Impact of amino acid substitutions at secondary structures in the BRCT domains of the tumor suppressor BRCA1: Implications for clinical annotation. J. Biol. Chem. 2019, 294, 5980–5992. [Google Scholar] [CrossRef] [Green Version]
- Anantha, R.W.; Simhadri, S.; Foo, T.K.; Miao, S.; Liu, J.; Shen, Z.; Ganesan, S.; Xia, B. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. eLife 2017, 6, e21350. [Google Scholar] [CrossRef]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, E.; Bates, S.E. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer Ther. 2007, 7, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Leung-Pineda, V.; Ryan, C.E.; Piwnica-Worms, H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol. Cell Biol. 2006, 26, 7529–7538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, J.; Huang, T.T.; Murai, J.; Haynes, B.; Steeg, P.S.; Pommier, Y.; Lee, J.M. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 2020, 39, 5520–5535. [Google Scholar] [CrossRef]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Tumiati, M.; Hietanen, S.; Hynninen, J.; Pietila, E.; Farkkila, A.; Kaipio, K.; Roering, P.; Huhtinen, K.; Alkodsi, A.; Li, Y.; et al. A Functional Homologous Recombination Assay Predicts Primary Chemotherapy Response and Long-Term Survival in Ovarian Cancer Patients. Clin. Cancer Res. 2018, 24, 4482–4493. [Google Scholar] [CrossRef] [Green Version]
- Tamura, N.; Shaikh, N.; Muliaditan, D.; Soliman, T.N.; McGuinness, J.R.; Maniati, E.; Moralli, D.; Durin, M.A.; Green, C.M.; Balkwill, F.R.; et al. Specific Mechanisms of Chromosomal Instability Indicate Therapeutic Sensitivities in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2020, 80, 4946–4959. [Google Scholar] [CrossRef]
- Cope, L.; Wu, R.C.; Shih Ie, M.; Wang, T.L. High level of chromosomal aberration in ovarian cancer genome correlates with poor clinical outcome. Gynecol. Oncol. 2013, 128, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.C.; Birkbak, N.J.; Culhane, A.C.; Drapkin, R.; Fatima, A.; Tian, R.; Schwede, M.; Alsop, K.; Daniels, K.E.; Piao, H.; et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res. 2012, 18, 5806–5815. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gao, J.; Zhou, J.; Liu, H.; Xu, C. Olaparib induced senescence under P16 or P53 dependent manner in ovarian cancer. J. Gynecol. Oncol. 2019, 30, e26. [Google Scholar] [CrossRef] [PubMed]
- Vescarelli, E.; Gerini, G.; Megiorni, F.; Anastasiadou, E.; Pontecorvi, P.; Solito, L.; De Vitis, C.; Camero, S.; Marchetti, C.; Mancini, R.; et al. MiR-200c sensitizes Olaparib-resistant ovarian cancer cells by targeting Neuropilin 1. J. Exp. Clin. Cancer Res. 2020, 39, 3. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e12. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef]
- Del Campo, J.M.; Matulonis, U.A.; Malander, S.; Provencher, D.; Mahner, S.; Follana, P.; Waters, J.; Berek, J.S.; Woie, K.; Oza, A.M.; et al. Niraparib Maintenance Therapy in Patients With Recurrent Ovarian Cancer After a Partial Response to the Last Platinum-Based Chemotherapy in the ENGOT-OV16/NOVA Trial. J. Clin. Oncol. 2019, 37, 2968–2973. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.M.; McMellen, A.; Watson, Z.L.; Aguilera, J.; Ferguson, R.; Nurmemmedov, E.; Thakar, T.; Moldovan, G.L.; Kim, H.; Cittelly, D.M.; et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Mol. Carcinog. 2019, 58, 1770–1782. [Google Scholar] [CrossRef]
