
Regulation of the Innate Immune System as a Therapeutic Approach to Supporting Respiratory Function in ALS
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
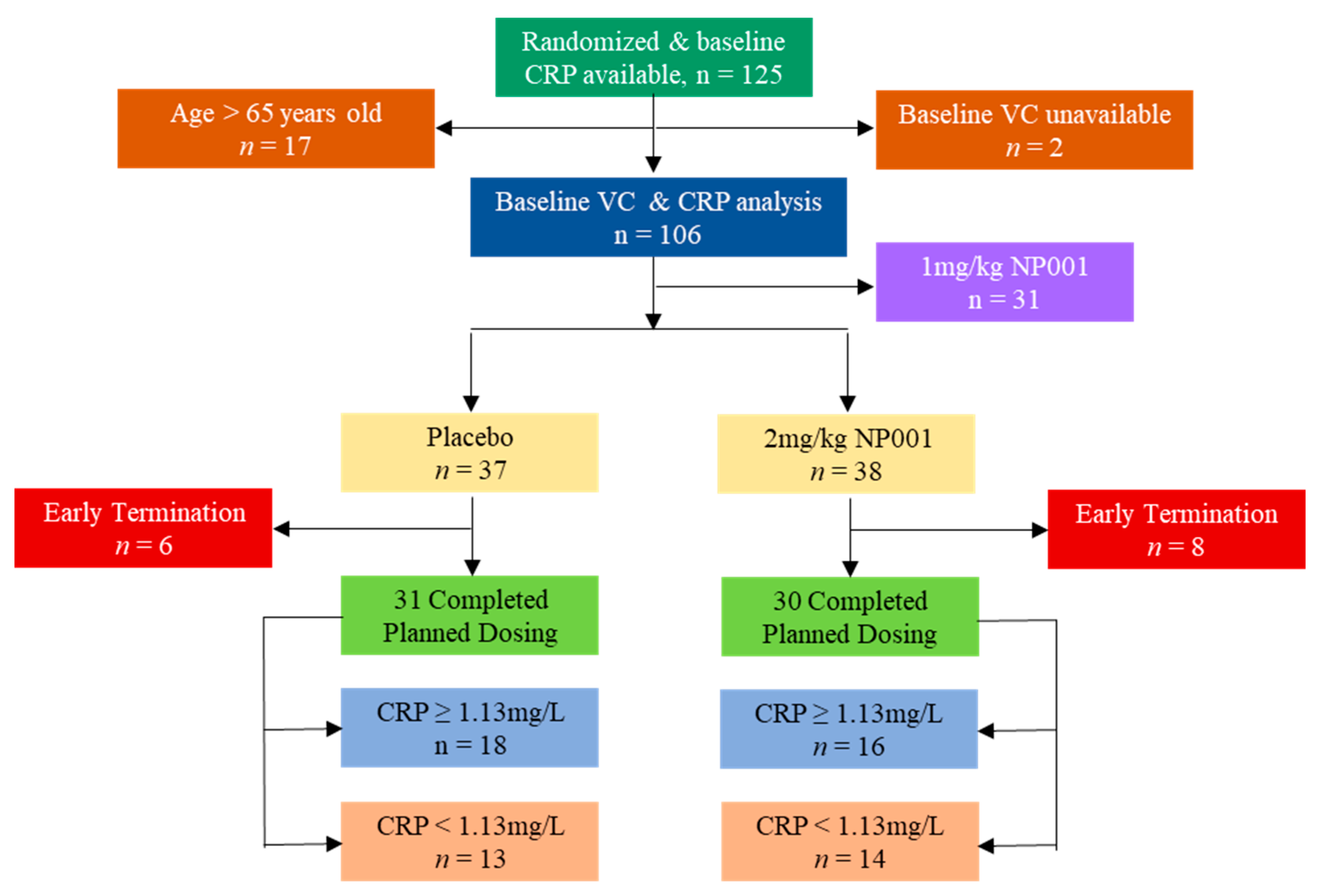
2.1. Description of ALS Phase 2A Trial and Participants
2.2. Analysis of Clinical Outcome Data
2.3. Plasma Factors Evaluated
2.4. Statistical Analyses
3. Results
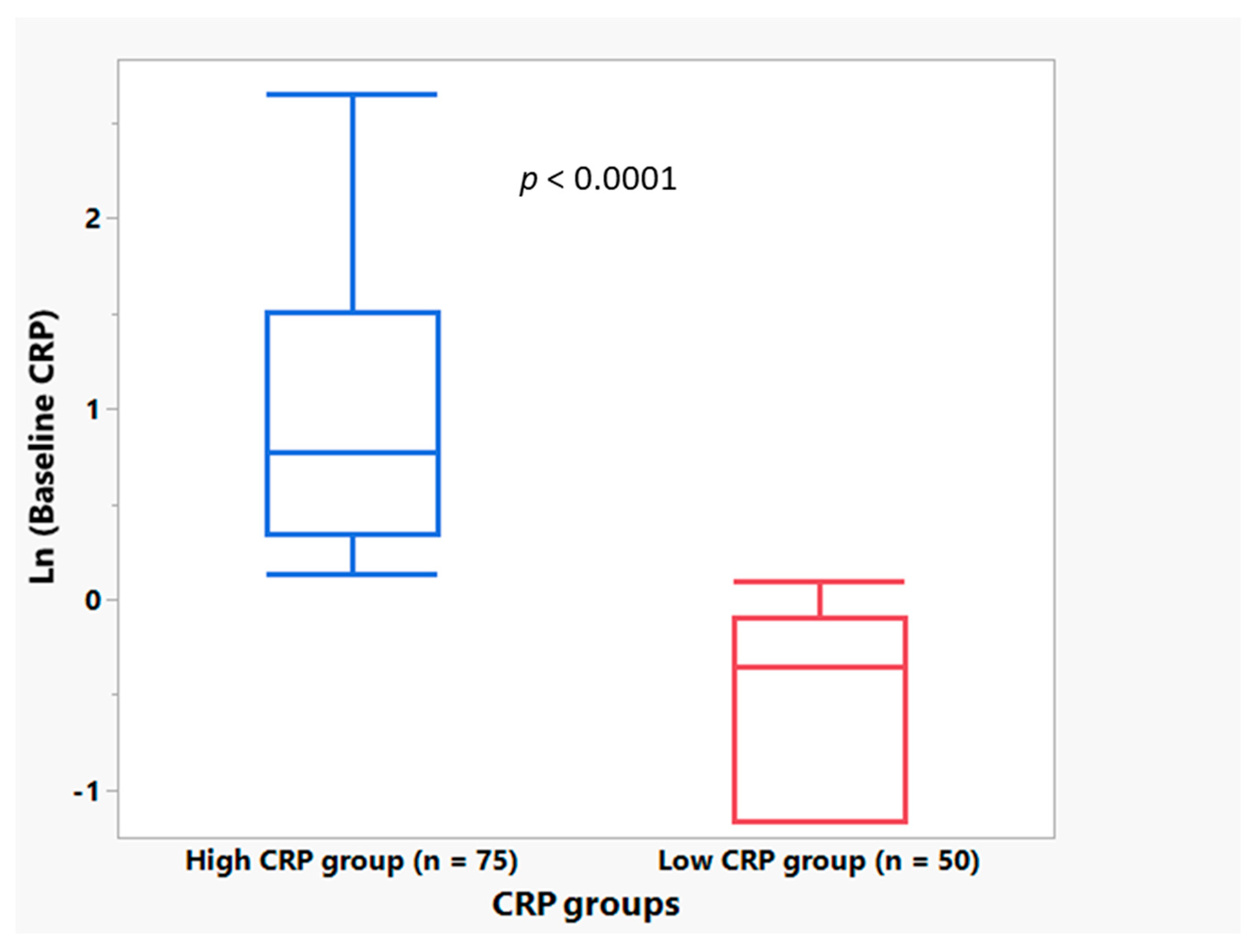
3.1. Plasma CRP Level Demographics
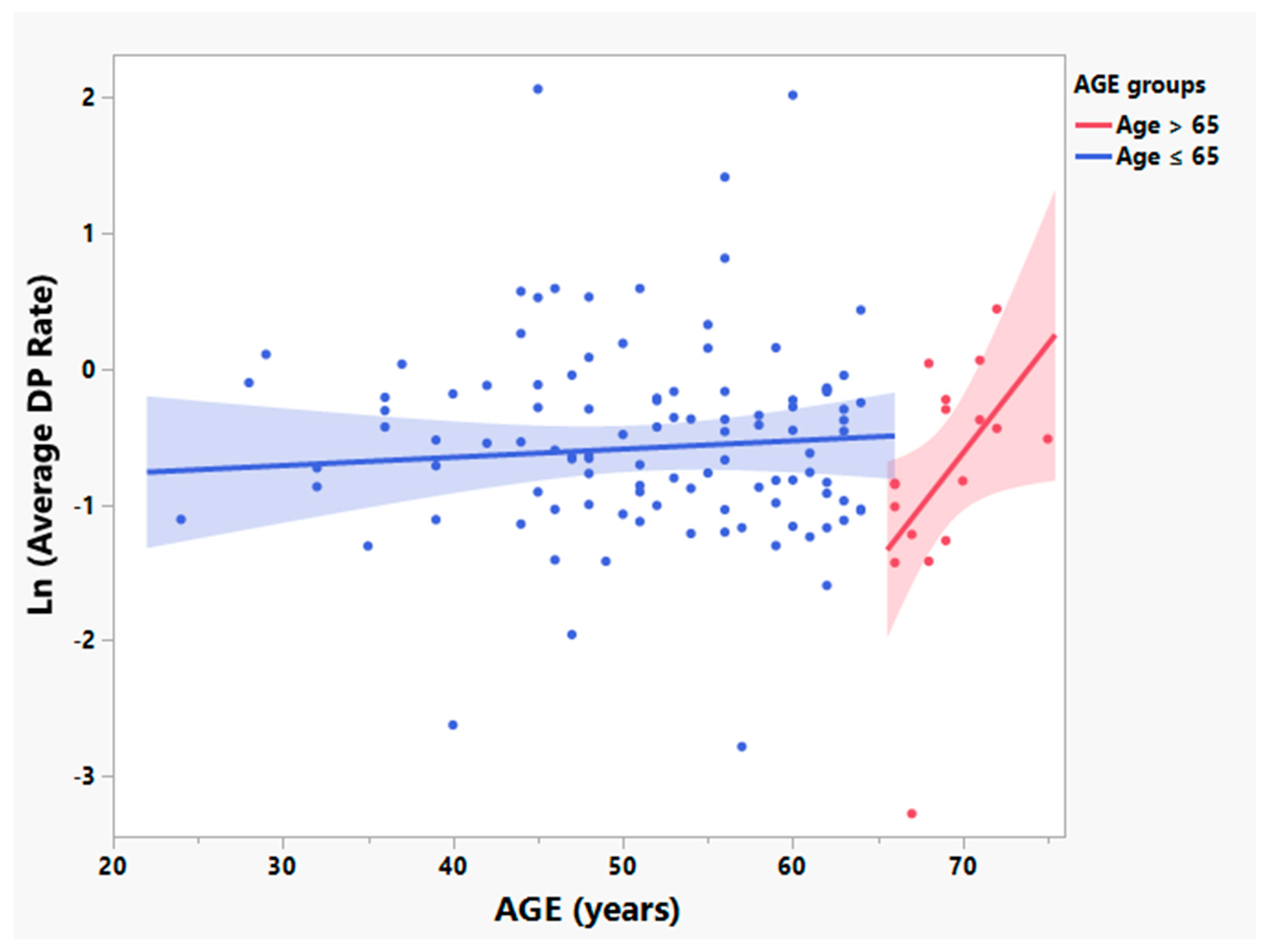
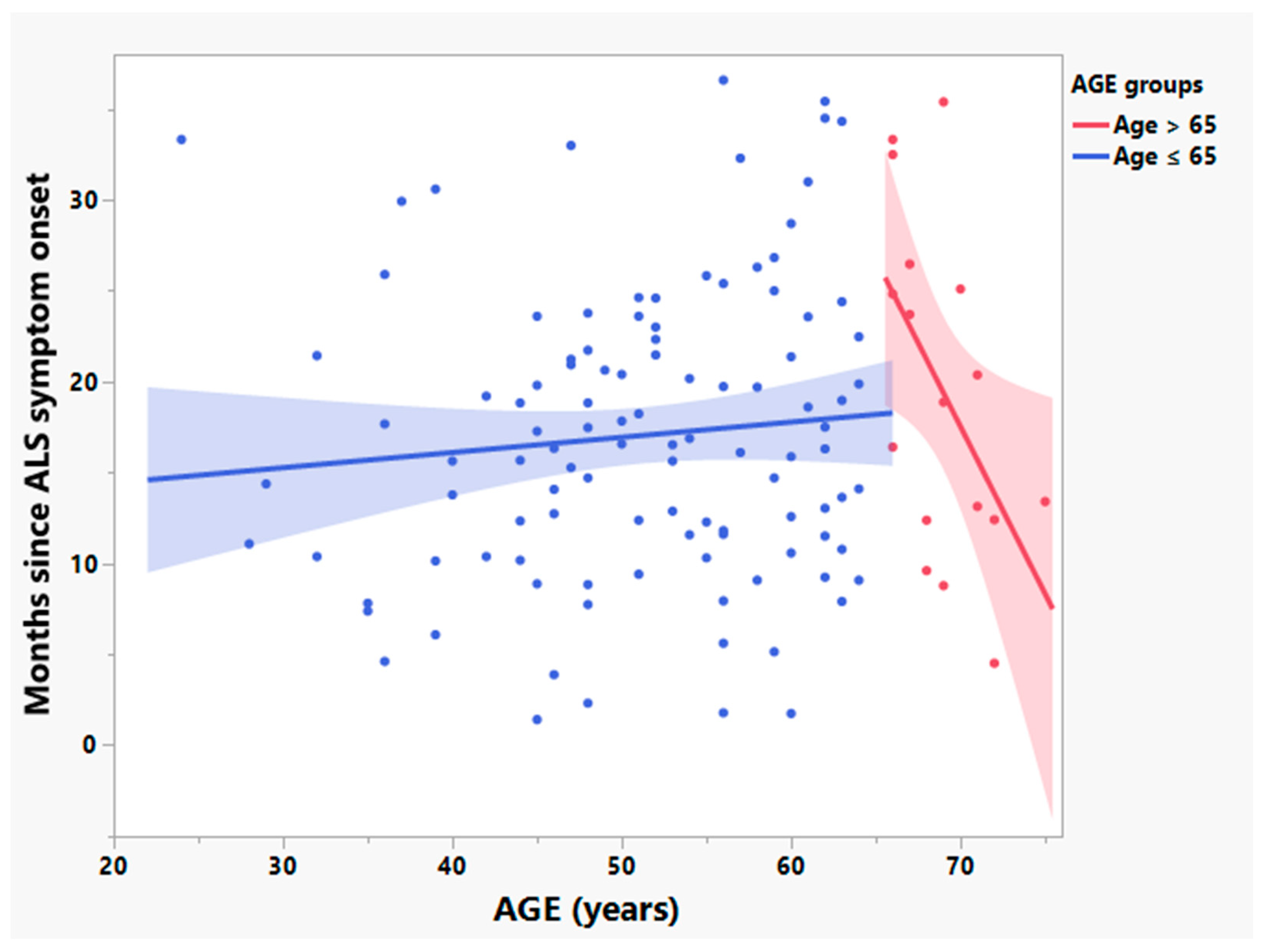
3.2. Participants > 65 Years Old Have Significantly Different Relationships between Age and Times since Symptom Onset and Rates of Disease Progression
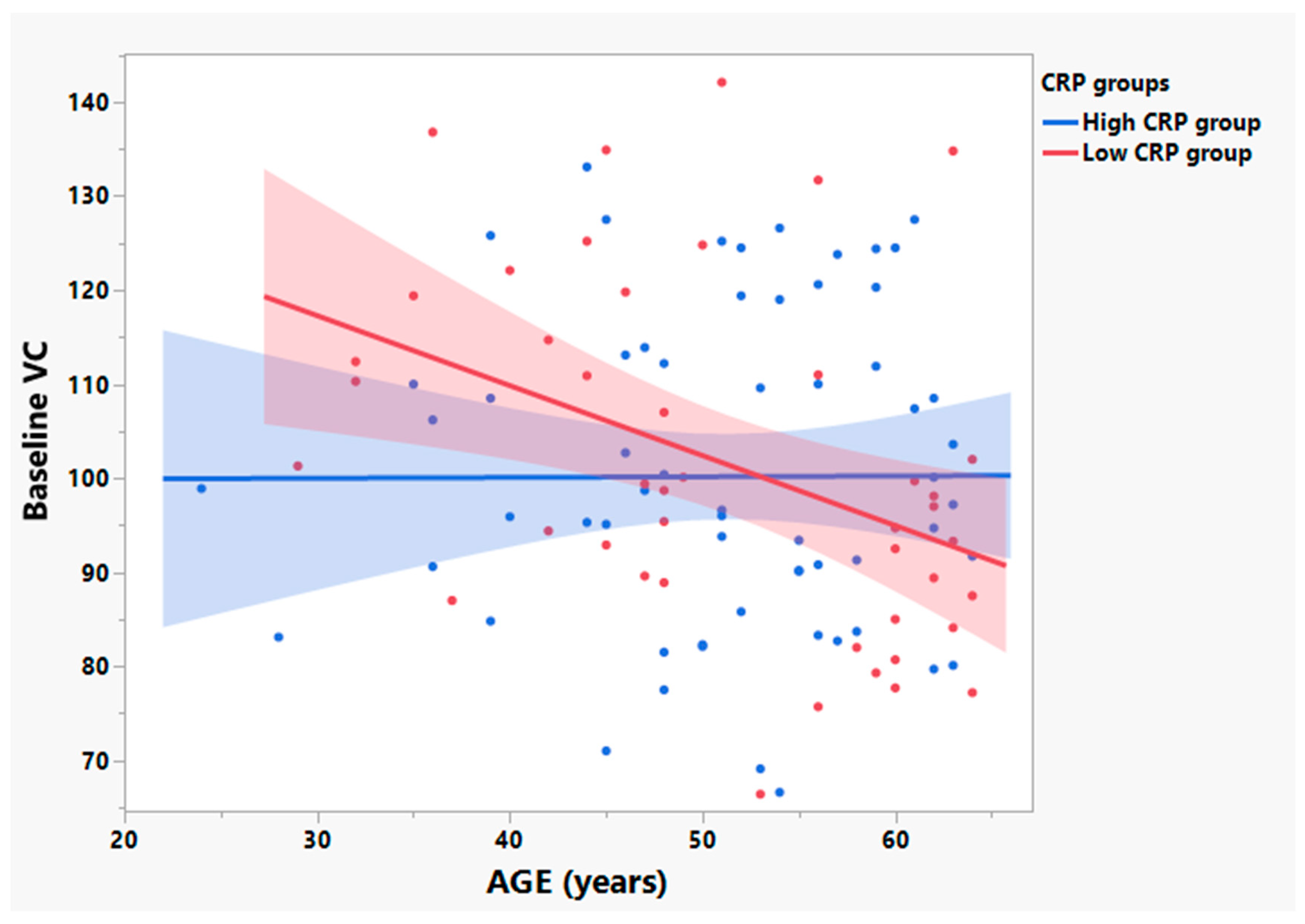
3.3. Participants with Low Baseline CRP Show Age Dependent Loss of VC in Those ≤ 65 Years Old
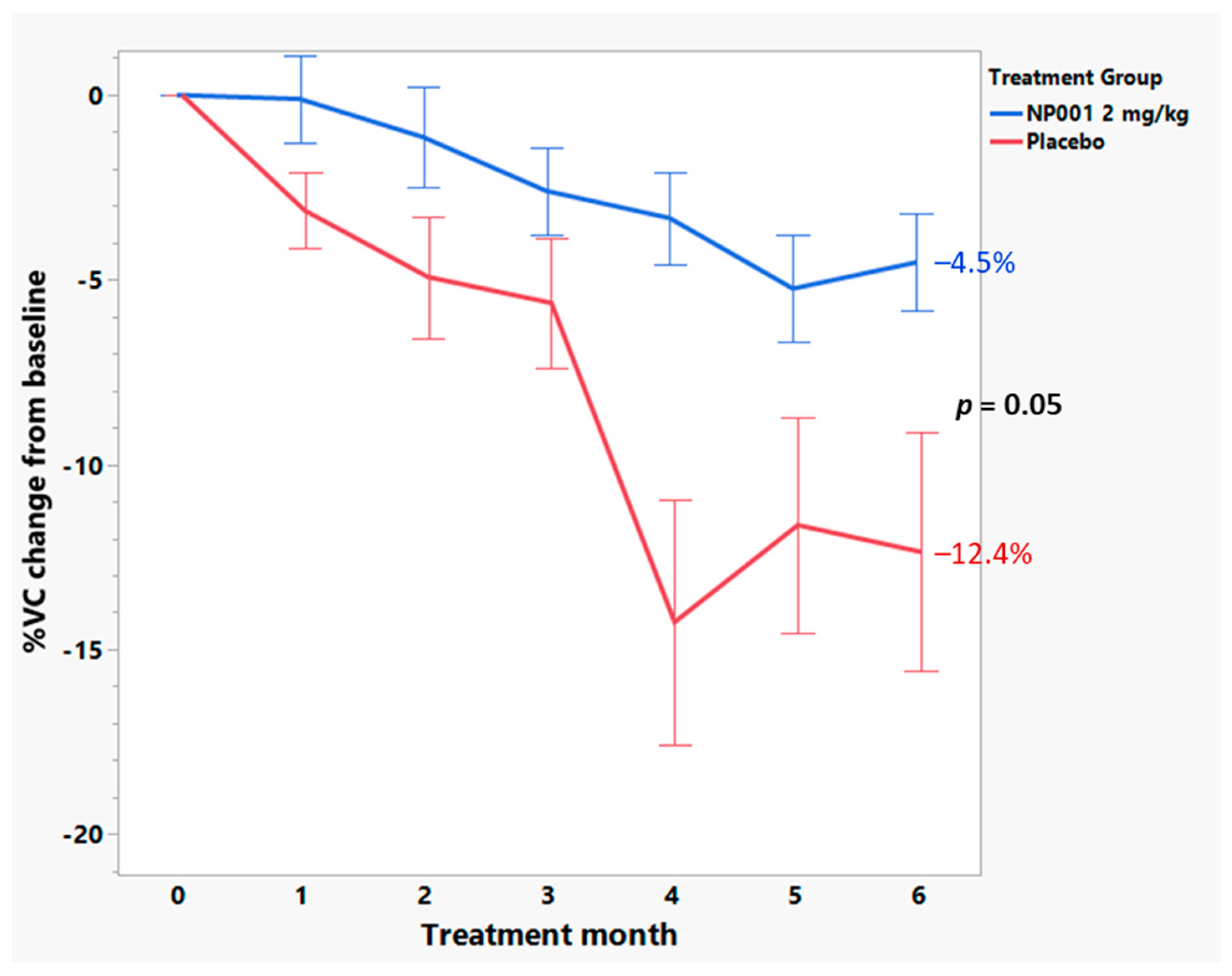
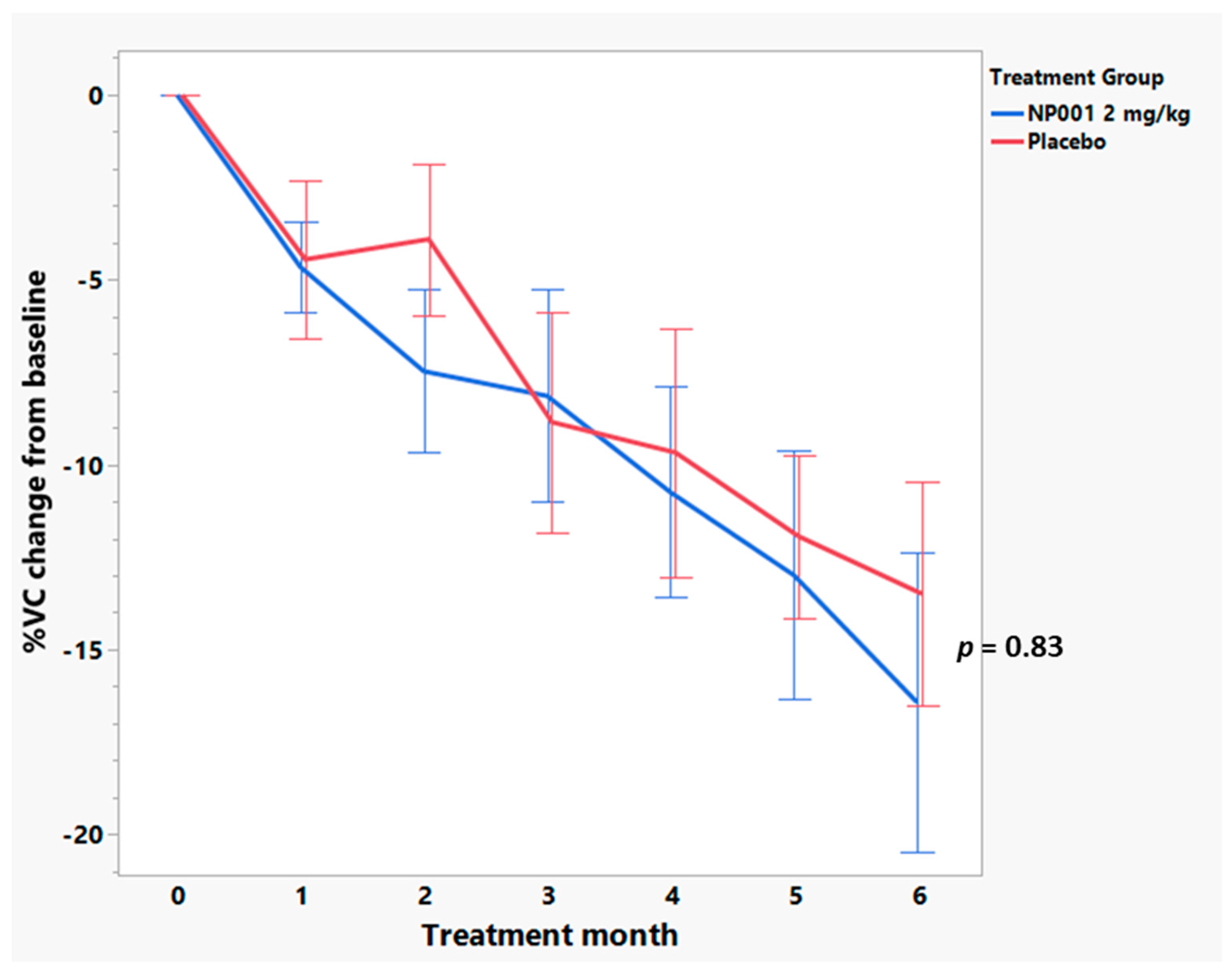
3.4. Baseline Plasma CRP Values Define a Subset of NP001 Treated ALS Patients Whose Loss in VC over the 6-Month Phase 2A Study Is Markedly Slowed vs. Placebo
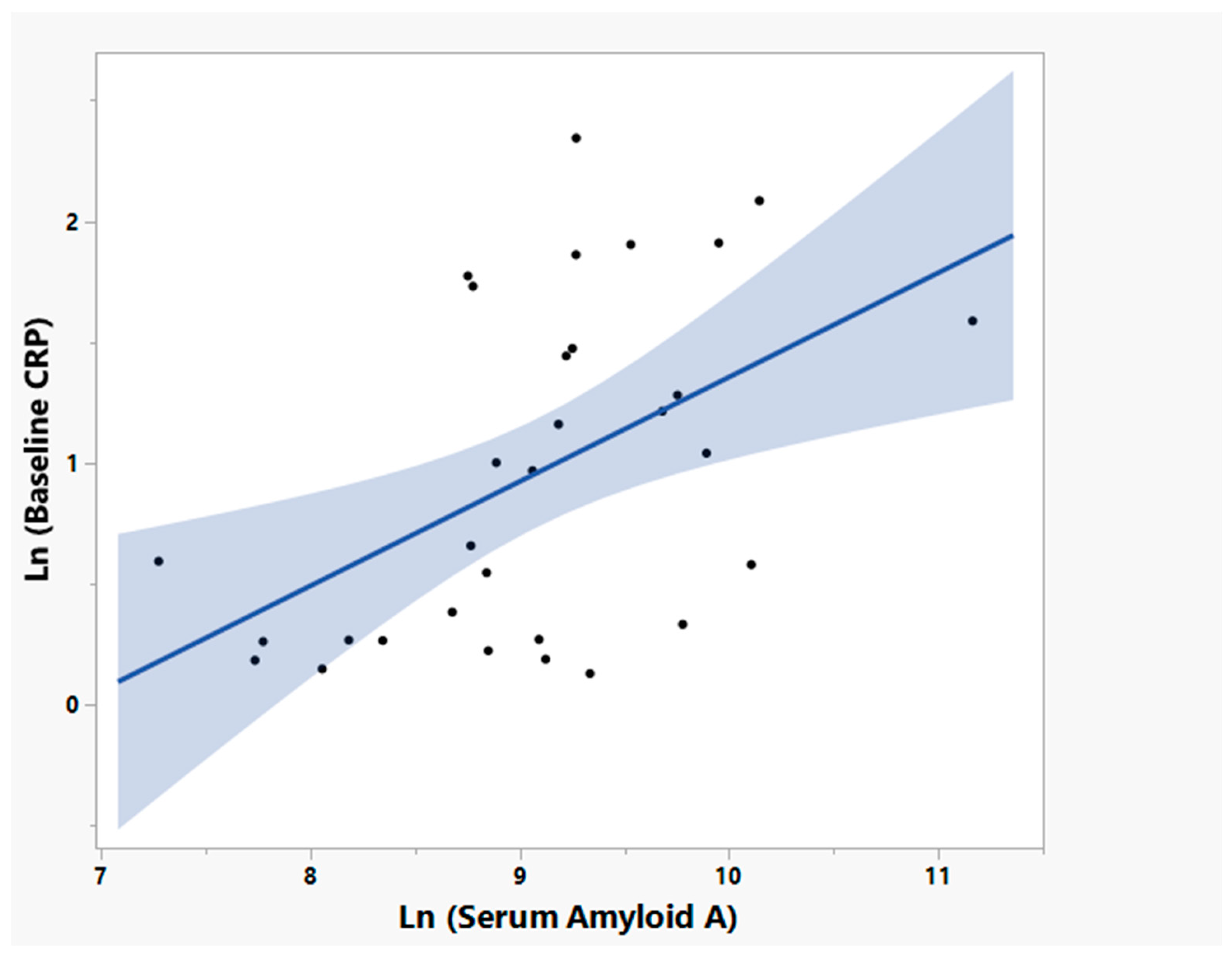
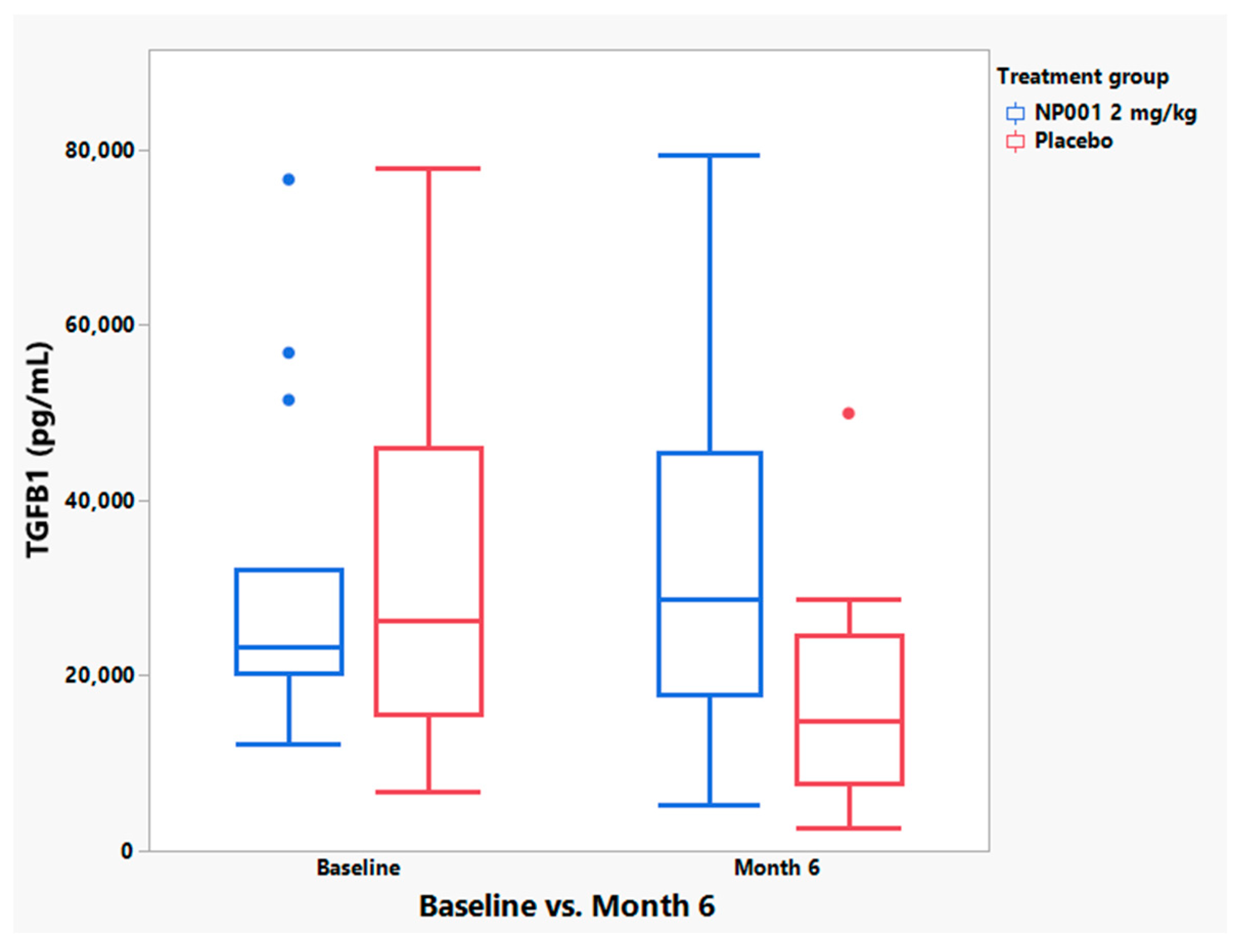
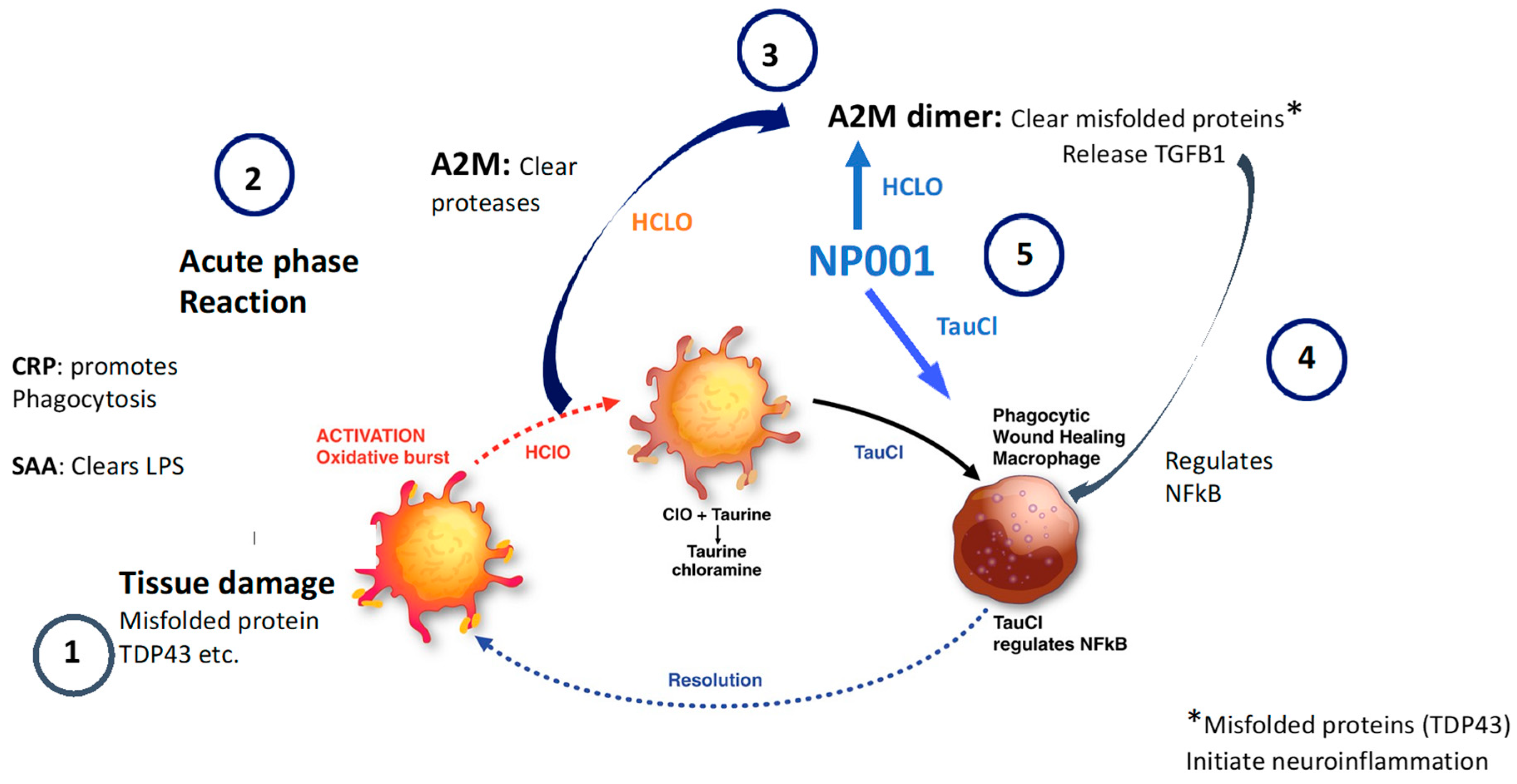
