

Autophagy Inhibitor Chloroquine Downmodulates Hepatic Stellate Cell Activation and Liver Damage in Bile-Duct-Ligated Mice
 , , and
, , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Ethical Declaration
2.3. Bile-Duct-Ligated Mice Model
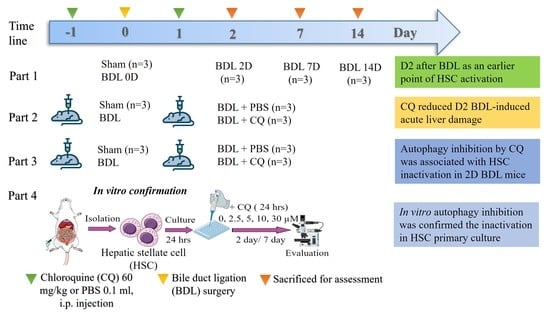
2.4. Experimental Design
- Part 1.
- Investigation on the early time point of HSC activation on BDL including 0D (Sham), 2D, 7D, and 14D groups. BDL mice were treated with CQ (Cas: 50-63-5) 60 mg/kg or control 0.1 mL PBS (Phosphate buffer saline is CQ solution) i.p. the day before and the day after surgery and sacrificed at 2D for Part 2 and Part 3. The dose of CQ was referenced from previous studies in mice [5,26,27].
- Part 2.
- Investigation on the effects of CQ on BDL liver injury on 2D included Sham, BDL + PBS, and BDL + CQ groups.
- Part 3.
- Investigation on the effects of CQ (60 mg/kg) on HSC activation in 2D BDL mice. (HSCs were isolated from the liver of Sham, BDL + PBS, and BDL + CQ mice groups.)
- Part 4.
- In vitro effects of autophagy inhibitor CQ on primary HSCs.
2.5. HSC Isolation
2.6. In Vitro Autophagy Inhibition
2.7. CCK8 Assay
2.8. Oil Red O Staining (ORO)
2.9. Real-Time RT-PCR
2.10. Immunocytochemistry (ICC)
2.11. Serum Test
2.12. Histopathology
2.13. Hematoxylin and Eosin (H&E)
2.14. Picrosirius Red
2.15. Immunohistochemistry (IHC)
2.16. Statistical Analysis
3. Results
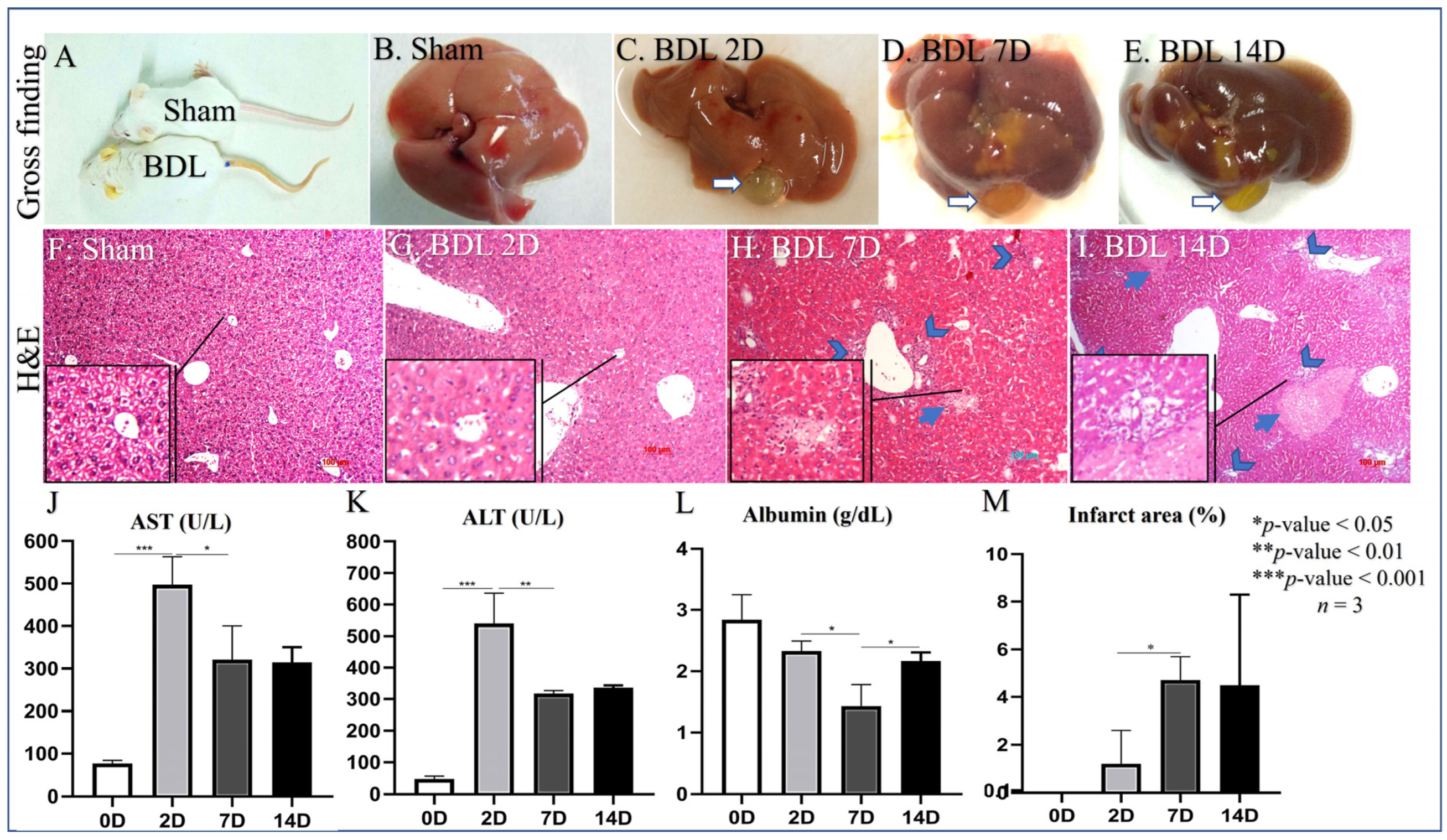
3.1. Liver Damage of Bile Duct Ligation
3.2. D2 after BDL as an Earlier Point of HSC Activation
3.3. CQ Reduced D2 BDL-Induced Acute Liver Damage
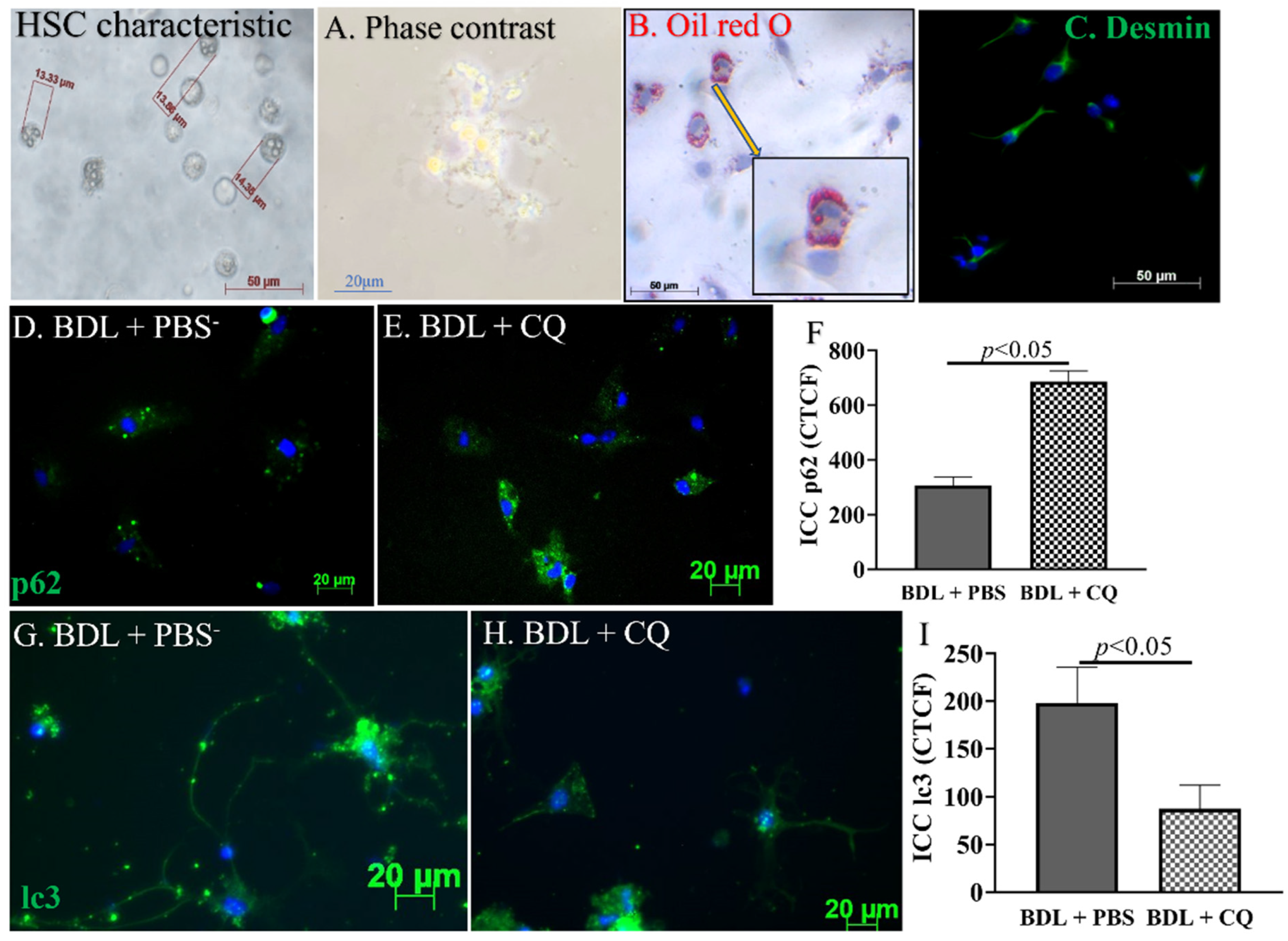
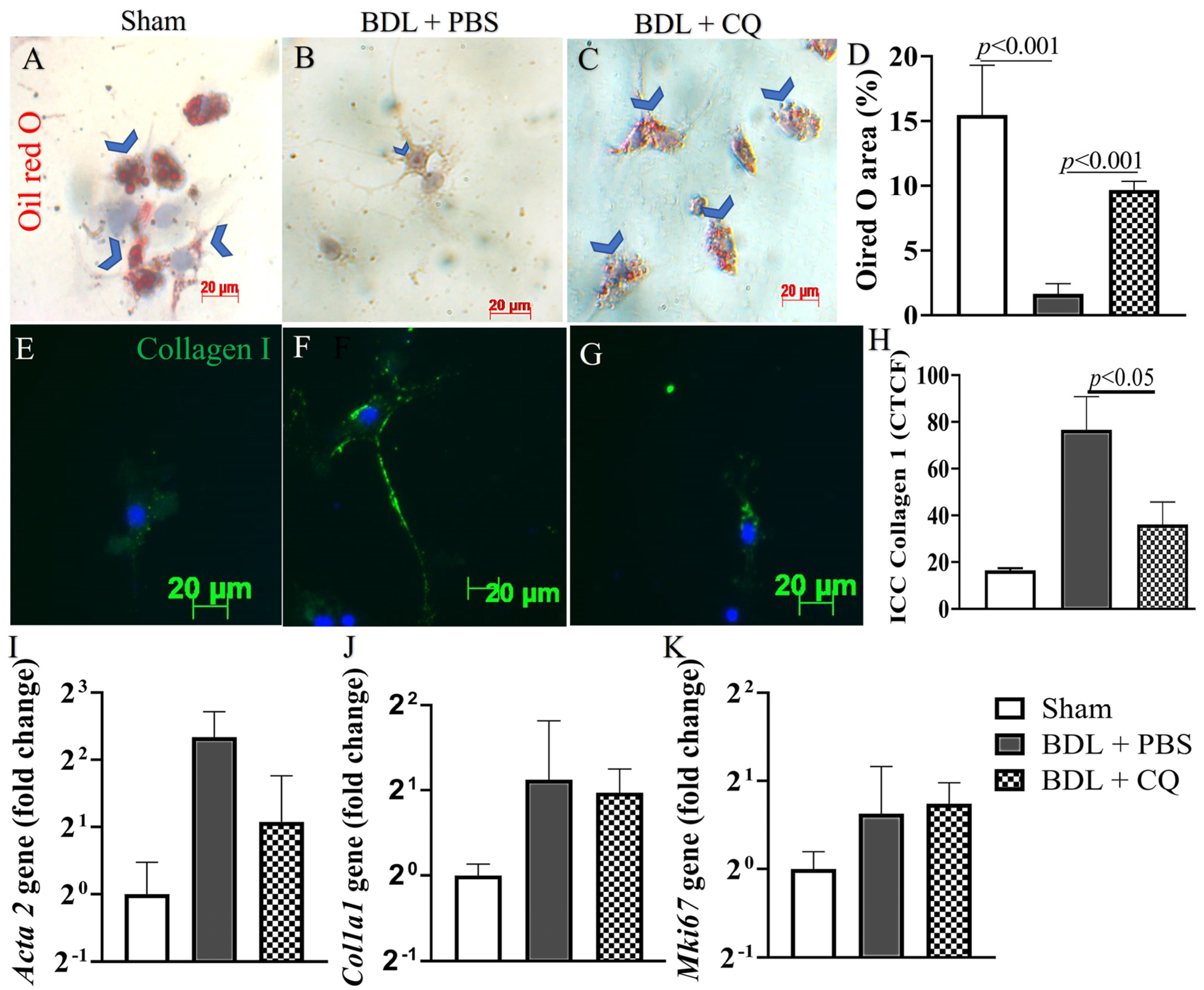
3.4. Autophagy Inhibition by CQ Was Associated with HSC Inactivation in 2D BDL Mice
