
Gene Suppression Therapies in Hereditary Cerebellar Ataxias: A Systematic Review of Animal Studies

Abstract
:1. Introduction
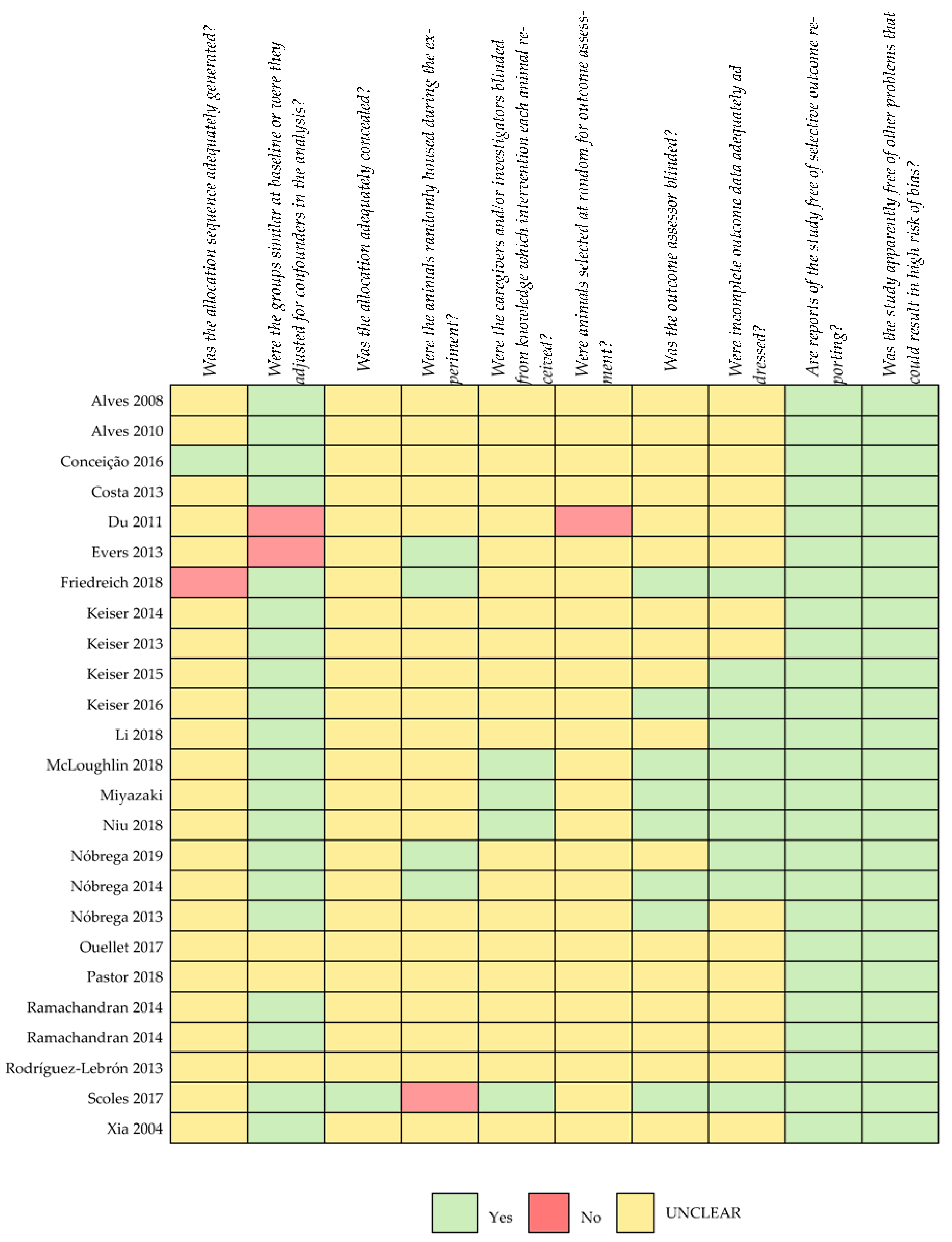
2. Materials and Methods
3. Results
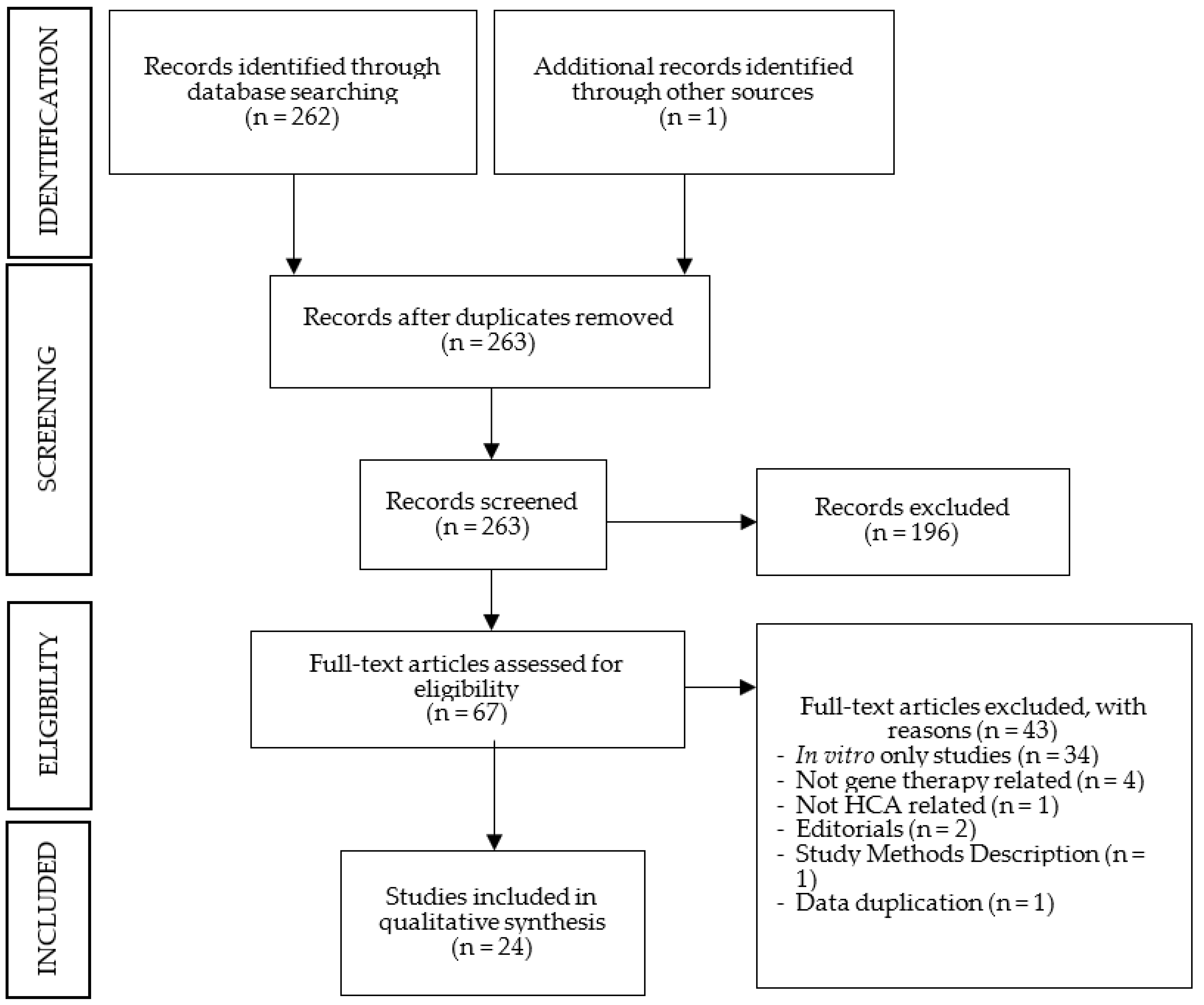
3.1. Search Outcome
3.2. Machado–Joseph Disease

| Author | Year | GST | Key Conclusions |
|---|---|---|---|
| Alves, S., et al. | 2008 | iRNA | The allele-specific silencing of mutant ataxin-3 was effective and selective in vivo and decreased the MJD-associated neuropathological phenotype. |
| Alves, S., et al. | 2010 | iRNA | WT ataxin-3 did not reduce the toxicity of mutant ataxin-3; WT overexpression did not protect against MJD neuropathology, and the knockdown of WT did not affect MJD neuropathology. The non-allele-specific silencing of ataxin-3 reduced neuropathology. |
| Rodríguez-Lebrón, E., et al. | 2012 | iRNA | iRNA was effective in suppressing ATXN3. Administration in a pre-symptomatic mouse model prevented the development of the neuropathological features and motor impairments found in the control group. |
| Costa, Mdo C., et al. | 2013 | iRNA | Despite the fact that iRNA was effective in suppressing ATXN3, at the end of the study, at 48 weeks of age, no improvement in motor impairment was detected; the authors suggested that the motor phenotype might not be solely due to cerebellar dysfunction or intervention was performed later than ideal. No adverse effects were detected. No differences in lifespan were detected between groups. |
| Nóbrega, C., et al. | 2013 | iRNA | iRNA proved to be effective in suppressing ATXN3. Its administration after symptom onset prevented the development of MJD-associated motor-behaviour and neuropathological abnormalities. |
| Nóbrega, C., et al. | 2014 | iRNA | The effective gene silencing of ATXN3 in pre-symptomatic mice led to the clearance of mutant ataxin-3 from neuronal nuclei and prevented the development of motor impairments. There were no differences between groups concerning glial or astrocytic activation. |
| Conceição, S., et al. | 2016 | iRNA | Intravenous administration was successful in crossing the BBB. iRNA was effective in mutant ataxin-3 knockdown in vivo. iRNA improved motor performance and recovered striatal- and cerebellar-associated neuropathology. No signs of toxicity were detected. |
| Li, Y.X., et al. | 2018 | iRNA | The downregulation of Relish expression in astrocytes delayed neurodegeneration and extended the lifespan in the SCA3 fly model. |
| Nóbrega, C., et al. | 2018 | iRNA | While evidence of neuronal dysfunction and gliosis was present at initial timepoints, 20 weeks post-injection, no differences between groups were found. No off-target effects or saturation of the endogenous iRNA processing machinery in the mouse striatum were detected. |
