
Induced Pluripotent Stem Cells and Their Applications in Amyotrophic Lateral Sclerosis
 , , , , , and
, , , , , and
Abstract
:1. Introduction
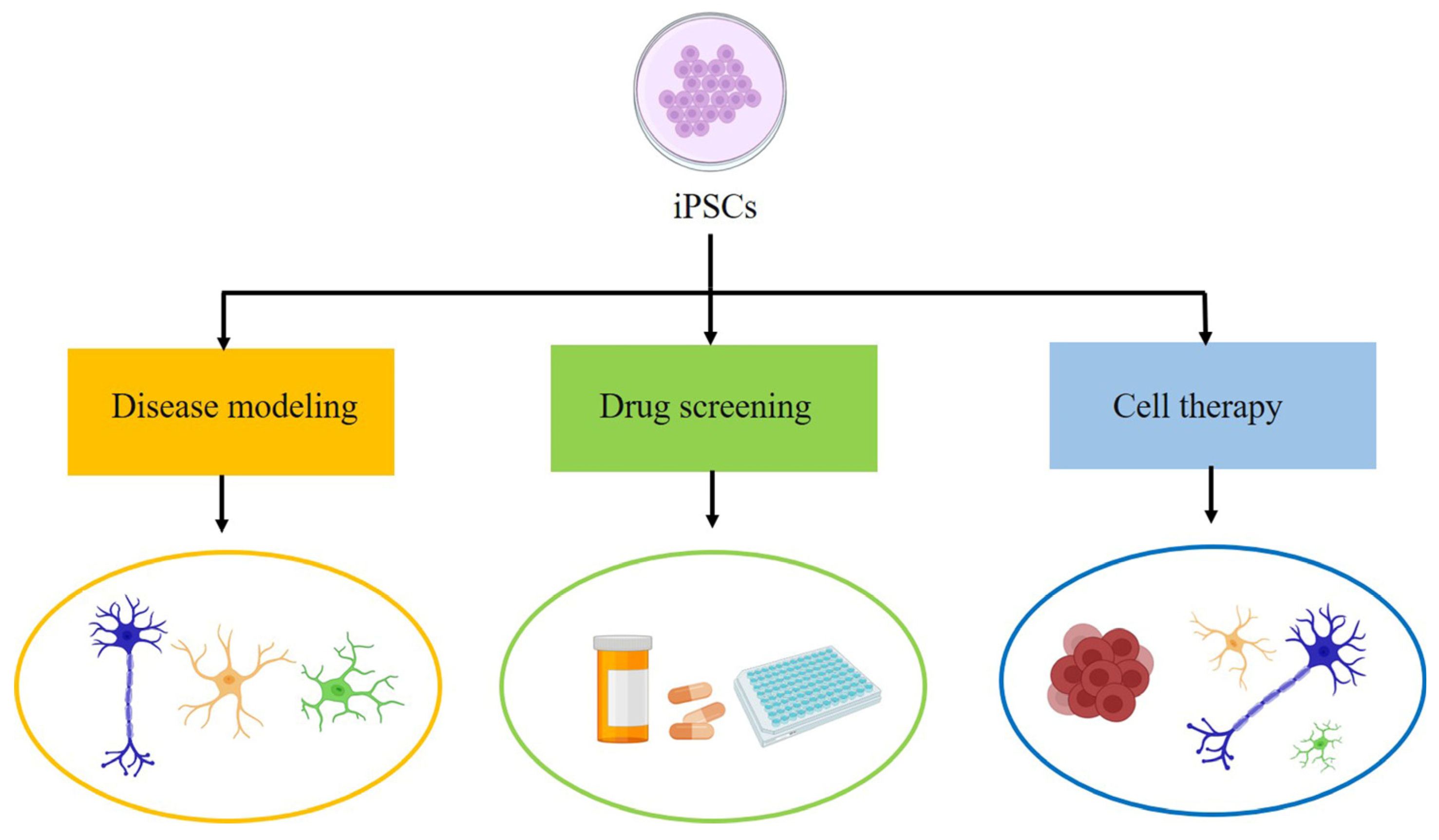
2. Induced Pluripotent Stem Cells
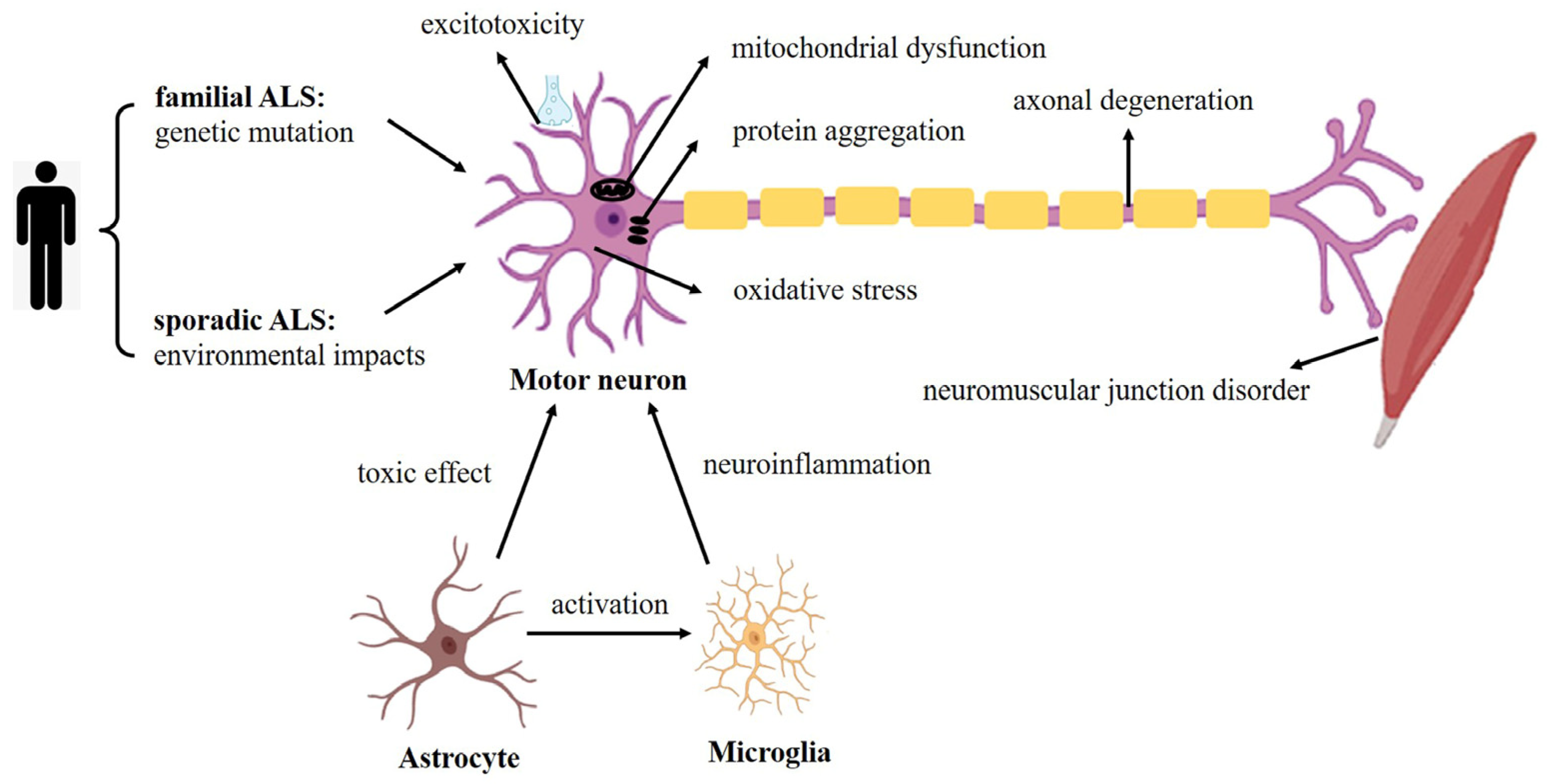
3. Modeling of ALS
3.1. iPSC-Derived Motor Neurons in ALS Modeling
3.2. iPSC-Derived Glial Cells in ALS Modeling
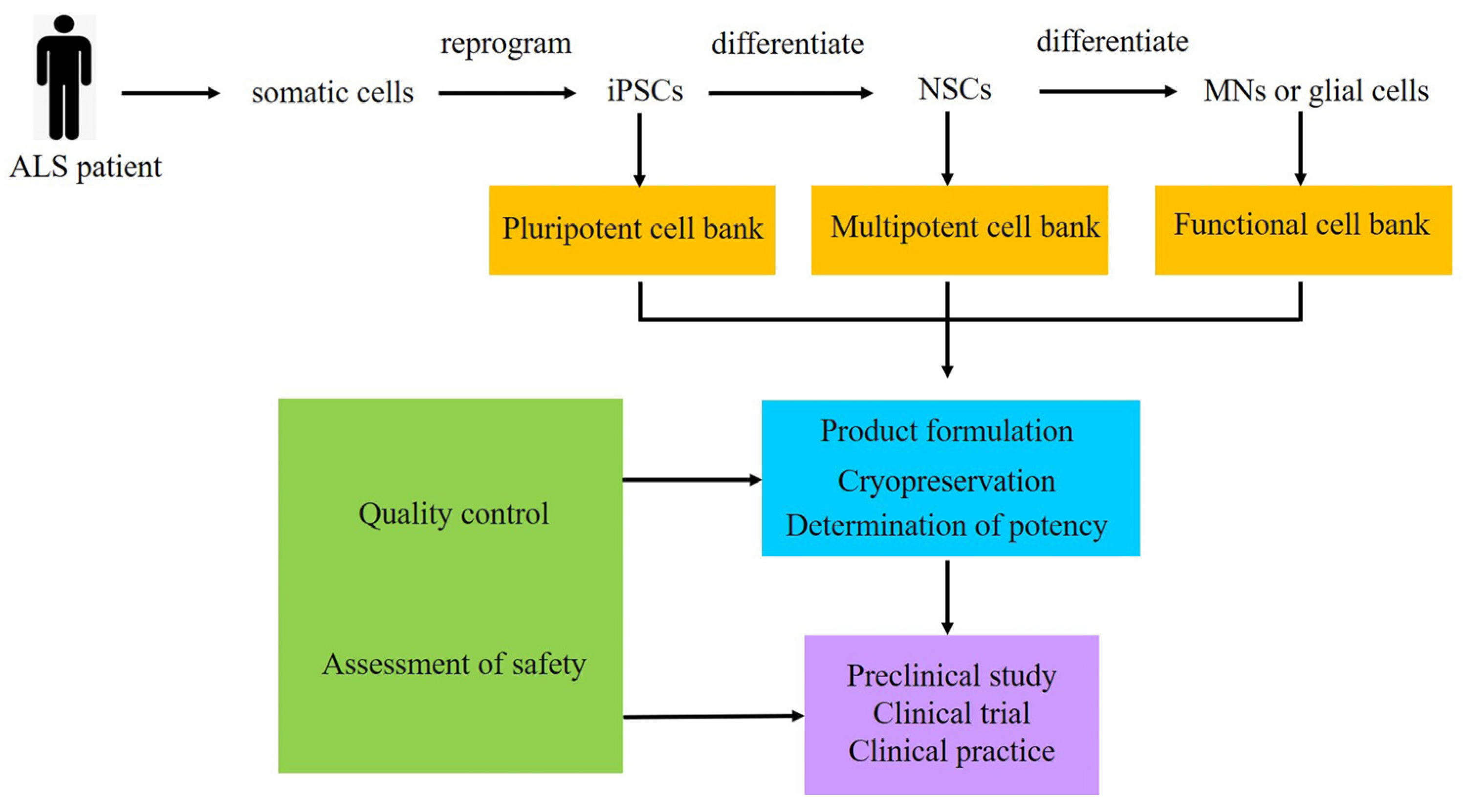
4. Cell Therapy of ALS
5. Drug Screening of ALS
6. Other Applications
7. Conclusions and Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| CNS | central nervous system |
| fALS | familial amyotrophic lateral sclerosis |
| sALS | sporadic amyotrophic lateral sclerosis |
| FDA | Food and Drug Administration |
| iPSCs | induced pluripotent stem cells |
| ESCs | embryonic stem cells |
| MNs | motor neurons |
| hiPSCs | human induced pluripotent stem cells |
| iPSCs-MNs | induced pluripotent stem cells derived motor neurons |
| ESCs-MNs | embryonic stem cells derived motor neurons |
| NPCs | neural progenitor cells |
| iPSCs-NPCs | induced pluripotent stem cells derived neural progenitor cells |
| NSCs | neural stem cells |
| iPSCs-NSCs | induced pluripotent stem cells derived neural stem cells |
| NMJ | neuromuscular junction |
| SOD1 | superoxide dismutase 1 |
| C9orf72 | chromosome nine open reading frame 72 |
| TARDBP | TAR DNA binding protein |
| FUS | fused in sarcoma |
| NfH | neurofilament heavy chain |
| NfL | neurofilament light chain |
| CSF | cerebrospinal fluid |
| HD-MEAs | high density microelectrode-arrays |
| DPR | dipeptide repeat |
| TDP-43 | TAR DNA binding protein 43 |
| RNAP II | RNA polymerase II |
| EAAT2 | excitatory amino acid transporter 2 |
| SG | stress granule |
| G3BP1 | GTPase activating protein (SH3 domain) binding protein 1 |
| Il-1α | interleukin 1α |
| TNF | tumor necrosis factor |
| C1q | complement component 1, subcomponent q |
| ROPI | ropinirole |
| HDAC | histone deacetylase |
| FTD | frontotemporal dementia |
| STAT3 | signal transducer and activator of transcription 3 |
| TGF-β | transforming growth factor-β |
References
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [PubMed] [Green Version]
- Pender, N.; Pinto-Grau, M.; Hardiman, O. Cognitive and behavioural impairment in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2020, 33, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, T.; Liu, L.; Yao, X.; Chen, L.; Fan, D.; Zhan, S.; Wang, S. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. 2020, 267, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Talbott, E.O.; Malek, A.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2016, 138, 225–238. [Google Scholar] [PubMed]
- Pham, J.; Keon, M.; Brennan, S.; Saksena, N. Connecting RNA-Modifying Similarities of TDP-43, FUS, and SOD1 with MicroRNA Dysregulation Amidst a Renewed Network Perspective of Amyotrophic Lateral Sclerosis Proteinopathy. Int. J. Mol. Sci. 2020, 21, 3464. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Kato, S.; Kato, M.; Abe, Y.; Matsumura, T.; Nishino, T.; Aoki, M.; Itoyama, Y.; Asayama, K.; Awaya, A.; Hirano, A.; et al. Redox system expression in the motor neurons in amyotrophic lateral sclerosis (ALS): Immunohistochemical studies on sporadic ALS, superoxide dismutase 1 (SOD1)-mutated familial ALS, and SOD1-mutated ALS animal models. Acta Neuropathol. 2005, 110, 101–112. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.; Rashid, S. Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Costa, J.; Gomes, C.; de Carvalho, M. Diagnosis, pathogenesis and therapeutic targets in amyotrophic lateral sclerosis. CNS Neurol. Disord. Drug Targets 2010, 9, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Van Harten, A.C.M.; Phatnani, H.; Przedborski, S. Non-cell-autonomous pathogenic mechanisms in amyotrophic lateral sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Sehgal, A.; Bhardwaj, S.; Singh, S.; Buhas, C.; Judea-Pusta, C.; Uivarosan, D.; Munteanu, M.A.; Bungau, S. Multifaceted Role of Matrix Metalloproteinases in Neurodegenerative Diseases: Pathophysiological and Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 1413. [Google Scholar] [CrossRef] [PubMed]
