
Absence of HDAC3 by Matrix Stiffness Promotes Chromatin Remodeling and Fibroblast Activation in Idiopathic Pulmonary Fibrosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Immunofluorescence
2.3. Immunoblotting
2.4. Transmission Electron Microscopy
2.5. Quantitative PCR
2.6. Polyacrylamide Hydrogels
2.7. Statistical Analysis
3. Results
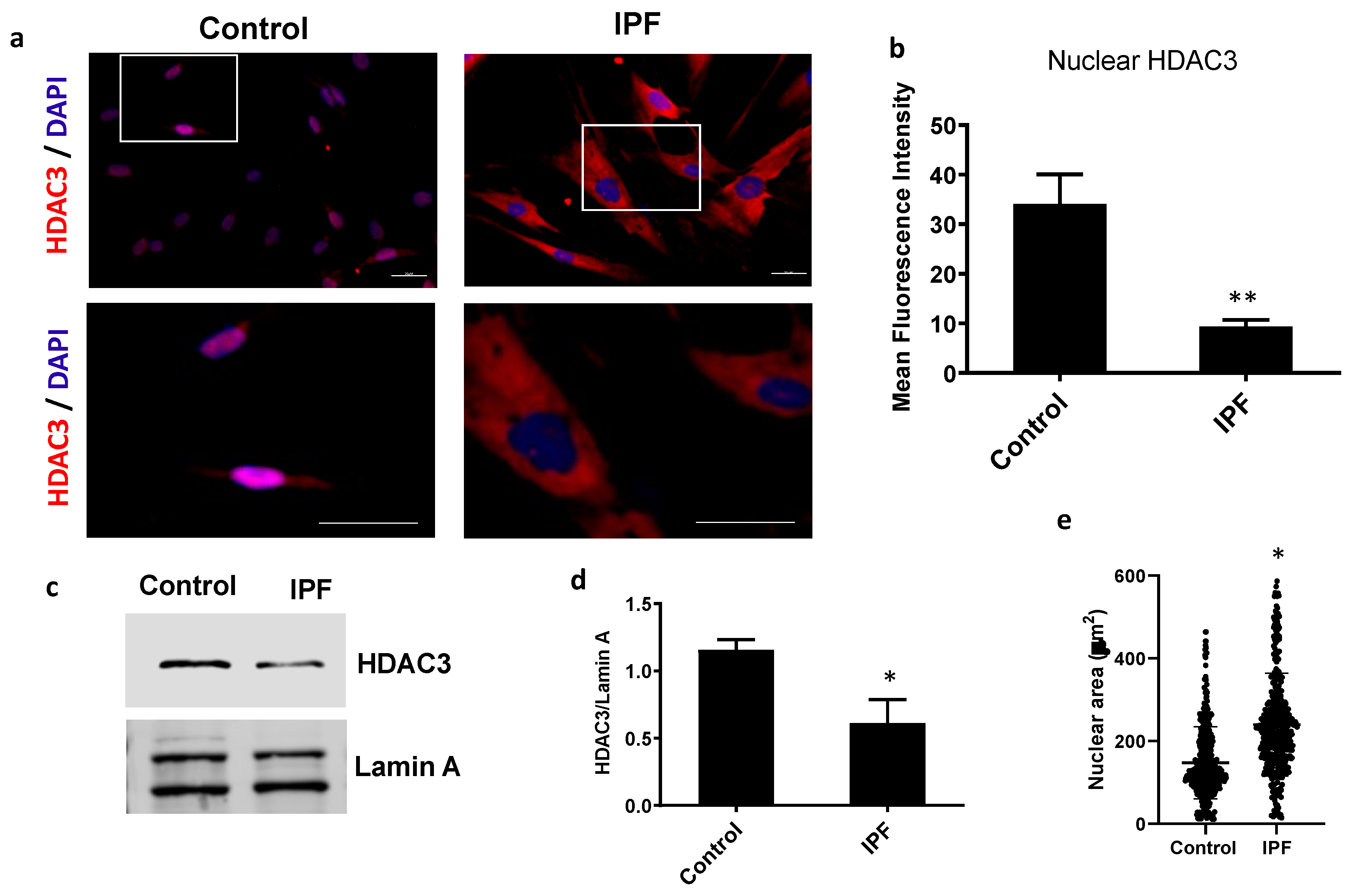
3.1. IPF Fibroblasts Have a Decrease in Nuclear HDAC3 Associated with Chromatin Changes
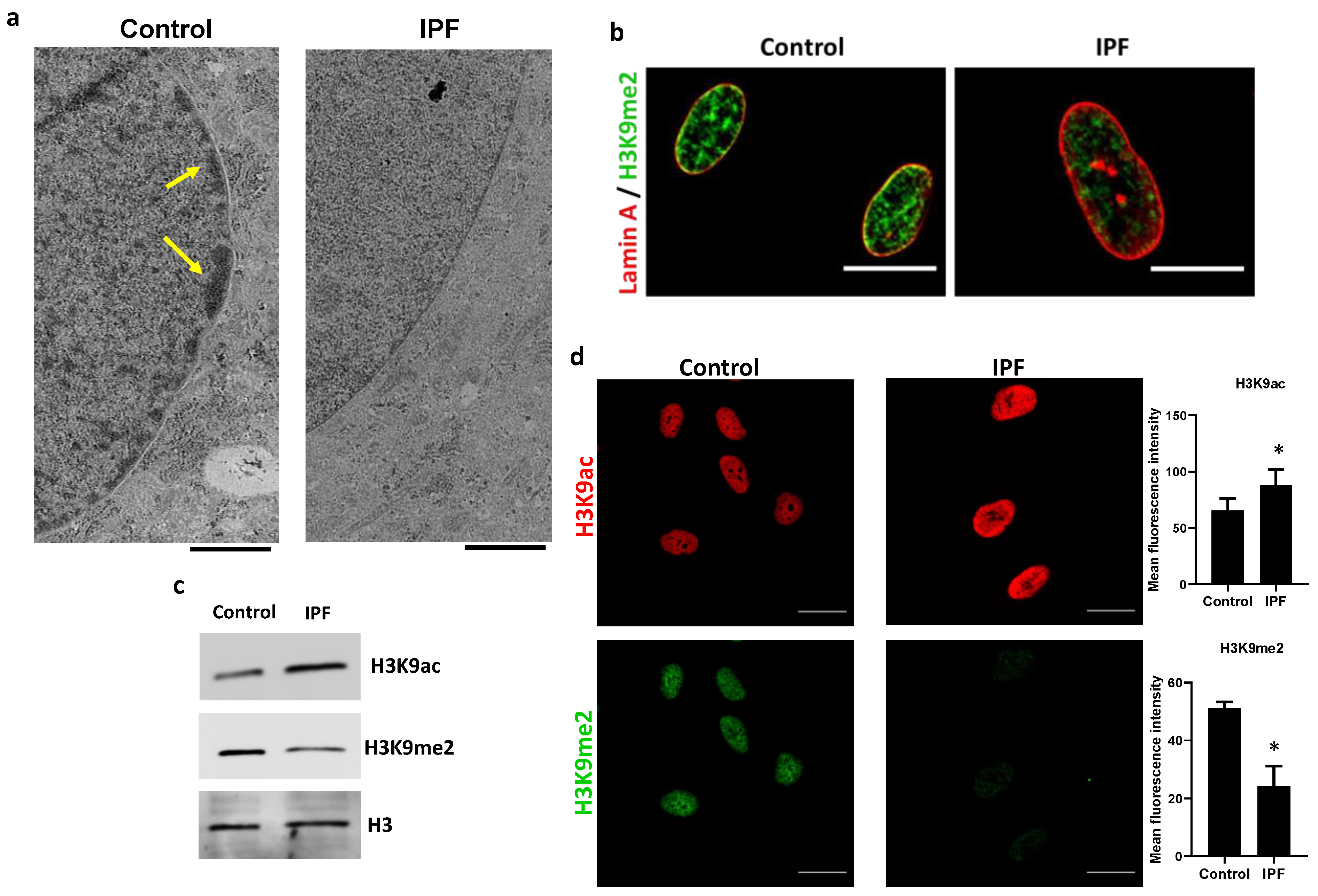
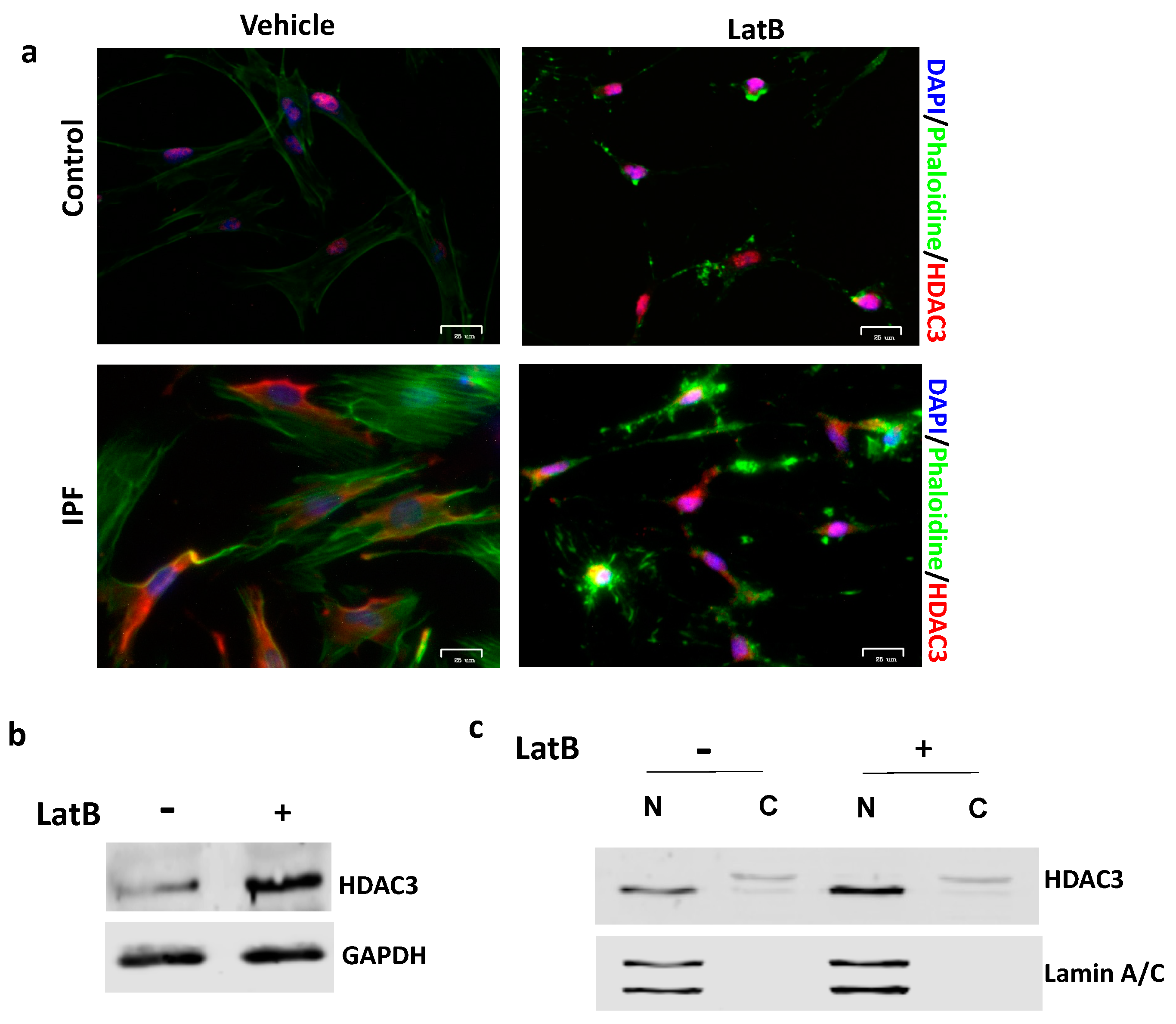
3.2. Nuclear HDAC3 Mislocalization Decreases Heterochromatin Histone Marks and Promotes Profibrotic Gene Expression
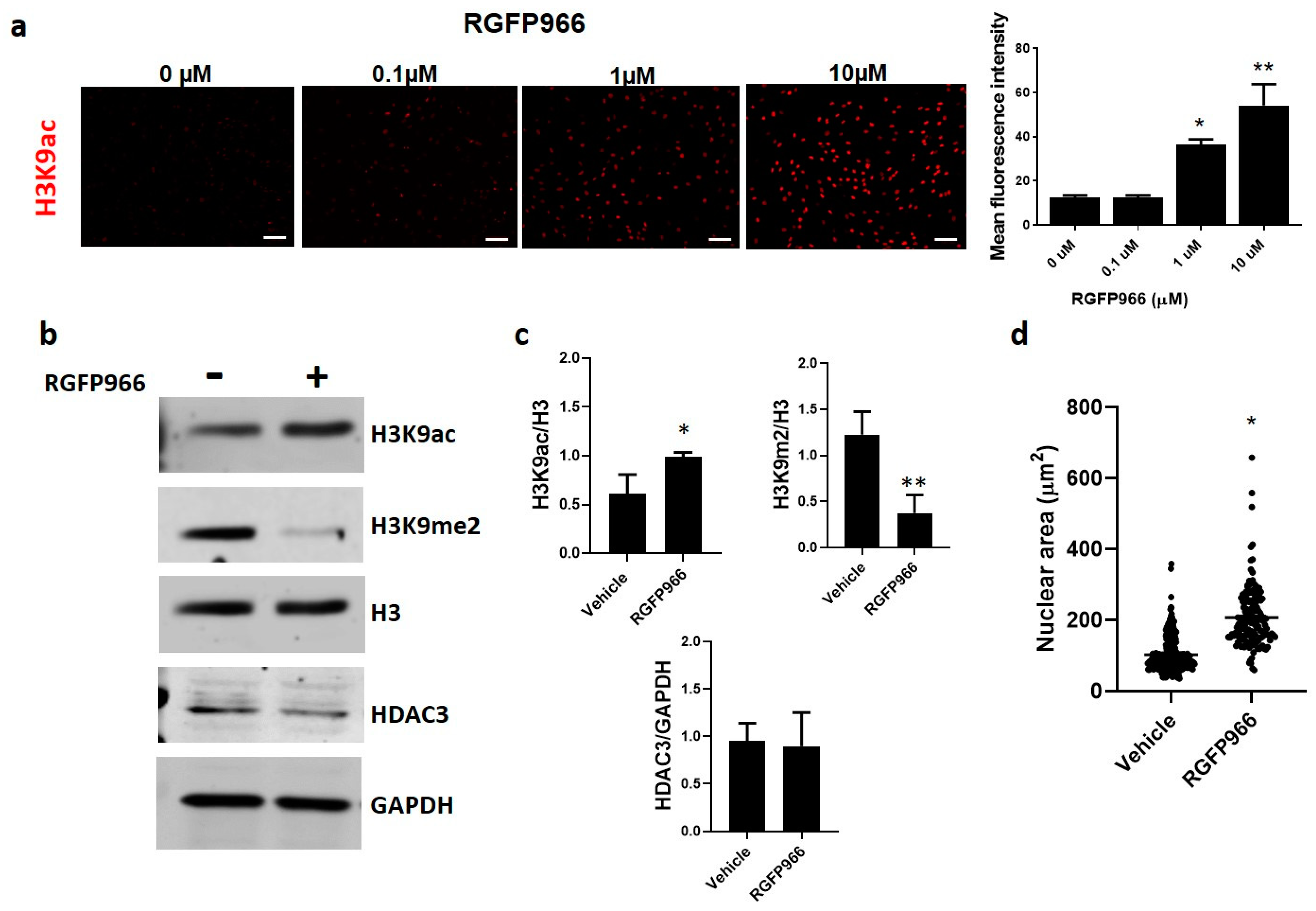
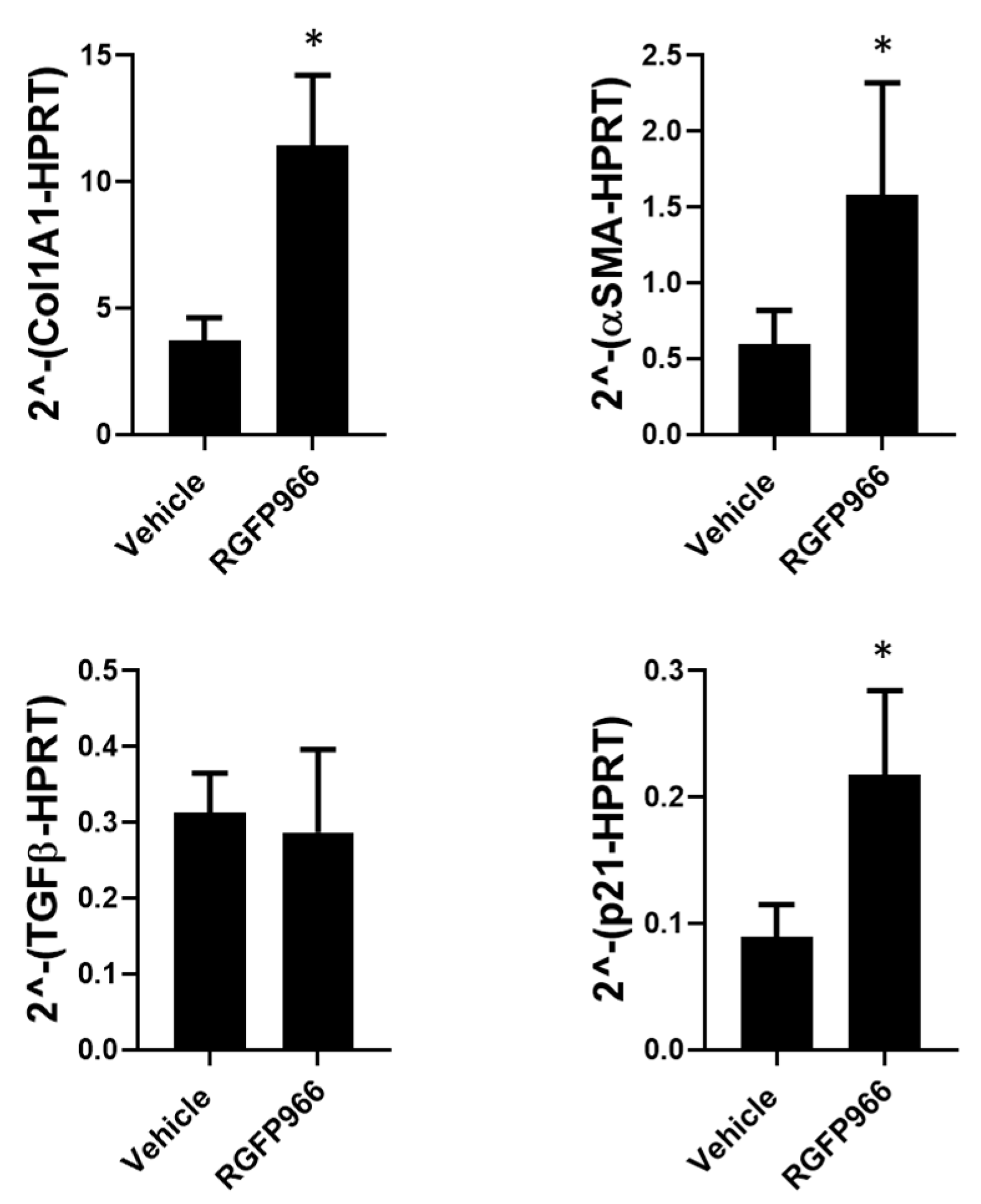
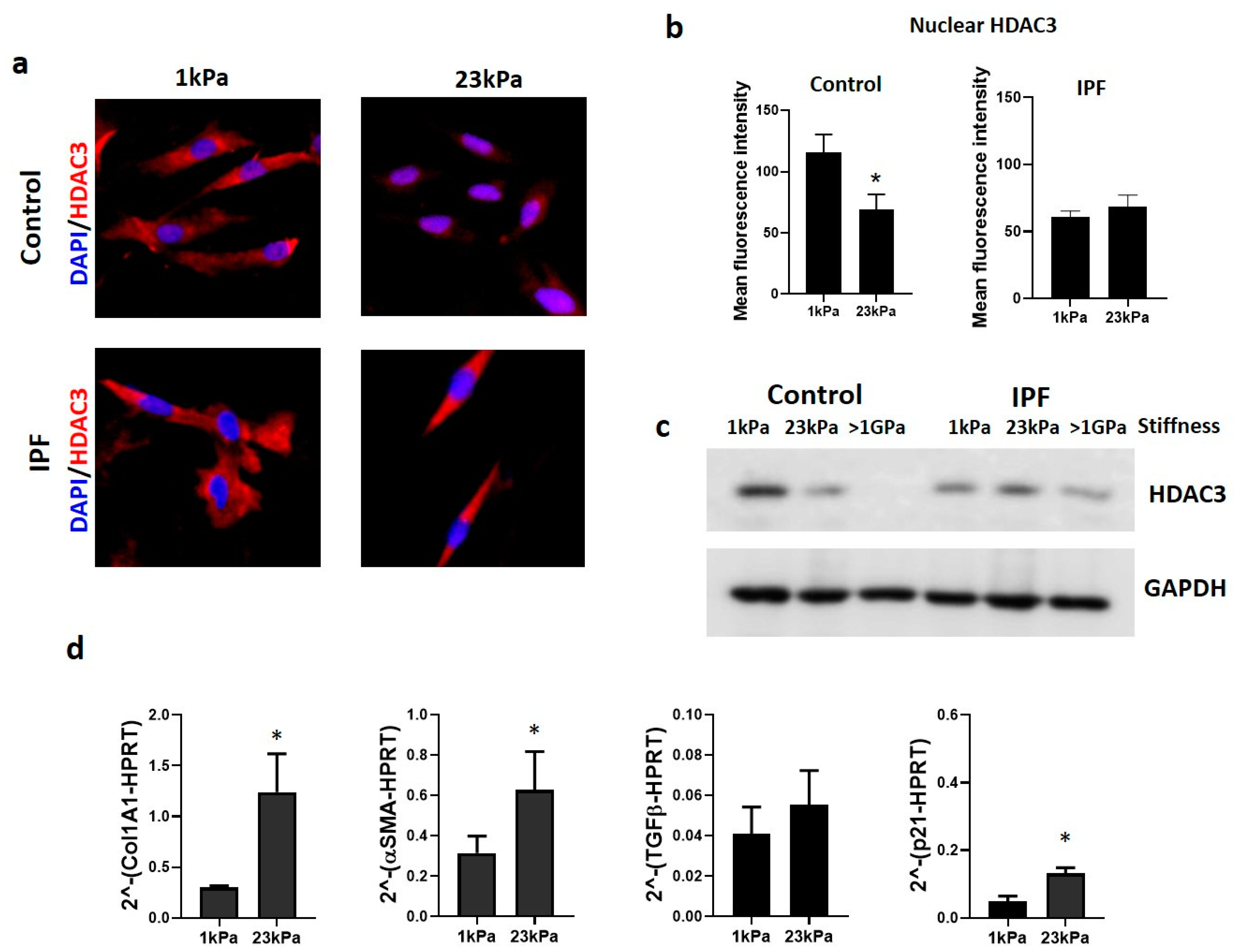
3.3. Extracellular Matrix Stiffness Causes a Decrease in HDAC3, Promoting Fibrosis-Related Gene Expression
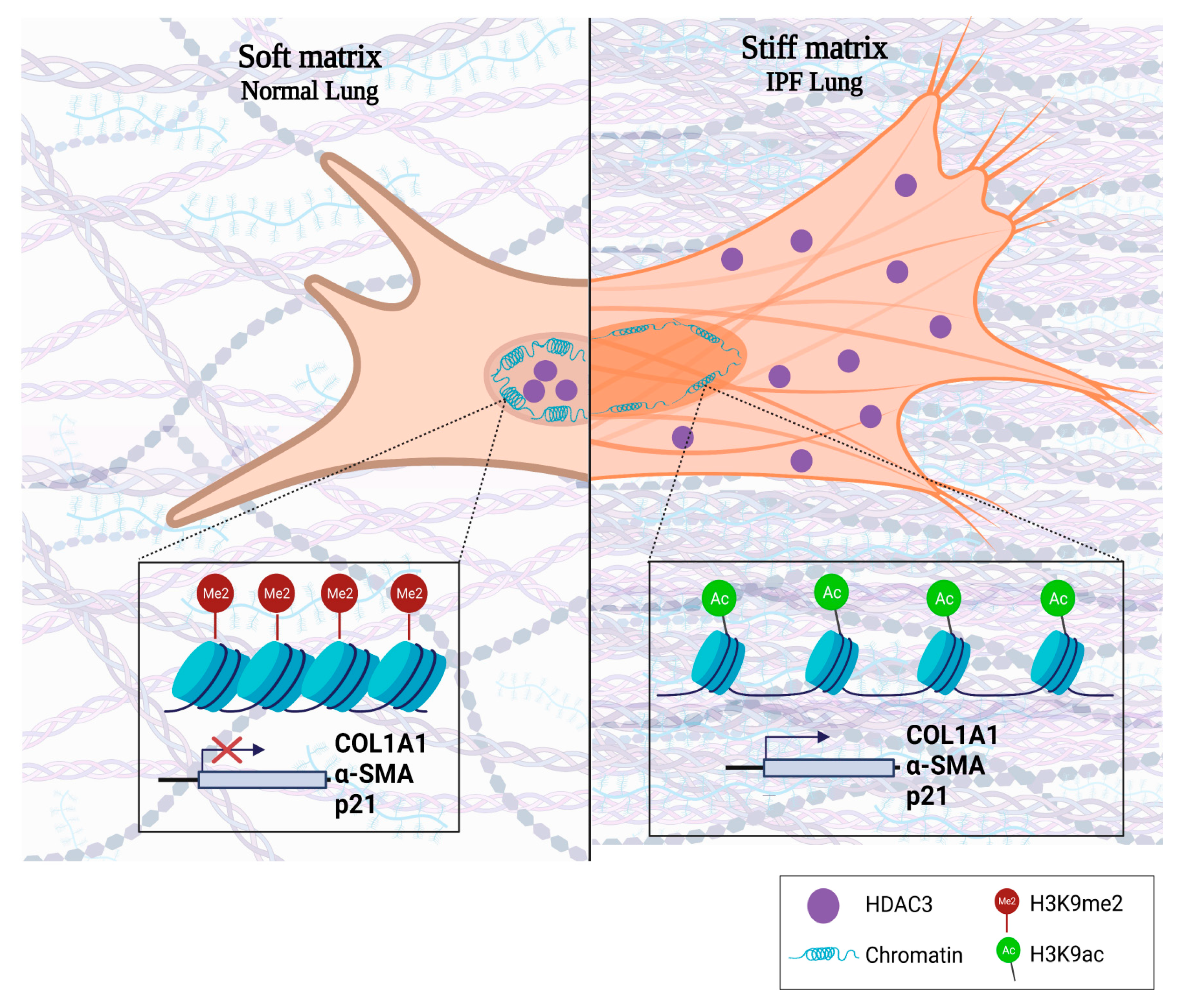
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, T.E.; Pardo, A.; Selman, M. Idiopathic Pulmonary Fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.; Selman, M. The Interplay of the Genetic Architecture, Aging, and Environmental Factors in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Fibroageing: An Ageing Pathological Feature Driven by Dysregulated Extracellular Matrix-Cell Mechanobiology. Ageing Res. Rev. 2021, 70, 101393. [Google Scholar] [CrossRef] [PubMed]
- Romero, Y.; Aquino-Gálvez, A. Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution. Int. J. Mol. Sci. 2021, 22, 8335. [Google Scholar] [CrossRef] [PubMed]
- Vigetti, D.; Viola, M.; Karousou, E.; Deleonibus, S.; Karamanou, K.; De Luca, G.; Passi, A. Epigenetics in Extracellular Matrix Remodeling and Hyaluronan Metabolism. FEBS J. 2014, 281, 4980–4992. [Google Scholar] [CrossRef]
