
VEGF Stimulates Activation of ERK5 in the Absence of C-Terminal Phosphorylation Preventing Nuclear Localization and Facilitating AKT Activation in Endothelial Cells

 , , , , , , and
, , , , , , and 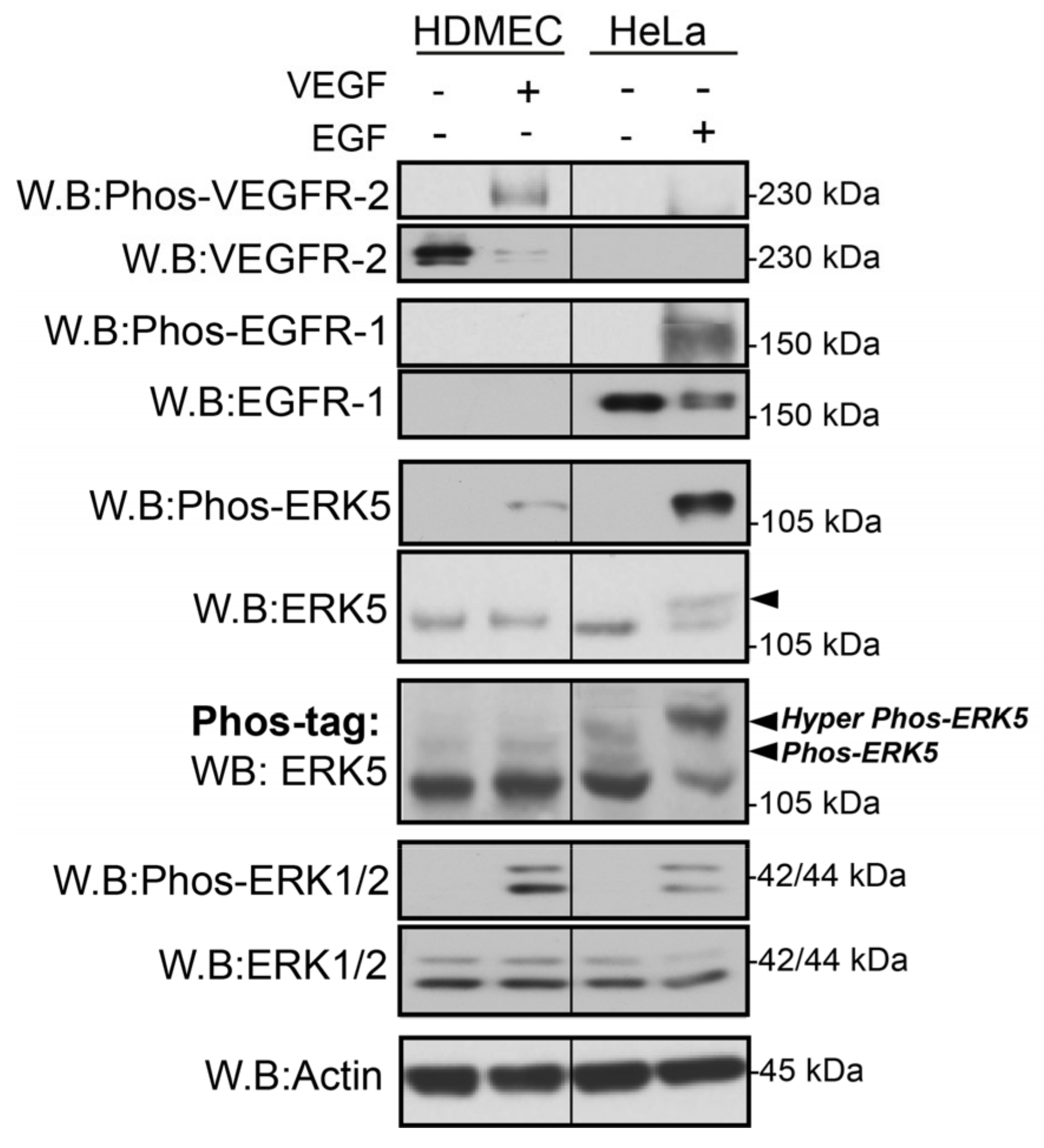{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Agonists and Inhibitors
2.2. Antibodies and Reagents
2.3. Cell Culture
2.4. siRNA Knockdown
2.5. Agonist Stimulation
2.6. RIPA Protein Extraction and Western Blot
2.7. Immunofluorescence
2.8. qRT-PCR
2.9. MEF2 Reporter Assay
2.10. Statistical Analysis
3. Results
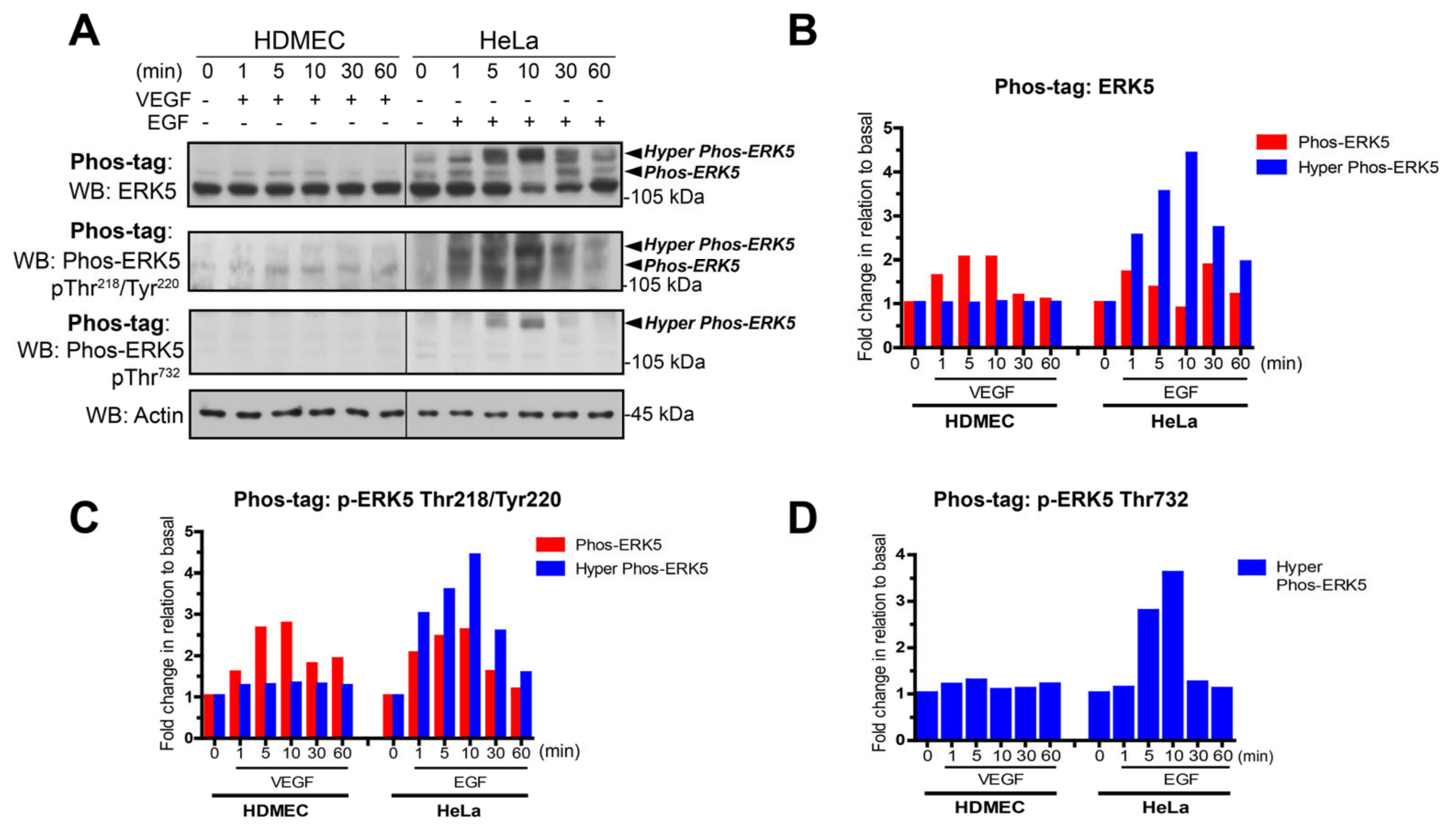
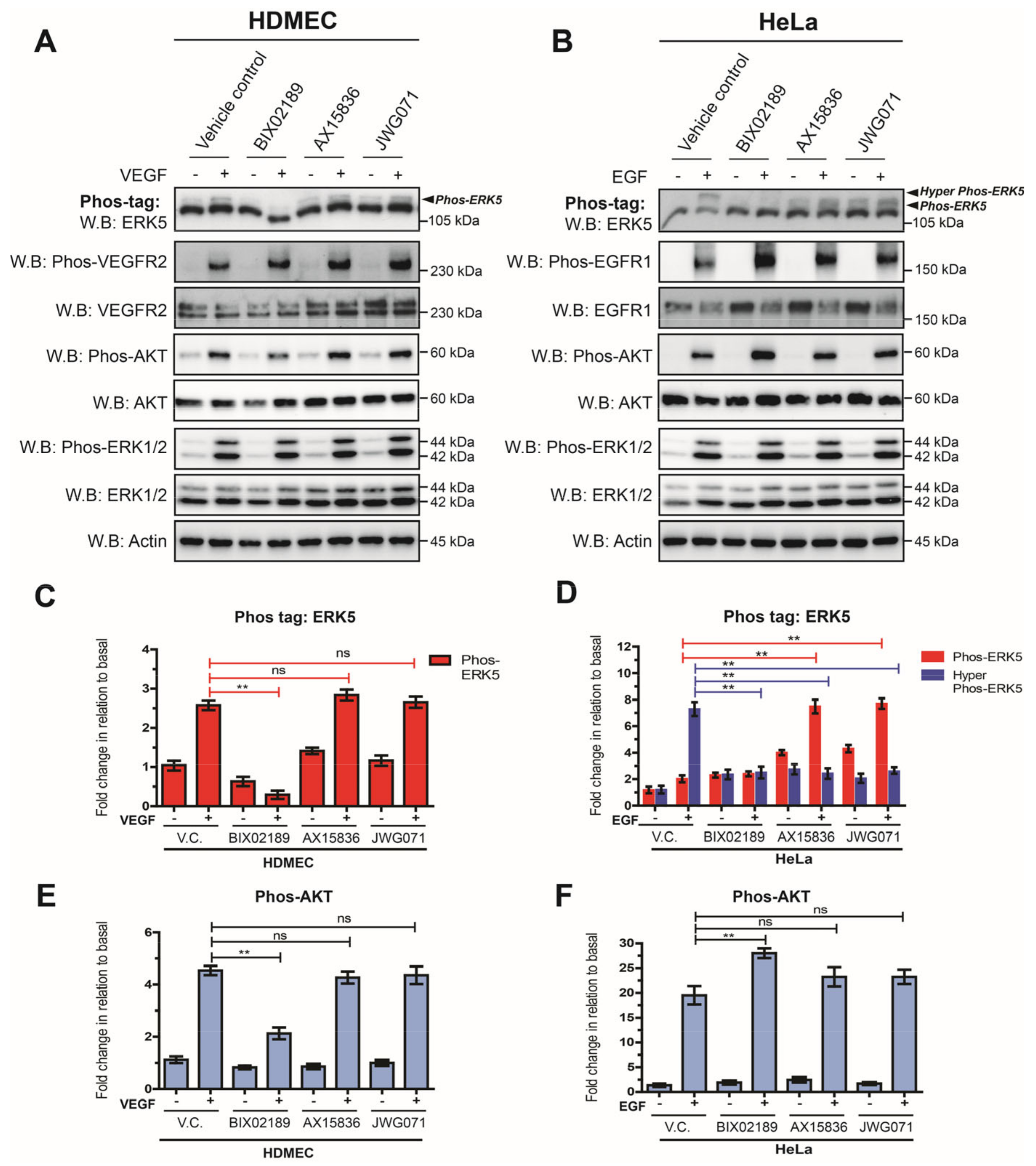