| Evers, M.M., et al. | 2013 | ASOs | The intracerebral injection of ASOs was effective in skipping targeted exons. No overt toxicity was observed in vivo. |
| McLoughlin, H.S., et al. | 2018 | ASOs | ASOs achieved the efficient silencing of mutant ATXN3 and prevented the nuclear accumulation of ataxin-3 protein. Administration in post-symptomatic mice fully recovered locomotor activity. No signs of an adverse immune response to treatment were detected. |
3.2.1. Suppression Efficacy
3.2.2. Neuropathology
3.2.3. Motor Behaviour
3.2.4. Wild-Type Protein
3.2.5. Safety Profile
3.3. Spinocerebellar Ataxia Type 1
| Authors | Year | GST | Key Conclusions |
|---|---|---|---|
| Xia, H., et al. | 2004 | iRNA | iRNA reduced ataxin-1 transcript levels, resulting in improved motor coordination, restored cerebellar morphology and resolved characteristic ataxin-1 inclusions in the Purkinje cells of SCA1 mice. |
| Keiser, M.S., et al. | 2013 | iRNA | The silencing of mutant ataxin-1 using miRNAs recovered behavioural deficits and improved neuropathology. It is suggested that behavioural recovery does not require the full recovery of all neuropathological aspects. |
| Keiser, M.S., et al. | 2014 | iRNA | Reduced ataxin-1 transcript and protein levels without overt neurotoxicity were achieved. It preserved cerebellar lobule integrity for over a year and preserved rotarod performance for months (30 w). |
| Keiser, M.S., et al. | 2015 | iRNA | Anti-ataxin-1 iRNA resulted in the reduction in ATXN1 mRNA, and no signs of toxicity were detected. |
| Keiser, M.S., et al. | 2016 | iRNA | The iRNA-mediated suppression of ataxin-1 mRNA altered disease progression, reversed motor symptoms and normalized cerebellar pathology when delivered before and after symptom onset. |
| Friedrich, J., et al. | 2018 | ASOs | Following a single ASO injection at 5 weeks of age, ataxin-1 transcripts remained reduced until 18 w, but ataxin-1 protein only remained at reduced levels in pons at 18 w. Nonetheless, mice demonstrated recover of neuropathological and motor behaviour phenotypes. |
3.3.1. Suppression Efficacy
3.3.2. Neuropathology
3.3.3. Motor Behaviour
3.3.4. Safety Profile
3.4. Spinocerebellar Ataxia Type 7
| Authors | Year | GST | Key Conclusions |
|---|---|---|---|
| Ramachandran, P.A.S., et al. | 2014 | iRNA | A sustained reduction in ataxin-7 expression led to significant and robust improvements in the ataxic and neuropathological phenotypes as well as delayed disease onset in SCA7 mice. No significant adverse effects were present. |
| Ramachandran, P.A.S., et al. | 2014 | iRNA | Preservation of normal retinal function 23 weeks post-retinal injection and no adverse toxicity with reduction in ataxin-7 transcript levels were reported. |