- Tsou, P.-S.; Varga, J.; O’Reilly, S. Advances in Epigenetics in Systemic Sclerosis: Molecular Mechanisms and Therapeutic Potential. Nat. Rev. Rheumatol. 2021, 17, 596–607. [Google Scholar] [CrossRef]
- Verdone, L. Histone Acetylation in Gene Regulation. Brief. Funct. Genom. Proteom. 2006, 5, 209–221. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-J.; Seto, E. HATs and HDACs: From Structure, Function and Regulation to Novel Strategies for Therapy and Prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Korfei, M.; Skwarna, S.; Henneke, I.; MacKenzie, B.; Klymenko, O.; Saito, S.; Ruppert, C.; von der Beck, D.; Mahavadi, P.; Klepetko, W.; et al. Aberrant Expression and Activity of Histone Deacetylases in Sporadic Idiopathic Pulmonary Fibrosis. Thorax 2015, 70, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Lyu, X.; Hu, M.; Peng, J.; Zhang, X.; Sanders, Y.Y. HDAC Inhibitors as Antifibrotic Drugs in Cardiac and Pulmonary Fibrosis. Ther. Adv. Chronic Dis. 2019, 10, 204062231986269. [Google Scholar] [CrossRef] [Green Version]
- Sanders, Y.Y.; Hagood, J.S.; Liu, H.; Zhang, W.; Ambalavanan, N.; Thannickal, V.J. Histone Deacetylase Inhibition Promotes Fibroblast Apoptosis and Ameliorates Pulmonary Fibrosis in Mice. Eur. Respir. J. 2014, 43, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Li, Q.; Lin, Y.; Zuo, H.; Cui, Y.; Chen, S.; Chen, Z.; Liu, H. Coordinated Expression of p300 and HDAC3 Upregulates Histone Acetylation during Dentinogenesis. J. Cell. Biochem. 2020, 121, 2478–2488. [Google Scholar] [CrossRef]
- Bossone, K.A.; Ellis, J.A.; Holaska, J.M. Histone Acetyltransferase Inhibition Rescues Differentiation of Emerin-deficient Myogenic Progenitors. Muscle Nerve 2020, 62, 128–136. [Google Scholar] [CrossRef]