- Fang, X. Potential role of gut microbiota and tissue barriers in Parkinson’s disease and amyotrophic lateral sclerosis. Int. J. Neurosci. 2016, 126, 771–776. [Google Scholar] [CrossRef]
- Zeng, Q.; Shen, J.; Chen, K.; Zhou, J.; Liao, Q.; Lu, K.; Yuan, J.; Bi, F. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci. Rep. 2020, 10, 12998. [Google Scholar] [CrossRef]
- Niccolai, E.; Di Pilato, V.; Nannini, G.; Baldi, S.; Russo, E.; Zucchi, E.; Martinelli, I.; Menicatti, M.; Bartolucci, G.; Mandrioli, J.; et al. The Gut Microbiota-Immunity Axis in ALS: A Role in Deciphering Disease Heterogeneity? Biomedicines 2021, 9, 753. [Google Scholar] [CrossRef]
- Nicholson, K.; Bjornevik, K.; Abu-Ali, G.; Chan, J.; Cortese, M.; Dedi, B.; Jeon, M.; Xavier, R.; Huttenhower, C.; Ascherio, A.; et al. The human gut microbiota in people with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Front. Degener. 2021, 22, 186–194. [Google Scholar] [CrossRef]
- Lenglet, T.; Camdessanche, J. Amyotrophic lateral sclerosis or not: Keys for the diagnosis. Rev. Neurol. 2017, 173, 280–287. [Google Scholar] [CrossRef]
- Huang, F.; Zhu, Y.; Hsiao-Nakamoto, J.; Tang, X.; Dugas, J.C.; Moscovitch-Lopatin, M.; Glass, J.D.; Brown, R.H., Jr.; Ladha, S.S.; Lacomis, D.; et al. Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1103–1116. [Google Scholar] [CrossRef]
- Niedermeyer, S.; Murn, M.; Choi, P. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D. Edaravone: A new drug approved for ALS. Cell 2017, 171, 725. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Witzel, S.; Maier, A.; Steinbach, R.; Grosskreutz, J.; Koch, J.C.; Sarikidi, A.; Petri, S.; Gunther, R.; Wolf, J.; Hermann, A.; et al. Safety and Effectiveness of Long-term Intravenous Administration of Edaravone for Treatment of Patients with Amyotrophic Lateral Sclerosis. JAMA Neurol. 2022, 79, 121–130. [Google Scholar] [CrossRef]
- Aschenbrenner, D.S. New Drug Approved For ALS. Am. J. Nurs. 2023, 123, 22–23. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chio, A.; Van Damme, P.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef] [Green Version]
- Ferraiuolo, L.; Maragakis, N. Mini-Review: Induced pluripotent stem cells and the search for new cell-specific ALS therapeutic targets. Neurosci. Lett. 2021, 755, 135911. [Google Scholar] [CrossRef]
- Ray, A.; Joshi, J.M.; Sundaravadivelu, P.K.; Raina, K.; Lenka, N.; Kaveeshwar, V.; Thummer, R.P. An Overview on Promising Somatic Cell Sources Utilized for the Efficient Generation of Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2021, 17, 1954–1974. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Okita, K.; Mochiduki, Y.; Takizawa, N.; Yamanaka, S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat. Biotechnol. 2008, 26, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Freed, C. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef]
- Haase, A.; Olmer, R.; Schwanke, K.; Wunderlich, S.; Merkert, S.; Hess, C.; Zweigerdt, R.; Gruh, I.; Meyer, J.; Wagner, S.; et al. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell 2009, 5, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kim, C.H.; Moon, J.I.; Chung, Y.G.; Chang, M.Y.; Han, B.S.; Ko, S.; Yang, E.; Cha, K.Y.; Lanza, R.; et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 2009, 4, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Yakubov, E.; Rechavi, G.; Rozenblatt, S.; Givol, D. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem. Biophys. Res. Commun. 2010, 394, 189–193. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhu, J.; Dai, Y.; Wang, L.; Liu, R.; Guo, X. Generation of an induced pluripotent stem cell line (ZZUi034-A) from a 65 year old Chinese female donor with sendai virus reprogramming protocol. Stem Cell Res. 2022, 62, 102788. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef]