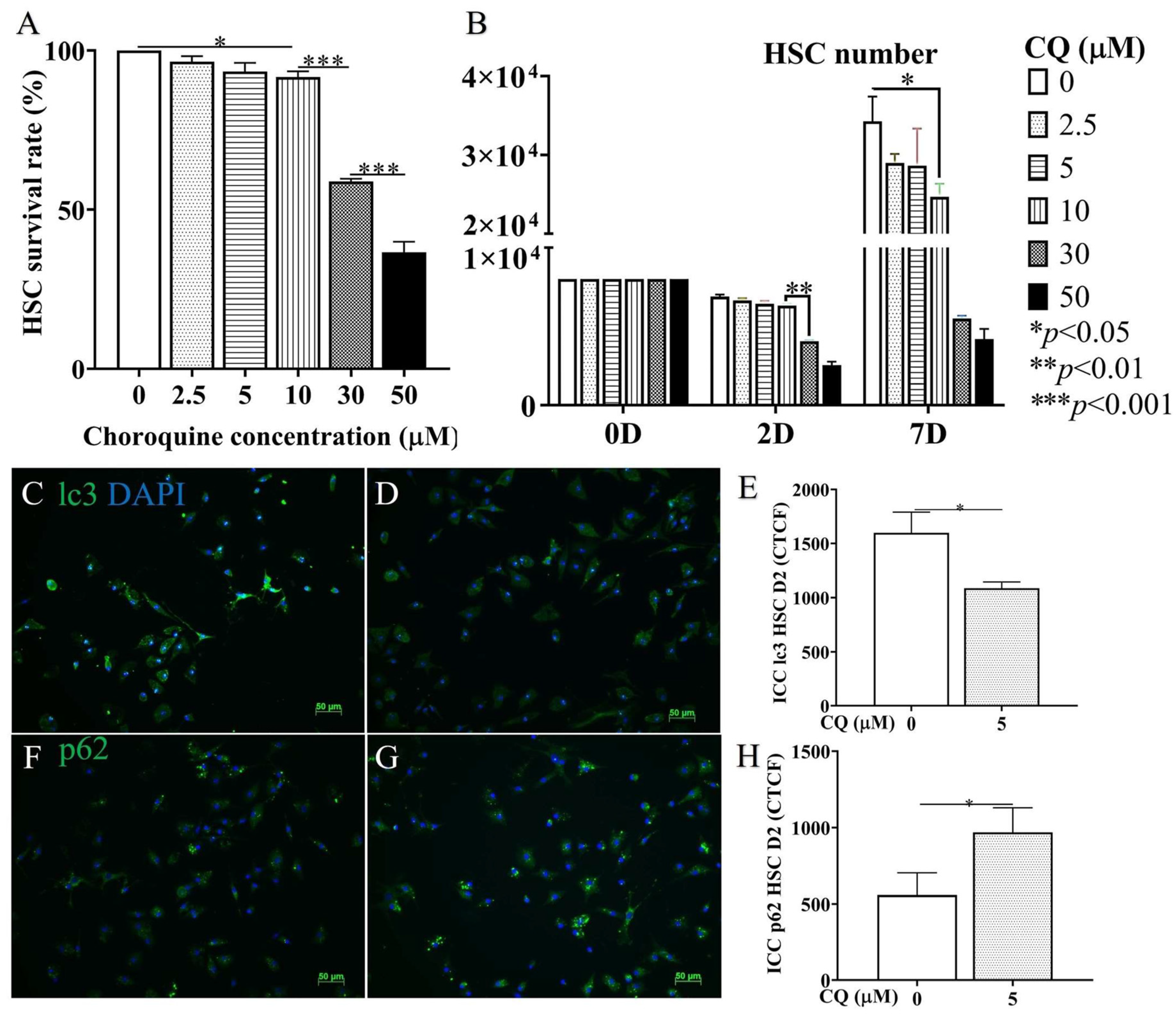
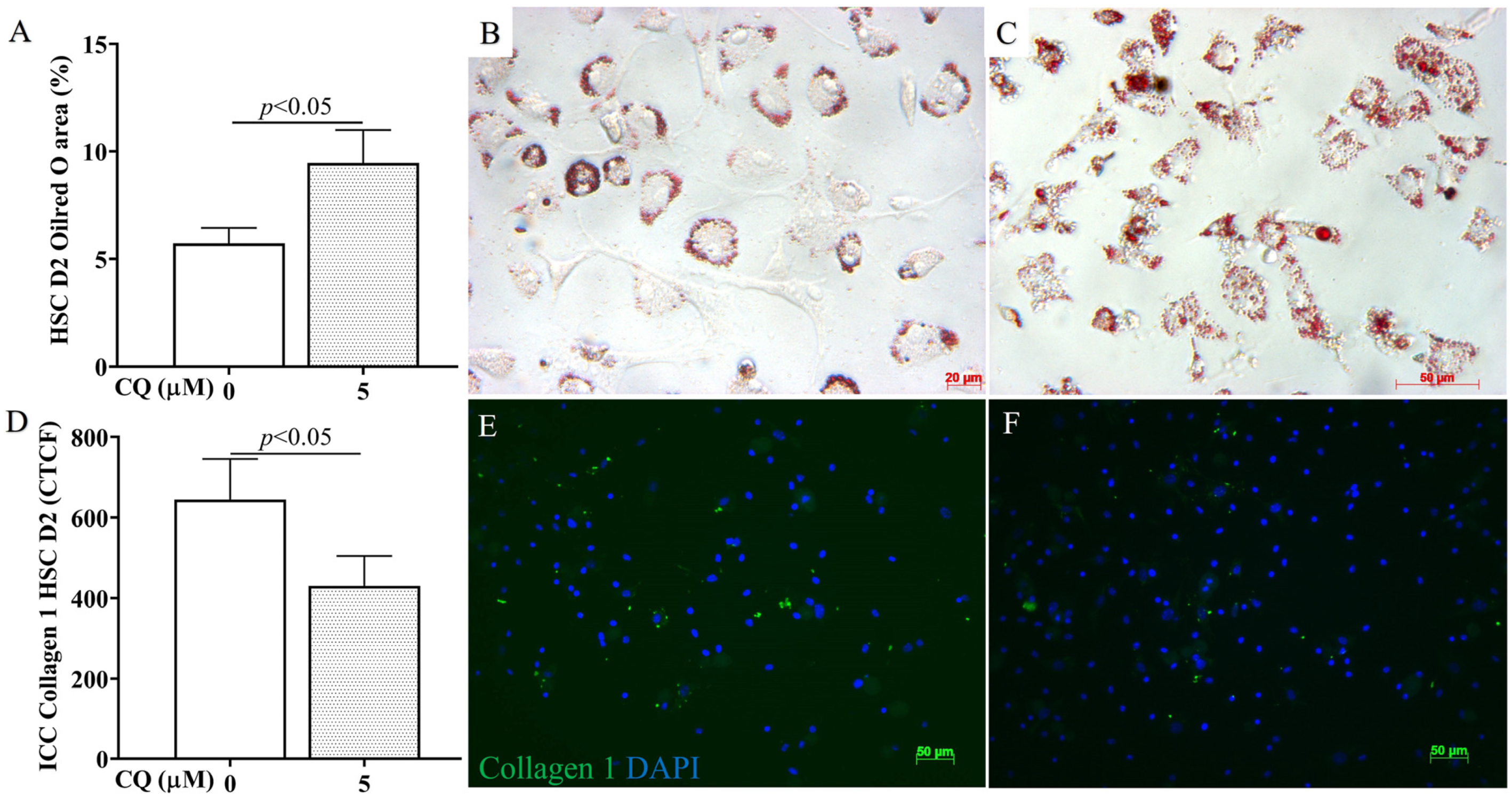
3.5. Confirmation of In Vitro Autophagy Inhibition and Activation in HSC Culture
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Pedro, J.M.B.-S.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. Embo J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Conway, O.; Akpinar, H.A.; Rogov, V.V.; Kirkin, V. Selective autophagy receptors in neuronal health and disease. J. Mol. Biol. 2020, 432, 2483–2509. Available online: https://www.sciencedirect.com/science/article/pii/S0022283619306096 (accessed on 19 January 2023). [CrossRef] [PubMed]
- Mallat, A.; Lodder, J.; Teixeira-Clerc, F.; Moreau, R.; Codogno, P.; Lotersztajn, S. Autophagy: A multifaceted partner in liver fibrosis. Biomed. Res. Int. 2014, 2014, 869390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Tacke, F. Relevance of autophagy in parenchymal and non-parenchymal liver cells for health and disease. Cells 2019, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Liu, A.; Dahmen, U.; Dirsch, O. Dual role of chloroquine in liver ischemia reperfusion injury: Reduction of liver damage in early phase, but aggravation in late phase. Cell Death Dis. 2013, 4, e694. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Wang, B.; Yang, J.; Zhuang, Y.; Wang, L.; Huang, X.; Chen, J. Chloroquine improved carbon tetrachloride-induced liver fibrosis through its inhibition of the activation of hepatic stellate cells: Role of autophagy. Biol. Pharm. Bull. 2014, 37, 1505–1509. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Xiao, X.; Li, D.; Tun, S.; Wang, Y.; Velkov, T.; Tang, S. Chloroquine ameliorates carbon tetrachloride-induced acute liver injury in mice via the concomitant inhibition of inflammation and induction of apoptosis. Cell Death Dis. 2018, 9, 1164. [Google Scholar] [CrossRef] [Green Version]