3.1. VEGF-Stimulated ERK5 Fails to Undergo C-Terminal Phosphorylation
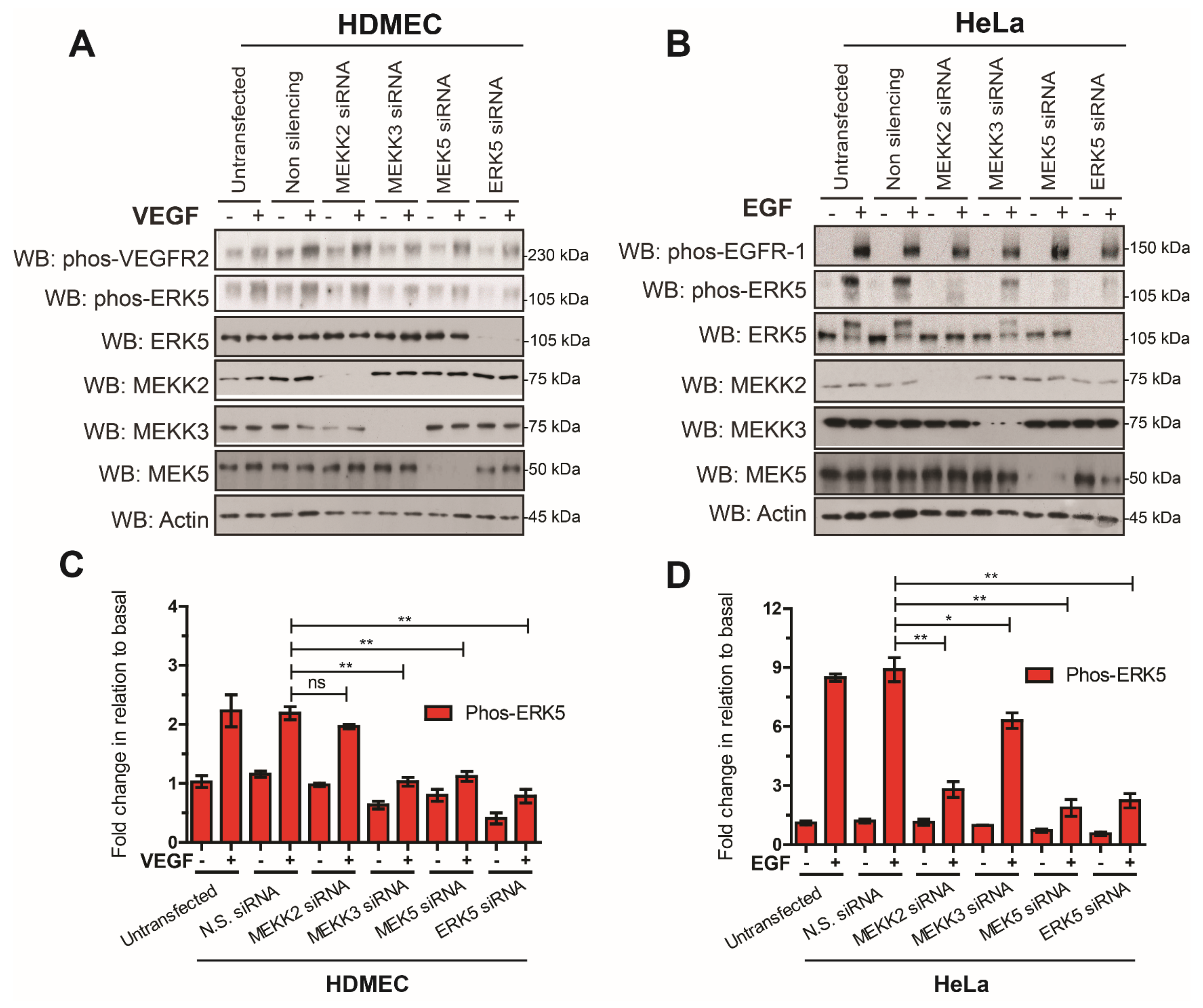
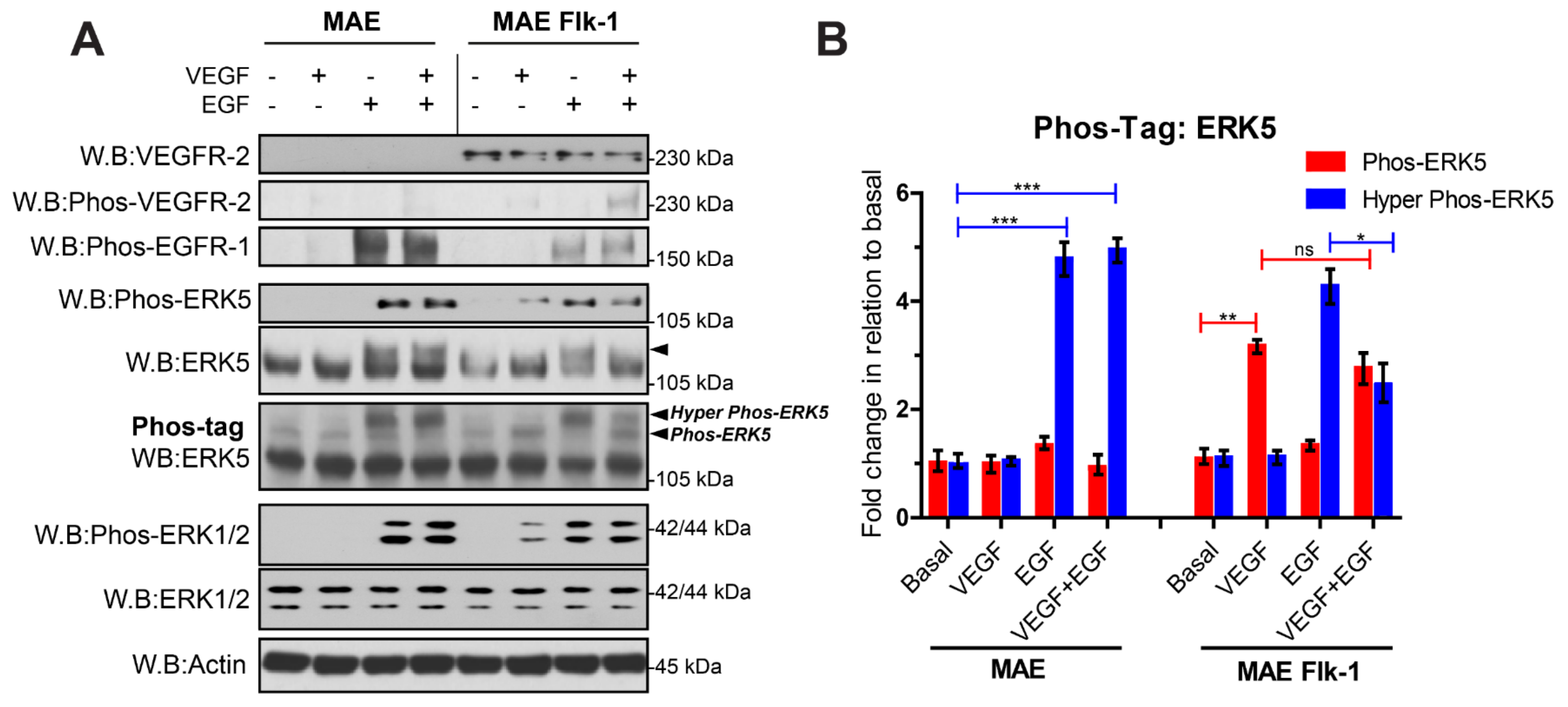
3.2. VEGFR-2 Activation Suppresses ERK5 C-Terminal Phosphorylation in Endothelial Cells
3.3. VEGF-Stimulated ERK5 Regulates AKT Activity in HDMECs
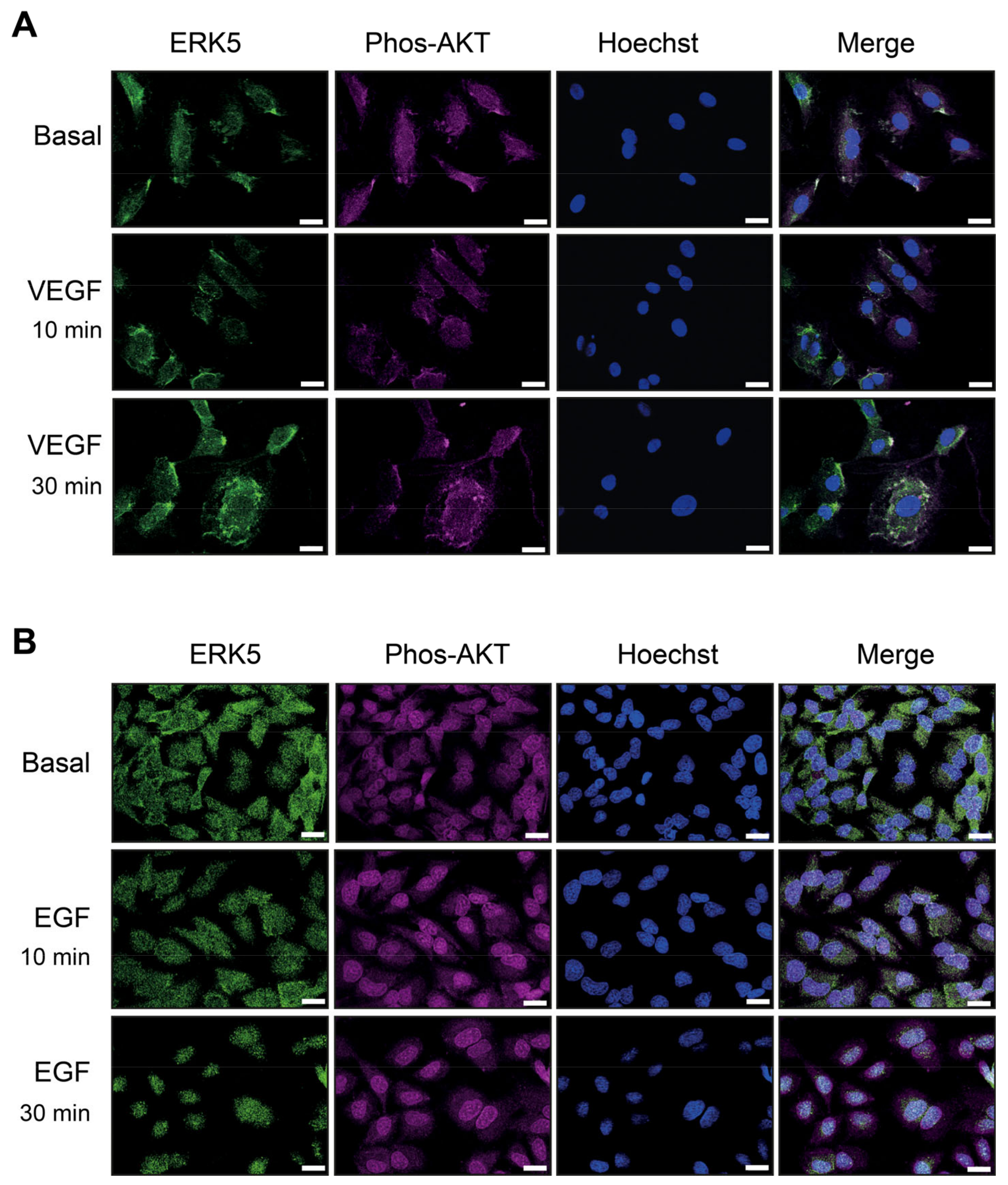
3.4. VEGF-Stimulated ERK5 Does Not Undergo Nuclear Translocation and Co-Localises with Phosphorylated AKT
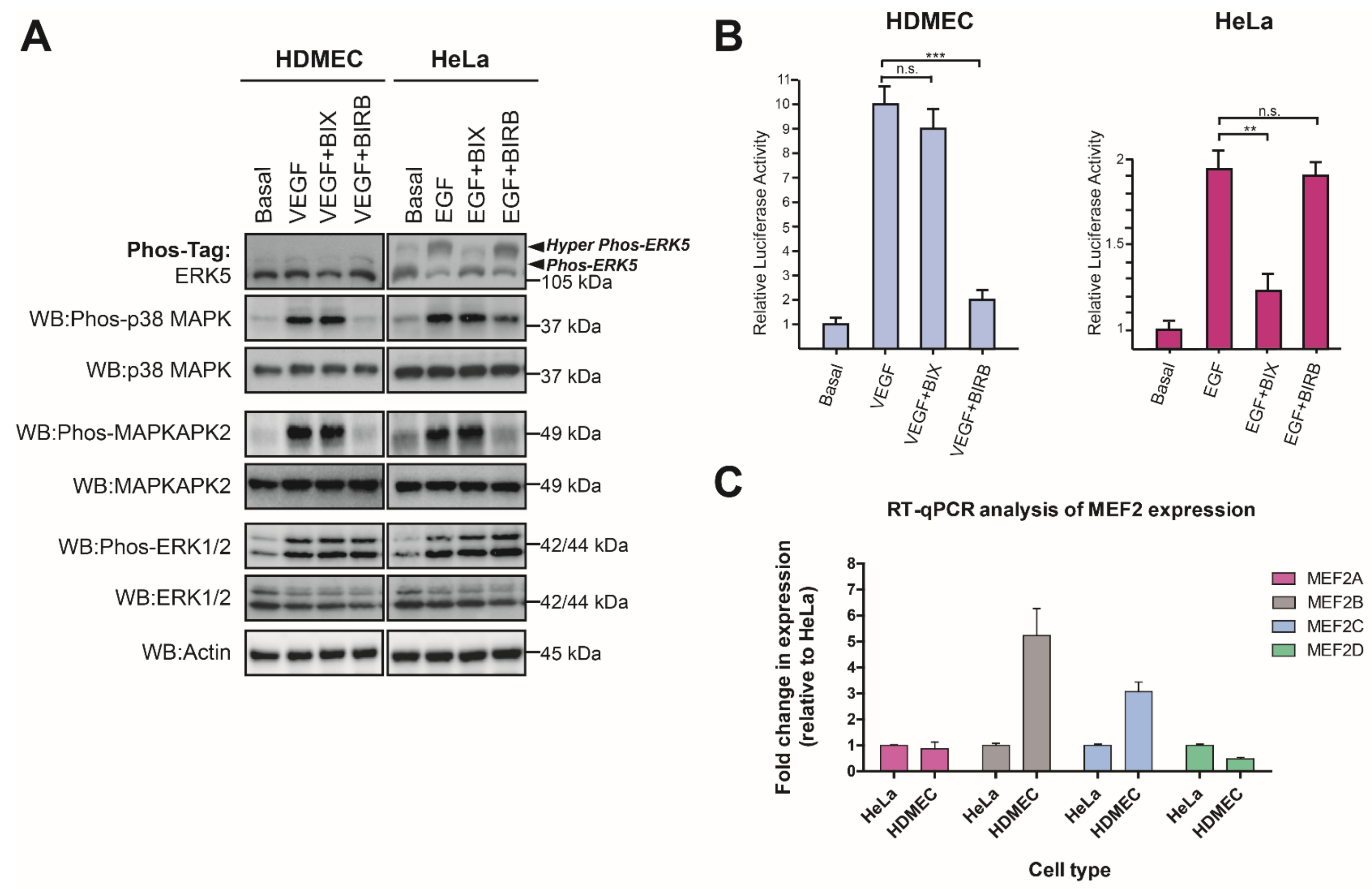
3.5. VEGF-Stimulated ERK5 Activity Does Not Regulate MEF2-Dependent Gene Expression in HDMECs
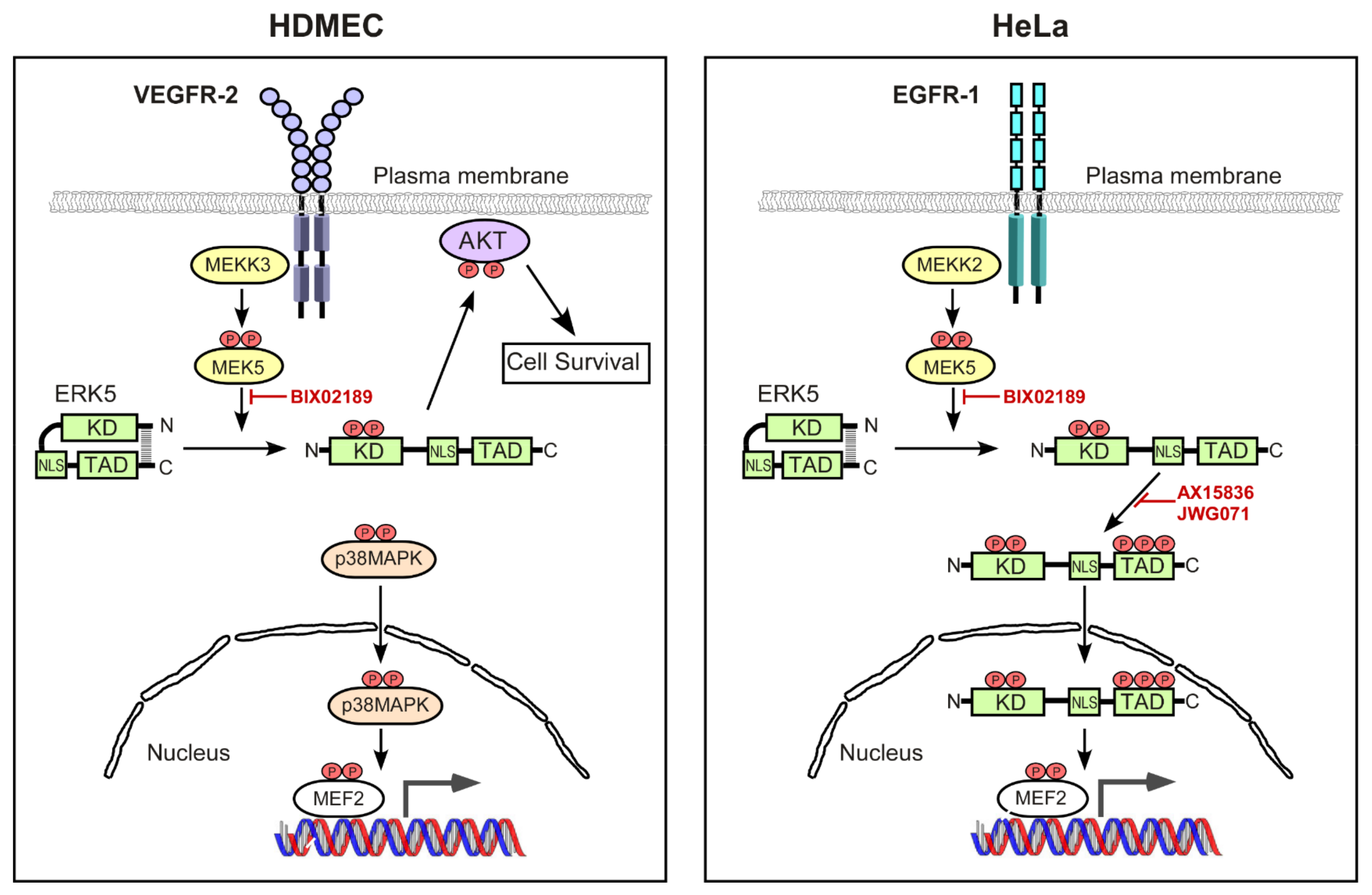