- Chen, K.; Long, Q.; Wang, T.; Zhao, D.; Zhou, Y.; Qi, J.; Wu, Y.; Li, S.; Chen, C.; Zeng, X.; et al. Gadd45a is a heterochromatin relaxer that enhances iPS cell generation. EMBO Rep. 2016, 17, 1641–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraguchi, D.; Nakamura, T. Pramef12 enhances reprogramming into naive iPS cells. Biochem. Biophys. Rep. 2022, 30, 101267. [Google Scholar] [CrossRef]
- Lee, S.; Huang, E. Modeling ALS and FTD with iPSC-derived neurons. Brain Res. 2017, 1656, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, N.J.; Roybon, L. Harnessing the Potential of Human Pluripotent Stem Cell-Derived Motor Neurons for Drug Discovery in Amyotrophic Lateral Sclerosis: From the Clinic to the Laboratory and Back to the Patient. Front. Drug Discov. 2021, 1, 773424. [Google Scholar] [CrossRef]
- Castillo Bautista, C.M.; Sterneckert, J. Progress and challenges in directing the differentiation of human iPSCs into spinal motor neurons. Front. Cell Dev. Biol. 2022, 10, 1089970. [Google Scholar] [CrossRef]
- Son, E.Y.; Ichida, J.K.; Wainger, B.J.; Toma, J.S.; Rafuse, V.F.; Woolf, C.J.; Eggan, K. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 2011, 9, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Ronchi, S.; Buccino, A.P.; Prack, G.; Kumar, S.S.; Schroter, M.; Fiscella, M.; Hierlemann, A. Electrophysiological Phenotype Characterization of Human iPSC-Derived Neuronal Cell Lines by Means of High-Density Microelectrode Arrays. Adv. Biol. 2021, 5, e2000223. [Google Scholar] [CrossRef]
- Toma, J.S.; Shettar, B.C.; Chipman, P.H.; Pinto, D.M.; Borowska, J.P.; Ichida, J.K.; Fawcett, J.P.; Zhang, Y.; Eggan, K.; Rafuse, V.F. Motoneurons derived from induced pluripotent stem cells develop mature phenotypes typical of endogenous spinal motoneurons. J. Neurosci. 2015, 35, 1291–1306. [Google Scholar] [CrossRef] [Green Version]
- Azadmanesh, J.; Borgstahl, G. A Review of the Catalytic Mechanism of Human Manganese Superoxide Dismutase. Antioxidants 2018, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.t.; Huang, C.L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, S.; Gascon, E.; Tran, H.; Chou, H.J.; Gendron, T.F.; Degroot, S.; Tapper, A.R.; Sellier, C.; Charlet-Berguerand, N.; Karydas, A.; et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013, 126, 385–399. [Google Scholar] [CrossRef] [Green Version]
- Westergard, T.; McAvoy, K.; Russell, K.; Wen, X.; Pang, Y.; Morris, B.; Pasinelli, P.; Trotti, D.; Haeusler, A. Repeat-associated non-AUG translation in C9orf72-ALS/FTD is driven by neuronal excitation and stress. EMBO Mol. Med. 2019, 11, e9423. [Google Scholar] [CrossRef]
- Sakae, N.; Bieniek, K.F.; Zhang, Y.J.; Ross, K.; Gendron, T.F.; Murray, M.E.; Rademakers, R.; Petrucelli, L.; Dickson, D.W. Poly-GR dipeptide repeat polymers correlate with neurodegeneration and Clinicopathological subtypes in C9ORF72-related brain disease. Acta Neuropathol. Commun. 2018, 6, 63. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Morotz, G.M.; Markovinovic, A.; Martin-Guerrero, S.M.; Preza, E.; Arias, N.; Mayl, K.; Aabdien, A.; Gesheva, V.; Nishimura, A.; et al. Disruption of ER-mitochondria tethering and signalling in C9orf72-associated amyotrophic lateral sclerosis and frontotemporal dementia. Aging Cell 2022, 21, e13549. [Google Scholar] [CrossRef]