3.5. NP001 May Function through Augmentation of the Normally Self-Regulated Innate Immune System
4. Discussion
5. Limitations of the Current Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, J.A.; Meng, L.; Kulke, S.F.; Rudnicki, S.A.; Wolff, A.A.; Bozik, M.E.; Malik, F.I.; Shefner, J.M. Association between Decline in Slow Vital Capacity and Respiratory Insufficiency, Use of Assisted Ventilation, Tracheostomy, or Death in Patients with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2018, 75, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiot, A.; Zaidi, S.; Iltis, C.; Ribon, M.; Berriat, F.; Schiaffino, L.; Jolly, A.; de la Grange, P.; Mallat, M.; Bohl, D.; et al. Modifying macrophages at the periphery has the capacity to change microglial reactivity and to extend ALS survival. Nat. Neurosci. 2020, 23, 1339–1351. [Google Scholar] [CrossRef]
- Shiraishi, W.; Yamasaki, R.; Hashimoto, Y.; Ko, S.; Kobayakawa, Y.; Isobe, N.; Matsushita, T.; Kira, J.-I. Clearance of peripheral nerve misfolded mutant protein by infiltrated macrophages correlates with motor neuron disease progression. Sci. Rep. 2021, 11, 16438. [Google Scholar] [CrossRef]