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radziejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: A family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef] [Green Version]
- Cano, E.; Mahadevan, L.C. Parallel signal processing among mammalian MAPKs. Trends Biochem. Sci. 1995, 20, 117–122. [Google Scholar] [CrossRef]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal transduction through MAP kinase cascades. Adv. Cancer Res. 1998, 74, 49–139. [Google Scholar]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Ulevitch, R.J.; Han, J. Primary structure of BMK1: A new mammalian map kinase. Biochem. Biophys. Res. Commun. 1995, 213, 715–724. [Google Scholar]
- Zhou, G.; Bao, Z.Q.; Dixon, J.E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Muller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell Signal. 2012, 24, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Paudel, R.; Fusi, L.; Schmidt, M. The MEK5/ERK5 Pathway in Health and Disease. Int. J. Mol. Sci. 2021, 22, 7594. [Google Scholar] [CrossRef]
- Abe, J.; Kusuhara, M.; Ulevitch, R.J.; Berk, B.C.; Lee, J.D. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J. Biol. Chem. 1996, 271, 16586–16590. [Google Scholar] [PubMed] [Green Version]
- Kato, Y.; Zhao, M.; Morikawa, A.; Sugiyama, T.; Chakravortty, D.; Koide, N.; Yoshida, T.; Tapping, R.I.; Yang, Y.; Yokochi, T.; et al. Big mitogen-activated kinase regulates multiple members of the MEF2 protein family. J. Biol. Chem. 2000, 275, 18534–18540. [Google Scholar] [CrossRef] [Green Version]
- Cavanaugh, J.E.; Ham, J.; Hetman, M.; Poser, S.; Yan, C.; Xia, Z. Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J. Neurosci. 2001, 21, 434–443. [Google Scholar]
- Carvajal-Vergara, X.; Tabera, S.; Montero, J.C.; Esparis-Ogando, A.; Lopez-Perez, R.; Mateo, G.; Gutierrez, N.; Parmo-Cabanas, M.; Teixido, J.; San Miguel, J.F.; et al. Multifunctional role of Erk5 in multiple