- Neuro, L.C.; Li, J.; Lim, R.G.; Kaye, J.A.; Dardov, V.; Coyne, A.N.; Wu, J.; Milani, P.; Cheng, A.; Thompson, T.G.; et al. An integrated multi-omic analysis of iPSC-derived motor neurons from C9ORF72 ALS patients. iScience 2021, 24, 103221. [Google Scholar]
- Bilican, B.; Serio, A.; Barmada, S.J.; Nishimura, A.L.; Sullivan, G.J.; Carrasco, M.; Phatnani, H.P.; Puddifoot, C.A.; Story, D.; Fletcher, J.; et al. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc. Natl. Acad. Sci. USA 2012, 109, 5803–5808. [Google Scholar] [CrossRef] [Green Version]
- Egawa, N.; Kitaoka, S.; Tsukita, K.; Naitoh, M.; Takahashi, K.; Yamamoto, T.; Adachi, F.; Kondo, T.; Okita, K.; Asaka, I.; et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 2012, 4, 145ra104. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649 e18. [Google Scholar] [CrossRef]
- Higelin, J.; Demestre, M.; Putz, S.; Delling, J.P.; Jacob, C.; Lutz, A.K.; Bausinger, J.; Huber, A.K.; Klingenstein, M.; Barbi, G.; et al. FUS Mislocalization and Vulnerability to DNA Damage in ALS Patients Derived hiPSCs and Aging Motoneurons. Front. Cell Neurosci. 2016, 10, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofweber, M.; Hutten, S.; Bourgeois, B.; Spreitzer, E.; Niedner-Boblenz, A.; Schifferer, M.; Ruepp, M.D.; Simons, M.; Niessing, D.; Madl, T.; et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell 2018, 173, 706–719.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reber, S.; Jutzi, D.; Lindsay, H.; Devoy, A.; Mechtersheimer, J.; Levone, B.R.; Domanski, M.; Bentmann, E.; Dormann, D.; Muhlemann, O.; et al. The phase separation-dependent FUS interactome reveals nuclear and cytoplasmic function of liquid-liquid phase separation. Nucleic Acids Res. 2021, 49, 7713–7731. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Liu, W.; Li, X.; Chen, Q.; Liu, T.; Gao, S.; Deng, M. The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the FUS-P525L mutation. Neurogenetics 2015, 16, 223–231. [Google Scholar] [CrossRef]
- Marrone, L.; Poser, I.; Casci, I.; Japtok, J.; Reinhardt, P.; Janosch, A.; Andree, C.; Lee, H.O.; Moebius, C.; Koerner, E.; et al. Isogenic FUS-eGFP iPSC Reporter Lines Enable Quantification of FUS Stress Granule Pathology that Is Rescued by Drugs Inducing Autophagy. Stem Cell Rep. 2018, 10, 375–389. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovas, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [Green Version]
- Picchiarelli, G.; Demestre, M.; Zuko, A.; Been, M.; Higelin, J.; Dieterle, S.; Goy, M.A.; Mallik, M.; Sellier, C.; Scekic-Zahirovic, J.; et al. FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. Neurosci. 2019, 22, 1793–1805. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Mu, Y.; Manley, J. Nuclear RNA transcript levels modulate nucleocytoplasmic distribution of ALS/FTD-associated protein FUS. Sci. Rep. 2022, 12, 8180. [Google Scholar] [CrossRef]
- Gubert, F.; Vasques, J.F.; Cozendey, T.D.; Domizi, P.; Toledo, M.F.; Kasai-Brunswick, T.H.; Loureiro, M.P.S.; Lima, J.M.B.; Gress, C.H.; Cabello, G.M.K.; et al. Generation of four patient-specific pluripotent induced stem cell lines from two Brazilian patients with amyotrophic lateral sclerosis and two healthy subjects. Stem Cell Res. 2019, 37, 101448. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, M.F.; Martinez, F.J.; Wright, S.; Ramos, C.; Volfson, D.; Mason, M.; Garnes, J.; Dang, V.; Lievers, J.; Shoukat-Mumtaz, U.; et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell Neurosci. 2013, 56, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, C.J.; Dariolli, R.; Jorge, F.M.; Monteiro, M.R.; Maximino, J.R.; Martins, R.S.; Strauss, B.E.; Krieger, J.E.; Callegaro, D.; Chadi, G. Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell Neurosci. 2015, 9, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Song, J.; Huang, H.; Chen, H.; Qian, K. Modeling hallmark pathology using motor neurons derived from the family and sporadic amyotrophic lateral sclerosis patient-specific iPS cells. Stem Cell Res. Ther. 2018, 9, 315. [Google Scholar] [CrossRef] [Green Version]