- Le, T.V.; Dinh, N.B.T.; Dang, M.T.; Phan, N.C.L.; Dang, L.T.T.; Grassi, G.; Holterman, A.X.L.; Le, H.M.; Truong, N.H. Effects of autophagy inhibition by chloroquine on hepatic stellate cell activation in ccl4-induced acute liver injury mouse model. J. Gastroenterol. Hepatol. 2022, 37, 216–224. [Google Scholar] [CrossRef]
- Raquel Benedita Terrabuio, D.; Augusto Diniz, M.; Teofilo de Moraes Falcão, L.; Luiza Vilar Guedes, A.; Akeme Nakano, L.; Silva Evangelista, A.; Roberto Lima, F.; Pires Abrantes-Lemos, C.; José Carrilho, F.; Luiz Rachid Cancado, E. Chloroquine is effective for maintenance of remission in autoimmune hepatitis: Controlled, double-blind, randomized trial. Hepatol. Commun. 2019, 3, 116–128. Available online: https://aasldpubs.onlinelibrary.wiley.com/doi/abs/10.1002/hep4.1275 (accessed on 19 January 2023). [CrossRef] [Green Version]
- Hao, X.; Li, W. Chloroquine diphosphate suppresses liver cancer via inducing apoptosis in wistar rats using interventional therapy. Oncol. Lett. 2021, 21, 233. [Google Scholar] [CrossRef]
- Barthet, V.J.A.; Brucoli, M.; Ladds, M.J.G.W.; Nössing, C.; Kiourtis, C.; Baudot, A.D.; O’Prey, J.; Zunino, B.; Müller, M.; May, S.; et al. Autophagy suppresses the formation of hepatocyte-derived cancer-initiating ductular progenitor cells in the liver. Sci. Adv. 2021, 7, eabf9141. Available online: https://www.science.org/doi/abs/10.1126/sciadv.abf9141 (accessed on 19 January 2023). [CrossRef]