- Lin, Z.P.; Al Zouabi, N.N.; Xu, M.L.; Bowen, N.E.; Wu, T.L.; Lavi, E.S.; Huang, P.H.; Zhu, Y.L.; Kim, B.; Ratner, E.S. In silico screening identifies a novel small molecule inhibitor that counteracts PARP inhibitor resistance in ovarian cancer. Sci. Rep. 2021, 11, 8042. [Google Scholar] [CrossRef]
- Ware, M.D.; DeSilva, D.; Sinilnikova, O.M.; Stoppa-Lyonnet, D.; Tavtigian, S.V.; Mazoyer, S. Does nonsense-mediated mRNA decay explain the ovarian cancer cluster region of the BRCA2 gene? Oncogene 2006, 25, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, K.A.; Ali, R.; Doherty, R.; Toss, M.S.; Miligy, I.; Alblihy, A.; Dorjsuren, D.; Simeonov, A.; Jadhav, A.; Wilson, D.M., 3rd; et al. FEN1 Blockade for Platinum Chemo-Sensitization and Synthetic Lethality in Epithelial Ovarian Cancers. Cancers 2021, 13, 1866. [Google Scholar] [CrossRef]
- Stordal, B.; Timms, K.; Farrelly, A.; Gallagher, D.; Busschots, S.; Renaud, M.; Thery, J.; Williams, D.; Potter, J.; Tran, T.; et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol. Oncol. 2013, 7, 567–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciucci, A.; Buttarelli, M.; Fagotti, A.; Scambia, G.; Gallo, D. Preclinical models of epithelial ovarian cancer: Practical considerations and challenges for a meaningful application. Cell. Mol. Life Sci. 2022, 79, 364. [Google Scholar] [CrossRef]
- Jimenez-Sainz, J.; Krysztofiak, A.; Garbarino, J.; Rogers, F.; Jensen, R.B. The Pathogenic R3052W BRCA2 Variant Disrupts Homology-Directed Repair by Failing to Localize to the Nucleus. Front. Genet. 2022, 13, 884210. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Elattar, A.; Cerbinskaite, A.; Wilkinson, S.J.; Drew, Y.; Kyle, S.; Los, G.; Hostomsky, Z.; Edmondson, R.J.; Curtin, N.J. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin. Cancer Res. 2010, 16, 2344–2351. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Ma, J.; Wu, J.; Ye, L.; Cai, H.; Xia, B.; Yu, X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Hunt, C.R.; Chakraborty, S.; Pandita, R.K.; Yordy, J.; Ramnarain, D.B.; Horikoshi, N.; Pandita, T.K. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat. Res. 2014, 181, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahassi, E.M.; Ovesen, J.L.; Riesenberg, A.L.; Bernstein, W.Z.; Hasty, P.E.; Stambrook, P.J. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 2008, 27, 3977–3985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanaswamy, P.B.; Tkachuk, S.; Haller, H.; Dumler, I.; Kiyan, Y. CHK1 and RAD51 activation after DNA damage is regulated via urokinase receptor/TLR4 signaling. Cell Death Dis. 2016, 7, e2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foo, T.K.; Vincelli, G.; Huselid, E.; Her, J.; Zheng, H.; Simhadri, S.; Wang, M.; Huo, Y.; Li, T.; Yu, X.; et al. ATR/ATM-Mediated Phosphorylation of BRCA1 T1394 Promotes Homologous Recombinational Repair and G2-M Checkpoint Maintenance. Cancer Res. 2021, 81, 4676–4684. [Google Scholar] [CrossRef]
- Brill, E.; Yokoyama, T.; Nair, J.; Yu, M.; Ahn, Y.R.; Lee, J.M. Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high-grade serous ovarian cancer. Oncotarget 2017, 8, 111026–111040. [Google Scholar] [CrossRef] [PubMed]