| Niu, C., et al. | 2018 | ASOs | ASOs were effective in suppressing mutant ataxin-7 transcript and protein levels; visual function was improved despite initiating treatment after symptom onset. At the end of the study, ataxin-7 ASOs only ameliorated rod photoreceptor function. CAG repeat-targeting ASOs were less effective than Ataxin-7 ASOs. |
3.4.1. Suppression Efficacy
3.4.2. Neuropathology
3.4.3. Motor Behaviour
3.4.4. Retinal Degeneration
3.4.5. Safety Profile
3.5. Other HCAs
| Authors | Year | HCA | GST | Key Conclusions |
|---|---|---|---|---|
| Scoles, D.R., et al. | 2017 | SCA2 | ASOs | The intracerebral injection of ASO led to reduced ataxin-2 transcript and protein levels, resulting in delayed onset of SCA2 motor and neuropathological phenotypes without microglial activation. |
| Pastor, P.D.H., et al. | 2018 | SCA6 | iRNA | iRNA selectively inhibited alpha-1ACT mutant protein and kept alpha-1A normal levels. It also prevented the development of motor and morphological abnormalities. |
| Ouellet, D.L., et al. | 2016 | FRDA | CRISPr/Cas9 | CRISPR-Cas9 technology resulted in the excision of the GAA repeat in intron 1 using electroporation. |
| Du, L., et al. | 2011 | A-T | ASOs | Splicing correction efficiency of AMOs conjugated with CPPs was demonstrated in vitro. In vivo, systemic administration revealed efficient brain uptake, particularly in PC, without apparent signs of toxicity. |
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jayadev, S.; Bird, T.D. Hereditary ataxias: Overview. Genet. Med. Off. J. Am. Coll. Med. Genet. 2013, 15, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Durr, A.; Cossee, M.; Agid, Y.; Campuzano, V.; Mignard, C.; Penet, C.; Mandel, J.L.; Brice, A.; Koenig, M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef]
- Perlman, S.L.; Boder Deceased, E.; Sedgewick, R.P.; Gatti, R.A. Ataxia-telangiectasia. Handb. Clin. Neurol. 2012, 103, 307–332. [Google Scholar] [CrossRef]
- Kuo, S.H. Ataxia. Continuum 2019, 25, 1036–1054. [Google Scholar] [CrossRef]
- Jacobi, H.; Bauer, P.; Giunti, P.; Labrum, R.; Sweeney, M.G.; Charles, P.; Durr, A.; Marelli, C.; Globas, C.; Linnemann, C.; et al. The natural history of spinocerebellar ataxia type 1, 2, 3, and 6: A 2-year follow-up study. Neurology 2011, 77, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Gertz, M.A.; Mauermann, M.L.; Grogan, M.; Coelho, T. Advances in the treatment of hereditary transthyretin amyloidosis: A review. Brain Behav. 2019, 9, e01371. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Tabrizi, S.J. Gene suppression approaches to neurodegeneration. Alzheimer’s Res. Ther. 2017, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Han, H. RNA Interference to Knock Down Gene Expression. Methods Mol. Biol. 2018, 1706, 293–302. [Google Scholar] [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Gheibi-Hayat, S.M.; Jamialahmadi, K. Antisense Oligonucleotide (AS-ODN) Technology: Principle, Mechanism and Challenges. Biotechnol. Appl. Biochem. 2021, 68, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw, M.; Zawdie, B. Mechanism and Applications of CRISPR/Cas-9-Mediated Genome Editing. Biol. Targets Ther. 2021, 15, 353–361. [Google Scholar] [CrossRef]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Zhang, L.; Huang, X. Genome modification by CRISPR/Cas9. FEBS J. 2014, 281, 5186–5193. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Tang, K.; Wang, Q.; Estill, J.; Yao, L.; Wang, X.; Chen, Y.; Yang, K. The use of GRADE approach in systematic reviews of animal studies. J. Evid. Based Med. 2016, 9, 98–104. [Google Scholar] [CrossRef]
- Alves, S.; Nascimento-Ferreira, I.; Auregan, G.; Hassig, R.; Dufour, N.; Brouillet, E.; Pedroso de Lima, M.C.; Hantraye, P.; Pereira de Almeida, L.; Deglon, N. Allele-specific RNA silencing of mutant ataxin-3 mediates neuroprotection in a rat model of Machado-Joseph disease. PLoS ONE 2008, 3, e3341. [Google Scholar] [CrossRef]
- Alves, S.; Nascimento-Ferreira, I.; Dufour, N.; Hassig, R.; Auregan, G.; Nobrega, C.; Brouillet, E.; Hantraye, P.; Pedroso de Lima, M.C.; Deglon, N.; et al. Silencing ataxin-3 mitigates degeneration in a rat model of Machado-Joseph disease: No role for wild-type ataxin-3? Hum. Mol. Genet. 2010, 19, 2380–2394. [Google Scholar] [CrossRef] [Green Version]
- Conceicao, M.; Mendonca, L.; Nobrega, C.; Gomes, C.; Costa, P.; Hirai, H.; Moreira, J.N.; Lima, M.C.; Manjunath, N.; Pereira de Almeida, L. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials 2016, 82, 124–137. [Google Scholar] [CrossRef]
- Costa Mdo, C.; Luna-Cancalon, K.; Fischer, S.; Ashraf, N.S.; Ouyang, M.; Dharia, R.M.; Martin-Fishman, L.; Yang, Y.; Shakkottai, V.G.; Davidson, B.L.; et al. Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1898–1908. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.M.; Tran, H.D.; Zalachoras, I.; Pepers, B.A.; Meijer, O.C.; den Dunnen, J.T.; van Ommen, G.J.; Aartsma-Rus, A.; van Roon-Mom, W.M. Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: Removal of the CAG containing exon. Neurobiol. Dis. 2013, 58, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.X.; Sibon, O.C.M.; Dijkers, P.F. Inhibition of NF-kappaB in astrocytes is sufficient to delay neurodegeneration induced by proteotoxicity in neurons. J. Neuroinflamm. 2018, 15, 261. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, C.; Codesso, J.M.; Mendonca, L.; Pereira de Almeida, L. RNA Interference Therapy for Machado-Joseph Disease: Long-Term Safety Profile of Lentiviral Vectors Encoding Short Hairpin RNAs Targeting Mutant Ataxin-3. Hum. Gene Ther. 2019, 30, 841–854. [Google Scholar] [CrossRef]