- Ding, H.; Ge, G.; Tseng, Y.; Ma, Y.; Zhang, J.; Liu, J. Hepatic autophagy fluctuates during the development of non-alcoholic fatty liver disease. Ann. Hepatol. 2020, 19, 516–522. Available online: https://www.elsevier.es/en-revista-annals-hepatology-16-articulo-hepatic-autophagy-fluctuates-during-development-S1665268120300557 (accessed on 19 January 2023). [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Acharya, P.; Chouhan, K.; Weiskirchen, S.; Weiskirchen, R. Cellular mechanisms of liver fibrosis. Front. Pharmacol. 2021, 12, 671640. Available online: https://www.frontiersin.org/article/10.3389/fphar.2021.671640 (accessed on 19 January 2023). [CrossRef]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Asp. Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Thoen, L.F.; Guimarães, E.L.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Hernández-Gea, V.G.-N.Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Hernández-Gea, V.F.S.L. Autophagy fuels tissue fibrogenesis. Autophagy 2012, 8, 849–850. [Google Scholar] [CrossRef] [Green Version]
- Tonon, F.; Cemazar, M.; Kamensek, U.; Zennaro, C.; Pozzato, G.; Caserta, S.; Ascione, F.; Grassi, M.; Guido, S.; Ferrari, C.; et al. 5-azacytidine downregulates the proliferation and migration of hepatocellular carcinoma cells in vitro and in vivo by targeting mir-139-5p/rock2 pathway. Cancers 2022, 14, 1630. [Google Scholar] [CrossRef]
- De Smet, V.; Eysackers, N.; Merens, V.; Dastjerd, M.K.; Halder, G.; Verhulst, S.; Mannaerts, I.; van Grunsven, L.A. Initiation of hepatic stellate cell activation extends into chronic liver disease. Cell Death Dis. 2021, 12, 1110. [Google Scholar] [CrossRef]