- Zhang, R.; Hadlock, K.G.; Do, H.; Yu, S.; Honrada, R.; Champion, S.; Forshew, D.; Madison, C.; Katz, J.; Miller, R.G.; et al. Gene expression profiling in peripheral blood mononuclear cells from patients with sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2011, 230, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Swarup, V.; Phaneuf, D.; Dupre, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor kappaB-mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef]
- Beers, D.R.; Zhao, W.; Neal, D.W.; Thonhoff, J.R.; Thome, A.D.; Faridar, A.; Wen, S.; Wang, J.; Appel, S.H. Elevated acute phase proteins reflect peripheral inflammation and disease severity in patients with amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 15295. [Google Scholar] [CrossRef]
- Khalil, R.H.; Al-Humadi, N. Types of acute phase reactants and their importance in vaccination. Biomed. Rep. 2020, 12, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Vashist, S.K.; Venkatesh, A.G.; Marion Schneider, E.; Beaudoin, C.; Luppa, P.B.; Luong, J.H. Bioanalytical advances in assays for C-reactive protein. Biotechnol. Adv. 2016, 34, 272–290. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.; Agrawal, A. Evolution of C-Reactive Protein. Front. Immunol. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sproston, N.R.; Ashworth, J.J. Role of C-Reactive Protein at Sites of Inflammation and Infection. Front. Immunol. 2018, 9, 754. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, D.N.; Agrawal, A. Structure-Function Relationships of C-Reactive Protein in Bacterial Infection. Front. Immunol. 2019, 10, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, R.V.; Kuznetsova, V.; Connelly, A.N.; Hel, Z.; Szalai, A.J. C-Reactive Protein Promotes the Expansion of Myeloid Derived Cells with Suppressor Functions. Front. Immunol. 2019, 10, 2183. [Google Scholar] [CrossRef]
- Jones, N.R.; Pegues, M.A.; McCrory, M.A.; Kerr, S.W.; Jiang, H.; Sellati, R.; Berger, V.; Villalona, J.; Parikh, R.; McFarland, M.; et al. Collagen-induced arthritis is exacerbated in C-reactive protein-deficient mice. Arthritis Rheum. 2011, 63, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Y.; Tang, Z.M.; Wang, Z.; Lv, J.M.; Liu, X.L.; Liang, Y.L.; Cheng, B.; Gao, N.; Ji, S.-R.; Wu, Y. C-Reactive Protein Protects Against Acetaminophen-Induced Liver Injury by Preventing Complement Overactivation. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 289–307. [Google Scholar] [CrossRef]