- Godman, C.A.; Joshi, R.; Tierney, B.R.; Greenspan, E.; Rasmussen, T.P.; Wang, H.-W.; Shin, D.-G.; Rosenberg, D.W.; Giardina, C. HDAC3 Impacts Multiple Oncogenic Pathways in Colon Cancer Cells with Effects on Wnt and Vitamin D Signaling. Cancer Biol. Ther. 2008, 7, 1570–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenther, M.G.; Yu, J.; Kao, G.D.; Yen, T.J.; Lazar, M.A. Assembly of the SMRT–histone Deacetylase 3 Repression Complex Requires the TCP-1 Ring Complex. Genes Dev. 2002, 16, 3130–3135. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.-L.; Bonine-Summers, A.R.; et al. Hdac3 Is Essential for the Maintenance of Chromatin Structure and Genome Stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poleshko, A.; Shah, P.P.; Gupta, M.; Babu, A.; Morley, M.P.; Manderfield, L.J.; Ifkovits, J.L.; Calderon, D.; Aghajanian, H.; Sierra-Pagán, J.E.; et al. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell 2017, 171, 573–587.e14. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Mih, J.D.; Shea, B.S.; Kho, A.T.; Sharif, A.S.; Tager, A.M.; Tschumperlin, D.J. Feedback Amplification of Fibrosis through Matrix Stiffening and COX-2 Suppression. J. Cell Biol. 2010, 190, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Cretu, A.; Castagnino, P.; Assoian, R. Studying the Effects of Matrix Stiffness on Cellular Function Using Acrylamide-Based Hydrogels. J. Vis. Exp. 2010, 42, e2089. [Google Scholar] [CrossRef] [Green Version]
- Tse, J.R.; Engler, A.J. Preparation of Hydrogel Substrates with Tunable Mechanical Properties. Curr. Protoc. Cell Biol. 2010, 47. [Google Scholar] [CrossRef]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-Selective Inhibitor Enhances Extinction of Cocaine-Seeking Behavior in a Persistent Manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [Green Version]