- Mabuchi, A.; Mullaney, I.; Sheard, P.W.; Hessian, P.A.; Mallard, B.L.; Tawadrous, M.N.; Zimmermann, A.; Senoo, H.; Wheatley, A.M. Role of hepatic stellate cell/hepatocyte interaction and activation of hepatic stellate cells in the early phase of liver regeneration in the rat. J. Hepatol. 2004, 40, 910–916. Available online: https://www.sciencedirect.com/science/article/pii/S0168827804000558 (accessed on 19 January 2023). [CrossRef]
- Wang, X.; Tang, X.; Gong, X.; Albanis, E.; Friedman, S.L.; Mao, Z. Regulation of hepatic stellate cell activation and growth by transcription factor myocyte enhancer factor 2. Gastroenterology 2004, 127, 1174–1188. Available online: https://www.sciencedirect.com/science/article/pii/S0016508504012314 (accessed on 19 January 2023). [CrossRef] [PubMed]
- Sicklick, J.K.; Li, Y.-X.; Choi, S.S.; Qi, Y.; Chen, W.; Bustamante, M.; Huang, J.; Zdanowicz, M.; Camp, T.; Torbenson, M.S.; et al. Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab. Investig. 2005, 85, 1368–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Liu, J.; Yang, W.; Ling, W. Lipopolysaccharide mediates hepatic stellate cell activation by regulating autophagy and retinoic acid signaling. Autophagy 2017, 13, 1813–1827. [Google Scholar] [CrossRef] [Green Version]
- Dang, T.M.; Le, T.V.; Do, H.Q.; Nguyen, V.T.; Holterman, A.X.L.; Dang, L.T.T.; Phan, N.C.L.; Pham, P.V.; Hoang, S.N.; Le, L.T.; et al. Optimization of the isolation procedure and culturing conditions for hepatic stellate cells obtained from mouse. Biosci. Rep. 2021, 41, BSR20202514. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. Available online: https://pubmed.ncbi.nlm.nih.gov/18195085 (accessed on 19 January 2023). [CrossRef]