- Baxi, E.G.; Thompson, T.; Li, J.; Kaye, J.A.; Lim, R.G.; Wu, J.; Ramamoorthy, D.; Lima, L.; Vaibhav, V.; Matlock, A.; et al. Answer ALS, a large-scale resource for sporadic and familial ALS combining clinical and multi-omics data from induced pluripotent cell lines. Nat. Neurosci. 2022, 25, 226–237. [Google Scholar] [CrossRef]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G.; et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bristol, L.A.; Rothstein, J. Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann. Neurol. 1996, 39, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, G.; Lakatos, A.; Patani, R. Human Stem Cell-Derived Astrocytes: Specification and Relevance for Neurological Disorders. Curr. Stem Cell Rep. 2016, 2, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.H.; Aubourg, P.; Ransohoff, R.M.; et al. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- Madill, M.; McDonagh, K.; Ma, J.; Vajda, A.; McLoughlin, P.; O’Brien, T.; Hardiman, O.; Shen, S. Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms. Mol. Brain 2017, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Devlin, A.C.; Chouhan, A.K.; Selvaraj, B.T.; Stavrou, M.; Burr, K.; Brivio, V.; He, X.; Mehta, A.R.; Story, D.; et al. Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia 2020, 68, 1046–1064. [Google Scholar] [CrossRef] [Green Version]
- Rajpurohit, C.S.; Kumar, V.; Cheffer, A.; Oliveira, D.; Ulrich, H.; Okamoto, O.K.; Zatz, M.; Ansari, U.A.; Khanna, V.K.; Pant, A.B. Mechanistic Insights of Astrocyte-Mediated Hyperactive Autophagy and Loss of Motor Neuron Function in SOD1(L39R) Linked Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2020, 57, 4117–4133. [Google Scholar] [CrossRef]
- Feng, B.; Amponsah, A.E.; Guo, R.; Liu, X.; Zhang, J.; Du, X.; Zhou, Z.; He, J.; Ma, J.; Cui, H. Autophagy-Mediated Inflammatory Cytokine Secretion in Sporadic ALS Patient iPSC-Derived Astrocytes. Oxid. Med. Cell Longev. 2022, 2022, 6483582. [Google Scholar]
- Allison, R.L.; Adelman, J.W.; Abrudan, J.; Urrutia, R.A.; Zimmermann, M.T.; Mathison, A.J.; Ebert, A.D. Microglia Influence Neurofilament Deposition in ALS iPSC-Derived Motor Neurons. Genes 2022, 13, 241. [Google Scholar] [CrossRef]
- Kerk, S.Y.; Bai, Y.; Smith, J.; Lalgudi, P.; Hunt, C.; Kuno, J.; Nuara, J.; Yang, T.; Lanza, K.; Chan, N.; et al. Homozygous ALS-linked FUS P525L mutations cell-autonomously perturb transcriptome profile and chemoreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022, 17, 678–692. [Google Scholar] [CrossRef]
- Eitan, C.; Siany, A.; Barkan, E.; Olender, T.; van Eijk, K.R.; Moisse, M.; Farhan, S.M.K.; Danino, Y.M.; Yanowski, E.; Marmor-Kollet, H.; et al. Whole-genome sequencing reveals that variants in the Interleukin 18 Receptor Accessory Protein 3’UTR protect against ALS. Nat. Neurosci. 2022, 25, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Barres, B.A. The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrige, M.; Frank, E.; Herardot, E.; Martineau, S.; Darle, A.; Benabides, M.; Domingues, S.; Chose, O.; Habeler, W.; Lorant, J.; et al. The Future of Regenerative Medicine: Cell Therapy Using Pluripotent Stem Cells and Acellular Therapies Based on Extracellular Vesicles. Cells 2021, 10, 240. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillee, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, M.; Keyhanian, K.; Douthwright, C. Glial Cell Dysfunction in C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Cells 2021, 10, 249. [Google Scholar] [CrossRef]