- Nobrega, C.; Nascimento-Ferreira, I.; Onofre, I.; Albuquerque, D.; Deglon, N.; de Almeida, L.P. RNA interference mitigates motor and neuropathological deficits in a cerebellar mouse model of Machado-Joseph disease. PLoS ONE 2014, 9, e100086. [Google Scholar] [CrossRef] [Green Version]
- Nobrega, C.; Nascimento-Ferreira, I.; Onofre, I.; Albuquerque, D.; Hirai, H.; Deglon, N.; de Almeida, L.P. Silencing mutant ataxin-3 rescues motor deficits and neuropathology in Machado-Joseph disease transgenic mice. PLoS ONE 2013, 8, e52396. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lebron, E.; Costa Mdo, C.; Luna-Cancalon, K.; Peron, T.M.; Fischer, S.; Boudreau, R.L.; Davidson, B.L.; Paulson, H.L. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1909–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, J.; Kordasiewicz, H.B.; O’Callaghan, B.; Handler, H.P.; Wagener, C.; Duvick, L.; Swayze, E.E.; Rainwater, O.; Hofstra, B.; Benneyworth, M.; et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 2018, 3, e123193. [Google Scholar] [CrossRef]
- Keiser, M.S.; Boudreau, R.L.; Davidson, B.L. Broad therapeutic benefit after RNAi expression vector delivery to deep cerebellar nuclei: Implications for spinocerebellar ataxia type 1 therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 588–595. [Google Scholar] [CrossRef] [Green Version]
- Keiser, M.S.; Geoghegan, J.C.; Boudreau, R.L.; Lennox, K.A.; Davidson, B.L. RNAi or overexpression: Alternative therapies for Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2013, 56, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Keiser, M.S.; Kordower, J.H.; Gonzalez-Alegre, P.; Davidson, B.L. Broad distribution of ataxin 1 silencing in rhesus cerebella for spinocerebellar ataxia type 1 therapy. Brain J. Neurol. 2015, 138, 3555–3566. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.S.; Monteys, A.M.; Corbau, R.; Gonzalez-Alegre, P.; Davidson, B.L. RNAi prevents and reverses phenotypes induced by mutant human ataxin-1. Ann. Neurol. 2016, 80, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Mao, Q.; Eliason, S.L.; Harper, S.Q.; Martins, I.H.; Orr, H.T.; Paulson, H.L.; Yang, L.; Kotin, R.M.; Davidson, B.L. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat. Med. 2004, 10, 816–820. [Google Scholar] [CrossRef]