- Korolchuk, V.I.; Mansilla, A.; Menzies FMRubinsztein, D.C. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 2009, 33, 517–527. Available online: https://pubmed.ncbi.nlm.nih.gov/19250912 (accessed on 19 January 2023). [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. Available online: https://pubmed.ncbi.nlm.nih.gov/24264436 (accessed on 19 January 2023). [CrossRef] [Green Version]
- Abshagen, K.; König, M.; Hoppe, A.; Müller, I.; Ebert, M.; Weng, H.; Holzhütter, H.G.; Zanger, U.M.; Bode, J.; Vollmar, B.; et al. Pathobiochemical signatures of cholestatic liver disease in bile duct ligated mice. BMC Syst. Biol. 2015, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Van Campenhout, S.; Van Vlierberghe, H.; Devisscher, L. Common bile duct ligation as model for secondary biliary cirrhosis. Methods Mol. Biol. 2019, 1981, 237–247. [Google Scholar] [CrossRef]
- Hernández-Gea, V.H.M.; Rozenfeld, R.; Lim, M.P.; Nieto, N.; Werner, S.; Devi, L.A.; Friedman, S.L. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol. 2013, 59, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Han, W.; Shi, H.; Ren, F.; Chen, D.; Chen, Y.; Duan, Z. Augmenter of liver regeneration protects against carbon tetrachloride-induced liver injury by promoting autophagy in mice. Oncotarget 2017, 8, 12637–12648. [Google Scholar] [CrossRef] [Green Version]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of autophagy in oxidative stress. Int. J. Mol. Sci. 2020, 21, 3289. Available online: https://pubmed.ncbi.nlm.nih.gov/32384691 (accessed on 19 January 2023). [CrossRef]
- Zhang, J.J.N.; Ping, J.; Xu, L. Tgf-β1-induced autophagy activates hepatic stellate cells via the erk and jnk signaling pathways. Int. J. Mol. Med. 2021, 47, 256–266. [Google Scholar] [CrossRef]