- Nava, M.M.; Miroshnikova, Y.A.; Biggs, L.C.; Whitefield, D.B.; Metge, F.; Boucas, J.; Vihinen, H.; Jokitalo, E.; Li, X.; García Arcos, J.M.; et al. Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell 2020, 181, 800–817.e22. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gao, Q.; Zhang, L.; Ding, Y.; Wang, H.; Cao, W. Inhibiting HDAC3 (Histone Deacetylase 3) Aberration and the Resultant Nrf2 (Nuclear Factor Erythroid-Derived 2-Related Factor-2) Repression Mitigates Pulmonary Fibrosis. Hypertension 2021, 78, e15–e25. [Google Scholar] [CrossRef]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR Corepressors Are Activating Cofactors for Histone Deacetylase 3. Mol. Cell. Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, J.; Wang, J.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J. Both Corepressor Proteins SMRT and N-CoR Exist in Large Protein Complexes Containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.-L.; Ueki, N.; Marcelain, K.; Hayman, M.J. The Ski Protein Can Inhibit Ligand Induced RARalpha and HDAC3 Degradation in the Retinoic Acid Signaling Pathway. Biochem. Biophys. Res. Commun. 2009, 383, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, Y.; Bueno, M.; Ramirez, R.; Álvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. mTORC1 Activation Decreases Autophagy in Aging and Idiopathic Pulmonary Fibrosis and Contributes to Apoptosis Resistance in IPF Fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef]
- Karagianni, P.; Wong, J. HDAC3: Taking the SMRT-N-CoRrect Road to Repression. Oncogene 2007, 26, 5439–5449. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Shi, Y.; Wang, L.; Sriram, S. Role of HDAC3 on p53 Expression and Apoptosis in T Cells of Patients with Multiple Sclerosis. PLoS ONE 2011, 6, e16795. [Google Scholar] [CrossRef] [Green Version]