- Teupser, D.; Weber, O.; Rao, T.N.; Sass, K.; Thiery, J.; Fehling, H.J. No reduction of atherosclerosis in C-reactive protein (CRP)-deficient mice. J. Biol. Chem. 2011, 286, 6272–6279. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.H.; Gona, P.; Sutherland, P.A.; Benjamin, E.J.; Wilson, P.W.; Larson, M.G.; Vasan, R.S.; Robins, S.J. Long-term C-reactive protein variability and prediction of metabolic risk. Am. J. Med. 2009, 122, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Qiu, S.; He, Y.; Li, L.; Wu, T.; Ding, N.; Li, F.; Zhao, A.Z.; Yang, G. Genetic ablation of C-reactive protein gene confers resistance to obesity and insulin resistance in rats. Diabetologia 2021, 64, 1169–1183. [Google Scholar] [CrossRef]
- Kharel, S.; Ojha, R.; Preethish-Kumar, V.; Bhagat, R. C-reactive protein levels in patients with amyotrophic lateral sclerosis: A systematic review. Brain Behav. 2022, 12, e2532. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, M.; Zhang, J.; Huang, X. Association Between C-Reactive Protein and Risk of Amyotrophic Lateral Sclerosis: A Mendelian Randomization Study. Front. Genet. 2022, 13, 919031. [Google Scholar] [CrossRef]