- Ramachandran, P.; Iredale, J.P. Liver fibrosis: A bidirectional model of fibrogenesis and resolution. QJM Int. J. Med. 2012, 105, 813–817. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, I.; Watanabe, T.; Niioka, M.; Sugioka, Y.; Inagaki, Y. Reversibility of liver fibrosis. Fibrogenes. Cell. Mol. Basis 2005, 39, 143–159. [Google Scholar]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular reaction in liver diseases: Pathological mechanisms and translational significances. Hepatology 2019, 69, 420–430. Available online: https://aasldpubs.onlinelibrary.wiley.com/doi/abs/10.1002/hep.30150 (accessed on 19 January 2023). [CrossRef] [PubMed] [Green Version]
- Zhou, Y.Z.Q.; Kong, Y.; Guo, X.; Zhang, H.; Fan, H.; Liu, L. Insulin-like growth factor binding protein-related protein 1 activates primary hepatic stellate cells via autophagy regulated by the pi3k/akt/mtor signaling pathway. Dig. Dis. Sci. 2020, 65, 509–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.Y.H.Y.J.; Lv, X.; Liu, Z.H.; Shen, Y.; Ye, G.R.; Deng, Y.M.; Shu, J.C. Transforming growth factor-β1 reduces apoptosis via autophagy activation in hepatic stellate cells. Mol. Med. Rep. 2014, 10, 1282–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrone, F.; Craparo, E.F.; Cemazar, M.; Kamensek, U.; Drago, S.E.; Dapas, B.; Scaggiante, B.; Zanconati, F.; Bonazza, D.; Grassi, M.; et al. Targeted delivery of sirnas against hepatocellular carcinoma-related genes by a galactosylated polyaspartamide copolymer. J. Control Release 2021, 330, 1132–1151. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | Gene ID | |
|---|---|---|---|
| acta2 | F: GCATCCACGAAACCACCTA | NM_007392.3 | |
| R: CACGAGTAACAAATCAAAGC | |||
| col1a1 | F: CAATGGCACGGCTGTGTGCG | NM_007742.4 | |
| R: AGCACTCGCCCTCCCGTCTT | |||
| gapdh | F: AAGTTGTCATGGATGACC | NM_001289726.1 | |