- Shan, X.; Fu, Y.-S.; Aziz, F.; Wang, X.-Q.; Yan, Q.; Liu, J.-W. Ginsenoside Rg3 Inhibits Melanoma Cell Proliferation through Down-Regulation of Histone Deacetylase 3 (HDAC3) and Increase of p53 Acetylation. PLoS ONE 2014, 9, e115401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, J.; Huang, X.; Yang, Y.; Zhu, W.-G.; Gu, W.; Luo, J. Regulation of Histone Acetyltransferase TIP60 Function by Histone Deacetylase 3. J. Biol. Chem. 2014, 289, 33878–33886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The Histone Deacetylase Inhibitor MS-275 Promotes Differentiation or Apoptosis in Human Leukemia Cells through a Process Regulated by Generation of Reactive Oxygen Species and Induction of p21CIP1/WAF1 1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Jevtić, P.; Levy, D.L. Mechanisms of Nuclear Size Regulation in Model Systems and Cancer. Adv. Exp. Med. Biol. 2014, 773, 537–569. [Google Scholar]
- Okudela, K. An Association between Nuclear Morphology and Immunohistochemical Expression of p53 and p16INK4A in Lung Cancer Cells. Med. Mol. Morphol. 2014, 47, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Zeimet, A.G.; Fiegl, H.; Goebel, G.; Kopp, F.; Allasia, C.; Reimer, D.; Steppan, I.; Mueller-Holzner, E.; Ehrlich, M.; Marth, C. DNA Ploidy, Nuclear Size, Proliferation Index and DNA-Hypomethylation in Ovarian Cancer. Gynecol. Oncol. 2011, 121, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samarnthai, N.; Elledge, R.; Prihoda, T.J.; Huang, J.; Massarweh, S.; Yeh, I.-T. Pathologic Changes in Breast Cancer After Anti-Estrogen Therapy. Breast J. 2012, 18, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chu, J.S.; Kurpinski, K.; Li, X.; Bautista, D.M.; Yang, L.; Sung, K.-L.P.; Li, S. Biophysical Regulation of Histone Acetylation in Mesenchymal Stem Cells. Biophys. J. 2011, 100, 1902–1909. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Iyer, K.V.; Kumar, A.; Shivashankar, G.V. Cell Geometric Constraints Induce Modular Gene-Expression Patterns via Redistribution of HDAC3 Regulated by Actomyosin Contractility. Proc. Natl. Acad. Sci. USA 2013, 110, 11349–11354. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, H.M.; Pelzel, H.R.; Schlamp, C.L.; Nickells, R.W. Histone Deacetylase 3 (HDAC3) Plays an Important Role in Retinal Ganglion Cell Death after Acute Optic Nerve Injury. Mol. Neurodegener. 2014, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Sterner, D.E.; Berger, S.L. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambliss, A.B.; Wu, P.-H.; Chen, W.-C.; Sun, S.X.; Wirtz, D. Simultaneously Defining Cell Phenotypes, Cell Cycle, and Chromatin Modifications at Single-Cell Resolution. FASEB J. 2013, 27, 2667–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demmerle, J.; Koch, A.J.; Holaska, J.M. Emerin and Histone Deacetylase 3 (HDAC3) Cooperatively Regulate Expression and Nuclear Positions of MyoD, Myf5, and Pax7 Genes during Myogenesis. Chromosome Res. 2013, 21, 765–779. [Google Scholar] [CrossRef] [Green Version]
- Somech, R.; Shaklai, S.; Geller, O.; Amariglio, N.; Simon, A.J.; Rechavi, G.; Gal-Yam, E.N. The Nuclear-Envelope Protein and Transcriptional Repressor LAP2β Interacts with HDAC3 at the Nuclear Periphery, and Induces Histone H4 Deacetylation. J. Cell Sci. 2005, 118, 4017–4025. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.-F.; Zhou, Z.-Q.; Guo, J.; Gu, H.-W.; Su, M.-Z.; Yu, B.-C.; Zhou, F.; Han, B.-Y.; Jia, M.; Ji, M.-H.; et al. Histone Deacetylase 3 in Hippocampus Contributes to Memory Impairment after Chronic Constriction Injury of Sciatic Nerve in Mice. Pain 2021, 162, 382–395. [Google Scholar] [CrossRef]
- Ji, H.; Zhou, Y.; Zhuang, X.; Zhu, Y.; Wu, Z.; Lu, Y.; Li, S.; Zeng, Y.; Lu, Q.R.; Huo, Y.; et al. HDAC3 Deficiency Promotes Liver Cancer through a Defect in H3K9ac/H3K9me3 Transition. Cancer Res. 2019, 79, 3676–3688. [Google Scholar] [CrossRef]