- Pillay, N.; Tighe, A.; Nelson, L.; Littler, S.; Coulson-Gilmer, C.; Bah, N.; Golder, A.; Bakker, B.; Spierings, D.C.J.; James, D.I.; et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019, 35, 519–533.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.G.; Wang, Z.Q.; Tong, W.M.; Shen, Y. PARP1 Val762Ala polymorphism reduces enzymatic activity. Biochem. Biophys. Res. Commun. 2007, 354, 122–126. [Google Scholar] [CrossRef]
- Yang, G.; Liu, C.; Chen, S.H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef] [Green Version]
- Slade, D. PARP and PARG inhibitors in cancer treatment. Genes Dev. 2020, 34, 360–394. [Google Scholar] [CrossRef] [Green Version]
- Heikkinen, K.; Mansikka, V.; Karppinen, S.M.; Rapakko, K.; Winqvist, R. Mutation analysis of the ATR gene in breast and ovarian cancer families. Breast Cancer Res. 2005, 7, R495–R501. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Ma, S.; Tian, Y.; Zhang, X.; Lv, W.; Zhai, H. Computational studies on the binding mechanism between triazolone inhibitors and Chk1 by molecular docking and molecular dynamics. Mol. Biosyst. 2015, 11, 275–286. [Google Scholar] [CrossRef]
- Kelley, S.R.; Kamal, T.J.; Molitch, M.E. Mechanism of verapamil calcium channel blockade-induced hyperprolactinemia. Am. J. Physiol. 1996, 270, E96–E100. [Google Scholar] [CrossRef]
- Kannan, P.; Telu, S.; Shukla, S.; Ambudkar, S.V.; Pike, V.W.; Halldin, C.; Gottesman, M.M.; Innis, R.B.; Hall, M.D. The “specific” P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem. Neurosci. 2011, 2, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMullen, M.; Karakasis, K.; Loembe, B.; Dean, E.; Parr, G.; Oza, A.M. DUETTE: A phase II randomized, multicenter study to investigate the efficacy and tolerability of a second maintenance treatment in patients with platinum-sensitive relapsed epithelial ovarian cancer, who have previously received poly(ADP-ribose) polymerase (PARP) inhibitor maintenance treatment. Int. J. Gynecol. Cancer 2020, 30, 1824–1828. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, S.; Ikeda, K.; Horie-Inoue, K.; Sato, S.; Takeda, S.; Hasegawa, K.; Inoue, S. Identification of novel mutations of ovarian cancer-related genes from RNA-sequencing data for Japanese epithelial ovarian cancer patients. Endocr. J. 2020, 67, 219–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Ma, K.; Liu, X.; Chen, S.H.; Li, P.; Yu, Y.; Leung, A.K.L.; Yu, X. Truncated PARP1 mediates ADP-ribosylation of RNA polymerase III for apoptosis. Cell Discov. 2022, 8, 3. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Mutation | Gene Region | Amino Acid Change | Type of Mutation | Functional Impact | Computed Pathogenicity | CADD Score | Allele Fraction [% of Total Reads] | Maximal Population Allele Frequency | |
|---|---|---|---|---|---|---|---|---|---|---|
| PEO1 | PEO1-OR | |||||||||
| TP53 | c.731G > A | CDS | p.G244D | M | loss | pathogenic | 27 | 100% | 100% | 0% |
| BRCA1 | c.4837A > G | CDS | p.S1613G | M | normal | benign (!) | <10 | 100% | 100% | 49% (South Asian) |
| c.2082C > T | CDS | p.S694S | S | normal | benign | <10 | 100% | 100% | 49% (South Asian) | |
| c.2311T > C | CDS | p.L771L | S | normal | benign | <10 | 100% | 100% | 49% (South Asian) | |
| c.2612C > T | CDS | p.P871L | M | normal | benign (!) | 18 | 100% | 100% | 81% (African) | |