| R: TCACCATCTTCCAGGAGC | |||
| lrat | F: CTGACCAATGACAAGGAACGCACTC | NM_023624.4 | |
| R: CTAATCCCAAGACAGCCGAAGCAAGAC | |||
| Map1lc3b | F: TTCTTCCTCCTGGTGAATGG | NM_026160.5 | |
| R: GTGGGTGCCTACGTTCTCAT | |||
| Mki67 |
F: AATCCAACTCAAGTAAACGGGG R: TTGGCTTGCTTCCATCCTCA | NM_001081117.2 | |
| Antibody | Source | Dilution | |
| collagen I | Abcam, ab270993 | 1:200 | |
| cytokeratin 7 | Abcam, ab199718 | 1:500 | |
| desmin | Abcam, ab8592 | 1:300 | |
| lc-3b | Abcam, ab192890 | 1:200 | |
| α-sma | Abcam, ab15734 | 1:300 | |
| sqstm1/p62 | Abcam, ab91526 | 1:500 | |
| Goat Anti-Rabbit IgG H&L (Alexa Flour 488- conjugated) | Abcam, Ab150077 | 1:500 | |
| Goat anti-Rabbit IgG HRP-conjugated | Abcam, ab6721 | 1:500 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.V.; Phan-Thi, H.-T.; Huynh-Thi, M.-X.; Dang, T.M.; Holterman, A.X.L.; Grassi, G.; Nguyen-Luu, T.-U.; Truong, N.H. Autophagy Inhibitor Chloroquine Downmodulates Hepatic Stellate Cell Activation and Liver Damage in Bile-Duct-Ligated Mice. Cells 2023, 12, 1025. https://doi.org/10.3390/cells12071025
Le TV, Phan-Thi H-T, Huynh-Thi M-X, Dang TM, Holterman AXL, Grassi G, Nguyen-Luu T-U, Truong NH. Autophagy Inhibitor Chloroquine Downmodulates Hepatic Stellate Cell Activation and Liver Damage in Bile-Duct-Ligated Mice. Cells. 2023; 12(7):1025. https://doi.org/10.3390/cells12071025
Chicago/Turabian StyleLe, Trinh Van, Hong-Thuy Phan-Thi, My-Xuan Huynh-Thi, Thanh Minh Dang, Ai Xuan Le Holterman, Gabriele Grassi, Thao-Uyen Nguyen-Luu, and Nhung Hai Truong. 2023. "Autophagy Inhibitor Chloroquine Downmodulates Hepatic Stellate Cell Activation and Liver Damage in Bile-Duct-Ligated Mice" Cells 12, no. 7: 1025. https://doi.org/10.3390/cells12071025