- Cheng, N.; Liang, Y.; Du, X.; Ye, R.D. Serum amyloid A promotes LPS clearance and suppresses LPS-induced inflammation and tissue injury. EMBO Rep. 2018, 19, e45517. [Google Scholar] [CrossRef]
- Siddiqui, T.; Zia, M.K.; Ali, S.S.; Ahsan, H.; Khan, F.H. Insight into the interactions of proteinase inhibitor- alpha-2-macroglobulin with hypochlorite. Int. J. Biol. Macromol. 2018, 117, 401–406. [Google Scholar] [CrossRef]
- Wyatt, A.R.; Kumita, J.R.; Mifsud, R.W.; Gooden, C.A.; Wilson, M.R.; Dobson, C.M. Hypochlorite-induced structural modifications enhance the chaperone activity of human alpha2-macroglobulin. Proc. Natl. Acad. Sci. USA 2014, 111, E2081–E2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.G.; Block, G.; Katz, J.S.; Barohn, R.J.; Gopalakrishnan, V.; Cudkowicz, M.; Zhang, J.R.; McGrath, M.S.; Ludington, E.; Appel, S.H.; et al. Randomized phase 2 trial of NP001-a novel immune regulator: Safety and early efficacy in ALS. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e100. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.G.; Zhang, R.; Block, G.; Katz, J.; Barohn, R.; Kasarskis, E.; Forshew, D.; Gopalakrishnan, V.; McGrath, M.S. NP001 regulation of macrophage activation markers in ALS: A phase I clinical and biomarker study. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.G.; Zhang, R.; Bracci, P.M.; Azhir, A.; Barohn, R.; Bedlack, R.; Benatar, M.; Berry, J.D.; Cudkowicz, M.; Kasarskis, E.J.; et al. Phase 2B randomized controlled trial of NP001 in amyotrophic lateral sclerosis: Pre-specified and post hoc analyses. Muscle Nerve 2022, 66, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor. Neuron. Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef]
- Zhang, R.; Bracci, P.M.; Azhir, A.; Forrest, B.D.; McGrath, M.S. Macrophage-Targeted Sodium Chlorite (NP001) Slows Progression of Amyotrophic Lateral Sclerosis (ALS) through Regulation of Microbial Translocation. Biomedicines 2022, 10, 2907. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services FaDA. Amyotrophic Lateral Sclerosis: Developing Drugs for Treatment Guidance for Industry. In Services USDoHaH; FDA: Silver Spring, MD, USA, 2019. Available online: www.fda.gov (accessed on 24 September 2019).