- Sareen, D.; Gowing, G.; Sahabian, A.; Staggenborg, K.; Paradis, R.; Avalos, P.; Latter, J.; Ornelas, L.; Garcia, L.; Svendsen, C.N. Human induced pluripotent stem cells are a novel source of neural progenitor cells (iNPCs) that migrate and integrate in the rodent spinal cord. J. Comp. Neurol. 2014, 522, 2707–2728. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Funayama, M.; Tsukita, K.; Hotta, A.; Yasuda, A.; Nori, S.; Kaneko, S.; Nakamura, M.; Takahashi, R.; Okano, H.; et al. Focal transplantation of human iPSC-derived glial-rich neural progenitors improves lifespan of ALS mice. Stem Cell Rep. 2014, 3, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Popescu, I.R.; Nicaise, C.; Liu, S.; Bisch, G.; Knippenberg, S.; Daubie, V.; Bohl, D.; Pochet, R. Neural progenitors derived from human induced pluripotent stem cells survive and differentiate upon transplantation into a rat model of amyotrophic lateral sclerosis. Stem Cells Transl. Med. 2013, 2, 167–174. [Google Scholar] [CrossRef]
- Nizzardo, M.; Simone, C.; Rizzo, F.; Ruggieri, M.; Salani, S.; Riboldi, G.; Faravelli, I.; Zanetta, C.; Bresolin, N.; Comi, G.P.; et al. Minimally invasive transplantation of iPSC-derived ALDHhiSSCloVLA4+ neural stem cells effectively improves the phenotype of an amyotrophic lateral sclerosis model. Hum. Mol. Genet. 2014, 23, 342–354. [Google Scholar] [CrossRef] [Green Version]
- Nizzardo, M.; Bucchia, M.; Ramirez, A.; Trombetta, E.; Bresolin, N.; Comi, G.P.; Corti, S. iPSC-derived LewisX+CXCR4+beta1-integrin+ neural stem cells improve the amyotrophic lateral sclerosis phenotype by preserving motor neurons and muscle innervation in human and rodent models. Hum. Mol. Genet. 2016, 25, 3152–3163. [Google Scholar] [CrossRef] [PubMed]
- Rosati, J.; Ferrari, D.; Altieri, F.; Tardivo, S.; Ricciolini, C.; Fusilli, C.; Zalfa, C.; Profico, D.C.; Pinos, F.; Bernardini, L.; et al. Establishment of stable iPS-derived human neural stem cell lines suitable for cell therapies. Cell Death Dis. 2018, 9, 937. [Google Scholar] [CrossRef] [PubMed]
- Forostyak, S.; Forostyak, O.; Kwok, J.C.F.; Romanyuk, N.; Rehorova, M.; Kriska, J.; Dayanithi, G.; Raha-Chowdhury, R.; Jendelova, P.; Anderova, M.; et al. Transplantation of Neural Precursors Derived from Induced Pluripotent Cells Preserve Perineuronal Nets and Stimulate Neural Plasticity in ALS Rats. Int. J. Mol. Sci. 2020, 21, 9593. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Li, M.; Zhang, L.; Chen, Z.; Lu, P. Motor neuron replacement therapy for amyotrophic lateral sclerosis. Neural Regen. Res. 2022, 17, 1633–1639. [Google Scholar]
- Sun, Y.; Feng, L.; Liang, L.; Stacey, G.N.; Wang, C.; Wang, Y.; Hu, B. Neuronal cell-based medicines from pluripotent stem cells: Development, production, and preclinical assessment. Stem Cells Transl. Med. 2021, 10, S31–S40. [Google Scholar] [CrossRef]
- Ting, H.C.; Su, H.L.; Chen, M.F.; Harn, H.J.; Lin, S.Z.; Chiou, T.W.; Chang, C.Y. Robust Generation of Ready-to-Use Cryopreserved Motor Neurons from Human Pluripotent Stem Cells for Disease Modeling. Int. J. Mol. Sci. 2022, 23, 13462. [Google Scholar] [CrossRef]
- Hayashi, Y.; Ohnuma, K.; Furue, M. Pluripotent Stem Cell Heterogeneity. Adv. Exp. Med. Biol. 2019, 1123, 71–94. [Google Scholar] [PubMed]
- Bar, S.; Benvenisty, N. Epigenetic aberrations in human pluripotent stem cells. EMBO J. 2019, 38, e101033. [Google Scholar] [CrossRef]
- Giacomelli, E.; Vahsen, B.F.; Calder, E.L.; Xu, Y.; Scaber, J.; Gray, E.; Dafinca, R.; Talbot, K.; Studer, L. Human stem cell models of neurodegeneration: From basic science of amyotrophic lateral sclerosis to clinical translation. Cell Stem Cell 2022, 29, 11–35. [Google Scholar] [CrossRef]
- Ohnuki, M.; Takahashi, K. Present and future challenges of induced pluripotent stem cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140367. [Google Scholar] [CrossRef]