| c.3113A > G | CDS | p.E1038G | M | normal | benign (!) | 14 | 100% | 100% | 49% (South Asian) | |
| c.3548A > G | CDS | p.K1183R | M | normal | benign (!) | <10 | 100% | 100% | 49% (South Asian) | |
| c.4308T > C | CDS | p.S1436S | S | normal | benign | <10 | 100% | 100% | 49% (South Asian) | |
| c.-1074C > G | 5′ UTR | – | – | normal | benign | <10 | 100% | 100% | 82% (African) | |
| c.-134T > C | 5′ UTR | – | – | normal | benign | <10 | 100% | 100% | 49% (South Asian) | |
| BRCA2 | c.4965C > G | CDS | p.Y1655 * | STOP | loss | pathogenic | 33 | 100% | 100% | 0.008% (European) |
| c.4964A > T | CDS | p.Y1655F | M | normal | uncertain | <10 | 34% | 94% | 0% | |
| c.3807T > C | CDS | p.V1269V | S | normal | benign | <10 | 100% | 100% | 19% (African) | |
| c.4563A > G | CDS | p.L1521L | S | normal | benign | <10 | 100% | 100% | 100% (Jewish) | |
| c.6513G > C | CDS | p.V2171V | S | normal | benign | <10 | 100% | 100% | 100% (Jewish) | |
| c.7397T > C | CDS | p.V2466A | M | normal | benign | <10 | 100% | 100% | 100% (Jewish) | |
| c.* 105A > C | 3′ UTR | – | – | normal | benign | <10 | 100% | 100% | 23% (South Asian) | |
| PARP1 | c.2285T > C | CDS | p.V762A | M | loss | benign (!) | 27 | 100% | 100% | 43% (East Asian) |
| c.243C > T | CDS | p.D81D | S | normal | benign | 12 | 100% | 100% | 43% (East Asian) | |
| c.852T > C | CDS | p.A284A | S | normal | benign | <10 | 100% | 100% | 81% (East Asian) | |
| c.-17G > C | 5′ UTR | – | – | normal | benign | 12 | 100% | 100% | 43% (East Asian) | |
| ATR | c.632T > C | CDS | p.M211T | M | gain | benign (!) | 14 | 32% | 34% | 73% (African) |
| c.1776T > A | CDS | p.G592G | S | normal | benign | <10 | 31% | 31% | 79% (African) | |
| c.1815T > C | CDS | p.D605D | S | normal | benign (!) | <10 | 32% | 31% | 44% (Jewish) | |
| c.5208T > C | CDS | p.Y1736Y | S | normal | benign (!) | <10 | 29% | 30% | 45% (Jewish) | |
| c.7875G > A | CDS | p.Q2625Q | S | normal | benign | <10 | 32% | 30% | 97% (African) | |
| CHEK1 | c.1411A > G | CDS | p.I471V | M | gain | benign | 14 | 100% | 100% | 99.98% (East Asian) |
| c.* 28–3033C > G | 3′ UTR | – | – | normal | benign | <10 | 53% | 59% (LQ) | 42% (South Asian) | |
| ABCB1 | c.210A > G | CDS | p.G70G | S | normal | benign | <10 | 100% | 100% | 100% (East Asian) |
| H2AX | c.-1420G > A | 5′ UTR | – | – | normal | benign | <10 | 53% | 35% (LQ) | 64% (East Asian) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biegała, Ł.; Gajek, A.; Marczak, A.; Rogalska, A. Olaparib-Resistant BRCA2MUT Ovarian Cancer Cells with Restored BRCA2 Abrogate Olaparib-Induced DNA Damage and G2/M Arrest Controlled by the ATR/CHK1 Pathway for Survival. Cells 2023, 12, 1038. https://doi.org/10.3390/cells12071038
Biegała Ł, Gajek A, Marczak A, Rogalska A. Olaparib-Resistant BRCA2MUT Ovarian Cancer Cells with Restored BRCA2 Abrogate Olaparib-Induced DNA Damage and G2/M Arrest Controlled by the ATR/CHK1 Pathway for Survival. Cells. 2023; 12(7):1038. https://doi.org/10.3390/cells12071038
Chicago/Turabian StyleBiegała, Łukasz, Arkadiusz Gajek, Agnieszka Marczak, and Aneta Rogalska. 2023. "Olaparib-Resistant BRCA2MUT Ovarian Cancer Cells with Restored BRCA2 Abrogate Olaparib-Induced DNA Damage and G2/M Arrest Controlled by the ATR/CHK1 Pathway for Survival" Cells 12, no. 7: 1038. https://doi.org/10.3390/cells12071038