- Hanmandlu, A.; Zhu, L.; Mertens, T.C.J.; Collum, S.; Bi, W.; Xiong, F.; Wang, R.; Amirthalingam, R.T.; Ren, D.; Han, L.; et al. Transcriptomic and Epigenetic Profiling of Fibroblasts in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 53–63. [Google Scholar] [CrossRef]
- Liang, H.; Wang, Q.; Wang, D.; Zheng, H.; Kalvakolanu, D.V.; Lu, H.; Wen, N.; Chen, X.; Xu, L.; Ren, J.; et al. RGFP966, a Histone Deacetylase 3 Inhibitor, Promotes Glioma Stem Cell Differentiation by Blocking TGF-β Signaling via SMAD7. Biochem. Pharmacol. 2020, 180, 114118. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, Z. Fibroblast Senescence in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 593283. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, H.; Zhou, J.Q.; Krick, S.; Barnes, J.W.; Thannickal, V.J.; Sanders, Y.Y. Modulation of H4K16Ac Levels Reduces pro-Fibrotic Gene Expression and Mitigates Lung Fibrosis in Aged Mice. Theranostics 2022, 12, 530–541. [Google Scholar] [CrossRef]
- Huang, X.; Yang, N.; Fiore, V.F.; Barker, T.H.; Sun, Y.; Morris, S.W.; Ding, Q.; Thannickal, V.J.; Zhou, Y. Matrix Stiffness–Induced Myofibroblast Differentiation Is Mediated by Intrinsic Mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 340–348. [Google Scholar] [CrossRef] [Green Version]
- Damodaran, K.; Venkatachalapathy, S.; Alisafaei, F.; Radhakrishnan, A.V.; Sharma Jokhun, D.; Shenoy, V.B.; Shivashankar, G.V. Compressive Force Induces Reversible Chromatin Condensation and Cell Geometry-Dependent Transcriptional Response. Mol. Biol. Cell 2018, 29, 3039–3051. [Google Scholar] [CrossRef] [PubMed]