- Niu, C.; Prakash, T.P.; Kim, A.; Quach, J.L.; Huryn, L.A.; Yang, Y.; Lopez, E.; Jazayeri, A.; Hung, G.; Sopher, B.L.; et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci. Transl. Med. 2018, 10, eaap8677. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, P.S.; Bhattarai, S.; Singh, P.; Boudreau, R.L.; Thompson, S.; Laspada, A.R.; Drack, A.V.; Davidson, B.L. RNA interference-based therapy for spinocerebellar ataxia type 7 retinal degeneration. PLoS ONE 2014, 9, e95362. [Google Scholar] [CrossRef]
- Ramachandran, P.S.; Boudreau, R.L.; Schaefer, K.A.; La Spada, A.R.; Davidson, B.L. Nonallele specific silencing of ataxin-7 improves disease phenotypes in a mouse model of SCA7. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 1635–1642. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Kayali, R.; Bertoni, C.; Fike, F.; Hu, H.; Iversen, P.L.; Gatti, R.A. Arginine-rich cell-penetrating peptide dramatically enhances AMO-mediated ATM aberrant splicing correction and enables delivery to brain and cerebellum. Hum. Mol. Genet. 2011, 20, 3151–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouellet, D.L.; Cherif, K.; Rousseau, J.; Tremblay, J.P. Deletion of the GAA repeats from the human frataxin gene using the CRISPR-Cas9 system in YG8R-derived cells and mouse models of Friedreich ataxia. Gene Ther. 2017, 24, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Pastor, P.D.H.; Du, X.; Fazal, S.; Davies, A.N.; Gomez, C.M. Targeting the CACNA1A IRES as a Treatment for Spinocerebellar Ataxia Type 6. Cerebellum 2018, 17, 72–77. [Google Scholar] [CrossRef]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef]
- Vazquez-Mojena, Y.; Leon-Arcia, K.; Gonzalez-Zaldivar, Y.; Rodriguez-Labrada, R.; Velazquez-Perez, L. Gene Therapy for Polyglutamine Spinocerebellar Ataxias: Advances, Challenges, and Perspectives. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 2731–2744. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US) Institute for Laboratory Animal Research. Overview. In Guidance for the Description of Animal Research in Sci-Entific Publications; National Academies Press: Washington, DC, USA, 2011. Available online: https://www.ncbi.nlm.nih.gov/books/NBK84209/ (accessed on 22 March 2023).


{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, C.; Malheiro, S.; Correia, M.; Damásio, J. Gene Suppression Therapies in Hereditary Cerebellar Ataxias: A Systematic Review of Animal Studies. Cells 2023, 12, 1037. https://doi.org/10.3390/cells12071037
Santos C, Malheiro S, Correia M, Damásio J. Gene Suppression Therapies in Hereditary Cerebellar Ataxias: A Systematic Review of Animal Studies. Cells. 2023; 12(7):1037. https://doi.org/10.3390/cells12071037
Chicago/Turabian StyleSantos, Carolina, Sofia Malheiro, Manuel Correia, and Joana Damásio. 2023. "Gene Suppression Therapies in Hereditary Cerebellar Ataxias: A Systematic Review of Animal Studies" Cells 12, no. 7: 1037. https://doi.org/10.3390/cells12071037