- Hegazy, S.H.; Thomassen, J.Q.; Rasmussen, I.J.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Frikke-Schmidt, R. C-reactive protein levels and risk of dementia-Observational and genetic studies of 111,242 individuals from the general population. Alzheimers Dement. 2022, 18, 2262–2271. [Google Scholar] [CrossRef]
- Bright, F.; Chan, G.; van Hummel, A.; Ittner, L.M.; Ke, Y.D. TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Int. J. Mol. Sci. 2021, 22, 7781. [Google Scholar] [CrossRef] [PubMed]
- James, B.D.; Wilson, R.S.; Boyle, P.A.; Trojanowski, J.Q.; Bennett, D.A.; Schneider, J.A. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain 2016, 139, 2983–2993. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Son, J.; Lee, D.; Tsai, J.; Wang, D.; Chocron, E.S.; Jeong, S.; Kittrell, P.; Murchison, C.F.; Kennedy, R.E.; et al. Gut- and oral-dysbiosis differentially impact spinal- and bulbar-onset ALS, predicting ALS severity and potentially determining the location of disease onset. BMC Neurol. 2022, 22, 62. [Google Scholar] [CrossRef]
- Kim, W.; Kim, S.H.; Jang, J.H.; Kim, C.; Kim, K.; Suh, Y.G.; Joe, Y.; Chung, H.T.; Cha, Y.-N.; Surh, Y.-J. Role of heme oxygenase-1 in potentiation of phagocytic activity of macrophages by taurine chloramine: Implications for the resolution of zymosan A-induced murine peritonitis. Cell. Immunol. 2018, 327, 36–46. [Google Scholar] [CrossRef]
- Ramirez, H.; Patel, S.B.; Pastar, I. The Role of TGFβ Signaling in Wound Epithelialization. Adv. Wound Care 2014, 3, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schempp, H.; Reim, M.; Dornisch, K.; Elstner, E.F. Chlorite-hemoprotein interaction as key role for the pharmacological activity of the chlorite-based drug WF10. Arzneimittelforschung 2001, 51, 554–562. [Google Scholar] [CrossRef]
- Jin, X.; Liu, M.Y.; Zhang, D.F.; Zhong, X.; Du, K.; Qian, P.; Gao, H.; Wei, M.-J. Natural products as a potential modulator of microglial polarization in neurodegenerative diseases. Pharmacol. Res. 2019, 145, 104253. [Google Scholar] [CrossRef]
- Pronto-Laborinho, A.; Pinto, S.; Gromicho, M.; Pereira, M.; Swash, M.; de Carvalho, M. Interleukin-6 and amyotrophic lateral sclerosis. J. Neurol. Sci. 2019, 398, 50–53. [Google Scholar] [CrossRef]
- Pinto, S.; De Carvalho, M. Phrenic nerve study as outcome in clinical trials for amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 2021, 22 (Suppl. S1), 9–13. [Google Scholar] [CrossRef] [PubMed]
- Caraci, F.; Spampinato, S.F.; Morgese, M.G.; Tascedda, F.; Salluzzo, M.G.; Giambirtone, M.C.; Caruso, G.; Munafò, A.; Torrisi, S.A.; Leggio, G.M.; et al. Neurobiological links between depression and AD: The role of TGF-beta1 signaling as a new pharmacological target. Pharmacol. Res. 2018, 130, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Wyczalkowska-Tomasik, A.; Czarkowska-Paczek, B.; Zielenkiewicz, M.; Paczek, L. Inflammatory Markers Change with Age, but do not Fall Beyond Reported Normal Ranges. Arch. Immunol. Ther. Exp. 2016, 64, 249–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP001 2 mg/kg | Placebo | Overall | |
|---|---|---|---|
| Characteristics | (n = 16) | (n = 18) | (n = 34) |
| Sex, n (%) | |||
| Female | 5 (31.3%) | 5 (27.8%) | 10 (29.4%) |
| Male | 11 (68.8%) | 13 (72.2%) | 24 (70.6%) |
| Age at baseline, year | 51.9 ± 9.9 | 51.8 ± 5.9 | 51.9 ± 7.9 |
| Type of ALS, n (%) | |||
| Familial | 0 (0.0%) | 2 (11.1%) | 2 (5.9%) |
| Sporadic | 16 (100.0%) | 16 (88.9%) | 32 (94.1%) |
| Site of ALS onset, n (%) | |||
| Bulbar | 3 (18.8%) | 3 (16.7%) | 6 (17.6%) |
| Limb | 13 (81.3%) | 15 (83.3%) | 28 (82.4%) |
| El Escorial criteria for ALS, n (%) | |||
| Definite | 7 (43.8%) | 9 (50.0%) | 16 (47.1%) |
| Probable | 8 (50.0%) | 7 (38.9%) | 15 (44.1%) |
| Possible | 1 (6.3%) | 2 (11.1%) | 3 (8.8%) |
| Concurrent riluzole use, n (%) | |||
| Yes | 11 (68.8%) | 12 (66.7%) | 23 (67.6%) |
| No | 5 (31.3%) | 6 (33.3.%) | 11 (32.4%) |
| ALSFRS-R score at baseline 1, Mean ± SD | 37.7 ± 4.1 | 39.2 ± 3.9 | 38.5 ± 4.0 |
| Vital capacity at baseline, Mean ± SD | 113.5 ± 17.0 | 109.2 ± 15.3 | 111.2 ± 16.0 |
| Months since ALS symptom onset 2, Mean ± SD | 19.29 ± 7.63 | 13.76 ± 6.55 | 16.36 ± 7.52 |
| CRP at Baseline (mg/L) 3, Mean ± SD | 3.48 ± 2.57 | 2.95 ± 2.24 | 3.20 ± 2.38 |
| NP001 2 mg/kg | Placebo | Overall | |
|---|---|---|---|
| Characteristics | (n = 14) | (n = 13) | (n = 27) |
| Sex, n (%) | |||
| Female | 5 (35.7%) | 4 (30.8%) | 9 (33.3%) |
| Male | 9 (64.3%) | 9 (69.2%) | 18 (66.7%) |
| Age at baseline, year | 51.3 ± 8.9 | 51.8 ± 11.6 | 51.6 ± 10.1 |
| Type of ALS, n (%) | |||
| Familial | 1 (7.1%) | 2 (15.4%) | 3 (11.1%) |
| Sporadic | 13 (92.9%) | 11 (84.6%) | 24 (88.9%) |
| Site of ALS onset, n (%) | |||
| Bulbar | 2 (14.3%) | 2 (15.4%) | 4 (14.8%) |
| Limb | 12 (85.7%) | 11 (84.6%) | 23 (85.2%) |
| El Escorial criteria for ALS, n (%) | |||
| Definite | 7 (50.0%) | 7 (53.8%) | 14 (51.9%) |
| Probable | 7 (50.0%) | 6 (46.2%) | 13 (48.1%) |
| Concurrent riluzole use, n (%) | |||
| Yes | 10 (71.4%) | 9 (69.2%) | 19 (70.4%) |
| No | 4 (28.6%) | 4 (30.8%) | 8 (29.6%) |
| ALSFRS-R score at baseline 1, Mean ± SD | 38.4 ± 6.5 | 40.0 ± 3.2 | 39.2 ± 5.1 |
| Vital capacity at baseline, Mean ± SD | 110.3 ± 15.7 | 110.8 ± 19.0 | 110.6 ± 17.1 |
| Months since ALS symptom onset 2, Mean ± SD | 13.60 ± 7.18 | 16.56 ± 7.68 | 15.02 ± 7.43 |
| CRP at Baseline (mg/L) 3, Mean ± SD | 0.73 ± 0.26 | 0.62 ± 0.30 | 0.68 ± 0.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGrath, M.S.; Zhang, R.; Bracci, P.M.; Azhir, A.; Forrest, B.D. Regulation of the Innate Immune System as a Therapeutic Approach to Supporting Respiratory Function in ALS. Cells 2023, 12, 1031. https://doi.org/10.3390/cells12071031
McGrath MS, Zhang R, Bracci PM, Azhir A, Forrest BD. Regulation of the Innate Immune System as a Therapeutic Approach to Supporting Respiratory Function in ALS. Cells. 2023; 12(7):1031. https://doi.org/10.3390/cells12071031
Chicago/Turabian StyleMcGrath, Michael S., Rongzhen Zhang, Paige M. Bracci, Ari Azhir, and Bruce D. Forrest. 2023. "Regulation of the Innate Immune System as a Therapeutic Approach to Supporting Respiratory Function in ALS" Cells 12, no. 7: 1031. https://doi.org/10.3390/cells12071031