- Okano, H.; Yasuda, D.; Fujimori, K.; Morimoto, S.; Takahashi, S. Ropinirole, a New ALS Drug Candidate Developed Using iPSCs. Trends Pharmacol. Sci. 2020, 41, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuburaya, N.; Homma, K.; Higuchi, T.; Balia, A.; Yamakoshi, H.; Shibata, N.; Nakamura, S.; Nakagawa, H.; Ikeda, S.I.; Umezawa, N.; et al. A small-molecule inhibitor of SOD1-Derlin-1 interaction ameliorates pathology in an ALS mouse model. Nat. Commun. 2018, 9, 2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.Y.; Markmiller, S.; Vu, A.Q.; Javaherian, A.; Dowdle, W.E.; Jolivet, P.; Bushway, P.J.; Castello, N.A.; Baral, A.; Chan, M.Y.; et al. Small-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD. Neuron 2019, 103, 802–819.e11. [Google Scholar] [CrossRef] [PubMed]
- Kuta, R.; Larochelle, N.; Fernandez, M.; Pal, A.; Minotti, S.; Tibshirani, M.; St Louis, K.; Gentil, B.J.; Nalbantoglu, J.N.; Hermann, A.; et al. Depending on the stress, histone deacetylase inhibitors act as heat shock protein co-inducers in motor neurons and potentiate arimoclomol, exerting neuroprotection through multiple mechanisms in ALS models. Cell Stress Chaperones 2020, 25, 173–191. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Sakamoto, K. Niclosamide affects intracellular TDP-43 distribution in motor neurons, activates mitophagy, and attenuates morphological changes under stress. J. Biosci. Bioeng. 2021, 132, 640–650. [Google Scholar] [CrossRef]
- Imamura, K.; Izumi, Y.; Banno, H.; Uozumi, R.; Morita, S.; Egawa, N.; Ayaki, T.; Nagai, M.; Nishiyama, K.; Watanabe, Y.; et al. Induced pluripotent stem cell-based Drug Repurposing for Amyotrophic lateral sclerosis Medicine (iDReAM) study: Protocol for a phase I dose escalation study of bosutinib for amyotrophic lateral sclerosis patients. BMJ Open 2019, 9, e033131. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Izumi, Y.; Nagai, M.; Nishiyama, K.; Watanabe, Y.; Hanajima, R.; Egawa, N.; Ayaki, T.; Oki, R.; Fujita, K.; et al. Safety and tolerability of bosutinib in patients with amyotrophic lateral sclerosis (iDReAM study): A multicentre, open-label, dose-escalation phase 1 trial. EClinicalMedicine 2022, 53, 101707. [Google Scholar] [CrossRef]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef]
- Morimoto, S.; Takahashi, S.; Fukushima, K.; Saya, H.; Suzuki, N.; Aoki, M.; Okano, H.; Nakahara, J. Ropinirole hydrochloride remedy for amyotrophic lateral sclerosis—Protocol for a randomized, double-blind, placebo-controlled, single-center, and open-label continuation phase I/IIa clinical trial (ROPALS trial). Regen. Ther. 2019, 11, 143–166. [Google Scholar] [CrossRef]
- Sleutjes, B.; Stikvoort Garcia, D.J.L.; Kovalchuk, M.O.; Heuberger, J.; Groeneveld, G.J.; Franssen, H.; van den Berg, L.H. Acute retigabine-induced effects on myelinated motor axons in amyotrophic lateral sclerosis. Pharmacol. Res. Perspect. 2022, 10, e00983. [Google Scholar] [CrossRef]
- Wainger, B.J.; Macklin, E.A.; Vucic, S.; McIlduff, C.E.; Paganoni, S.; Maragakis, N.J.; Bedlack, R.; Goyal, N.A.; Rutkove, S.B.; Lange, D.J.; et al. Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 186–196. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lee, J.H.; Chung, A.Y.; Jo, Y.; Shin, J.H.; Park, H.C.; Kim, H.; Lopez-Gonzalez, R.; Ryu, J.R.; Sun, W. Prevention of mitochondrial impairment by inhibition of protein phosphatase 1 activity in amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 888. [Google Scholar] [CrossRef] [PubMed]