- Stowers, R.S.; Shcherbina, A.; Israeli, J.; Gruber, J.J.; Chang, J.; Nam, S.; Rabiee, A.; Teruel, M.N.; Snyder, M.P.; Kundaje, A.; et al. Matrix Stiffness Induces a Tumorigenic Phenotype in Mammary Epithelium through Changes in Chromatin Accessibility. Nat. Biomed. Eng. 2019, 3, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Meridew, J.A.; Link, P.A.; Ducharme, M.T.; Lydon, K.L.; Choi, K.M.; Caporarello, N.; Tan, Q.; Diaz Espinosa, A.M.; Xiong, Y.; et al. ZNF416 Is a Pivotal Transcriptional Regulator of Fibroblast Mechanoactivation. J. Cell Biol. 2021, 220, e202007152. [Google Scholar] [CrossRef]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikström, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic Extracellular Matrix Activates a Profibrotic Positive Feedback Loop. J. Clin. Investig. 2014, 124, 1622–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.J.; Batan, D.; Bishop, C.T.; Ramirez, D.; Aguado, B.A.; Schroeder, M.E.; Crocini, C.; Schwisow, J.; Moulton, K.; Macdougall, L.; et al. Extracellular Matrix Stiffness Controls Cardiac Valve Myofibroblast Activation through Epigenetic Remodeling. Bioeng. Transl. Med. 2022, 7, e10394. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toscano-Marquez, F.; Romero, Y.; Espina-Ordoñez, M.; Cisneros, J. Absence of HDAC3 by Matrix Stiffness Promotes Chromatin Remodeling and Fibroblast Activation in Idiopathic Pulmonary Fibrosis. Cells 2023, 12, 1020. https://doi.org/10.3390/cells12071020
Toscano-Marquez F, Romero Y, Espina-Ordoñez M, Cisneros J. Absence of HDAC3 by Matrix Stiffness Promotes Chromatin Remodeling and Fibroblast Activation in Idiopathic Pulmonary Fibrosis. Cells. 2023; 12(7):1020. https://doi.org/10.3390/cells12071020
Chicago/Turabian StyleToscano-Marquez, Fernanda, Yair Romero, Marco Espina-Ordoñez, and José Cisneros. 2023. "Absence of HDAC3 by Matrix Stiffness Promotes Chromatin Remodeling and Fibroblast Activation in Idiopathic Pulmonary Fibrosis" Cells 12, no. 7: 1020. https://doi.org/10.3390/cells12071020