- De Jongh, R.; Spijkers, X.M.; Pasteuning-Vuhman, S.; Vulto, P.; Pasterkamp, R.J. Neuromuscular junction-on-a-chip: ALS disease modeling and read-out development in microfluidic devices. J. Neurochem. 2021, 157, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Namboori, S.C.; Thomas, P.; Ames, R.; Hawkins, S.; Garrett, L.O.; Willis, C.R.G.; Rosa, A.; Stanton, L.W.; Bhinge, A. Single-cell transcriptomics identifies master regulators of neurodegeneration in SOD1 ALS iPSC-derived motor neurons. Stem Cell Rep. 2021, 16, 3020–3035. [Google Scholar] [CrossRef] [PubMed]
- Deneault, E.; Chaineau, M.; Nicouleau, M.; Castellanos Montiel, M.J.; Franco Flores, A.K.; Haghi, G.; Chen, C.X.; Abdian, N.; Shlaifer, I.; Beitel, L.K.; et al. A streamlined CRISPR workflow to introduce mutations and generate isogenic iPSCs for modeling amyotrophic lateral sclerosis. Methods 2022, 203, 297–310. [Google Scholar] [CrossRef]
- Imamura, K.; Yada, Y.; Izumi, Y.; Morita, M.; Kawata, A.; Arisato, T.; Nagahashi, A.; Enami, T.; Tsukita, K.; Kawakami, H.; et al. Prediction Model of Amyotrophic Lateral Sclerosis by Deep Learning with Patient Induced Pluripotent Stem Cells. Ann. Neurol. 2021, 89, 1226–1233. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell Type | ALS Subtypes | Key Findings |
|---|---|---|
| Motor neurons (or neurons) | SOD1 mutation | Neurofilament aggregation; disorders in mitochondria; decreased survival rate |
| C9orf72 mutation | RNA foci formation; accumulation of RAN translation products; stress granule formation; excitotoxicity | |
| TARDBP mutation | TDP-43 protein aggregation; shorter neurites; mitochondrial Ca2+ uptake disorder | |
| FUS mutation | FUS protein aggregation; DNA damage; cytoplasmic mislocation | |
| sporadic ALS | TDP-43 aggregation; higher neurofilament inclusion; mitochondria distribution impairment | |
| Astrocytes | SOD1 mutation | Impair autophagy |
| C9orf72 mutation | Increased oxidative stress and neurotoxicity | |
| sporadic ALS | Autophagy-mediated inflammatory cytokine secretion | |
| Microglia | FUS mutation | Disrupt intracellular calcium signaling |
| sporadic ALS | Influence neurofilament deposition |
| Name | Functions | Reference |
|---|---|---|
| Bosutinib | A Src/c-Abl inhibitor; increases the survival of ALS MNs, increases autophagy, reduces the amount of misfolded SOD1 protein. | [52] |
| Retigabine (ezogabine) | A Kv7 channel activator; blocks hyperexcitability, improves MN survival. | [80] |
| ROPI | A dopamine agonist; suppresses neurite retraction and cell death, inhibits oxidative stress, improves mitochondrial function, inhibits TDP-43 and FUS aggregation. | [111] |
| Compound #56 | An inhibitor of SOD1-Derlin-1 interaction; ameliorates ALS pathology in MNs, delays onset and prolongs survival of ALS model mice. | [112] |
| Mitoxantrone | Contains extended planararomatic moieties; prevents TDP-43, FUS from forming SGs. | [113] |
| SAHA, RGFP109 | Histone deacetylase inhibitors; reduce loss of nuclear FUS, rescue the DNA repair response (combined with arimoclomol). | [114] |
| Niclosamide | A STAT3 inhibitor; prevents TDP-43 mislocation, degrades TDP-43 aggregates, activates mitophagy, attenuates morphological changes. | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, H.; Huo, Z.; Chen, Y.; Zhao, Z.; Meng, F.; Wang, X.; Liu, S.; Zhang, H.; Zhou, F.; Liu, J.; et al. Induced Pluripotent Stem Cells and Their Applications in Amyotrophic Lateral Sclerosis. Cells 2023, 12, 971. https://doi.org/10.3390/cells12060971
Du H, Huo Z, Chen Y, Zhao Z, Meng F, Wang X, Liu S, Zhang H, Zhou F, Liu J, et al. Induced Pluripotent Stem Cells and Their Applications in Amyotrophic Lateral Sclerosis. Cells. 2023; 12(6):971. https://doi.org/10.3390/cells12060971
Chicago/Turabian StyleDu, Hongmei, Zijun Huo, Yanchun Chen, Zhenhan Zhao, Fandi Meng, Xuemei Wang, Shiyue Liu, Haoyun Zhang, Fenghua Zhou, Jinmeng Liu, and et al. 2023. "Induced Pluripotent Stem Cells and Their Applications in Amyotrophic Lateral Sclerosis" Cells 12, no. 6: 971. https://doi.org/10.3390/cells12060971