myeloma. Blood 2005, 105, 4492–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeishi, Y.; Abe, J.; Lee, J.D.; Kawakatsu, H.; Walsh, R.A.; Berk, B.C. Differential regulation of p90 ribosomal S6 kinase and big mitogen-activated protein kinase 1 by ischemia/reperfusion and oxidative stress in perfused guinea pig hearts. Circ. Res. 1999, 85, 1164–1172. [Google Scholar] [PubMed] [Green Version]
- Sohn, S.J.; Sarvis, B.K.; Cado, D.; Winoto, A. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 2002, 277, 43344–43351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, T.H.; Hayashi, M.; Tapping, R.I.; Kato, Y.; Lee, J.D. MEKK3 directly regulates MEK5 activity as part of the big mitogen-activated protein kinase 1 (BMK1) signaling pathway. J. Biol. Chem. 1999, 274, 36035–36038. [Google Scholar]
- Sun, W.; Kesavan, K.; Schaefer, B.C.; Garrington, T.P.; Ware, M.; Johnson, N.L.; Gelfand, E.W.; Johnson, G.L. MEKK2 associates with the adapter protein Lad/RIBP and regulates the MEK5-BMK1/ERK5 pathway. J. Biol. Chem. 2001, 276, 5093–5100. [Google Scholar] [CrossRef] [Green Version]
- Mody, N.; Leitch, J.; Armstrong, C.; Dixon, J.; Cohen, P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001, 502, 21–24. [Google Scholar]
- Morimoto, H.; Kondoh, K.; Nishimoto, S.; Terasawa, K.; Nishida, E. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J. Biol. Chem. 2007, 282, 35449–35456. [Google Scholar] [CrossRef] [Green Version]
- Buschbeck, M.; Ullrich, A. The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J. Biol. Chem. 2005, 280, 2659–2667. [Google Scholar] [CrossRef] [Green Version]
- Raviv, Z.; Kalie, E.; Seger, R. MEK5 and ERK5 are localized in the nuclei of resting as well as stimulated cells, while MEKK2 translocates from the cytosol to the nucleus upon stimulation. J. Cell Sci. 2004, 117, 1773–1784. [Google Scholar] [CrossRef] [Green Version]
- Kondoh, K.; Terasawa, K.; Morimoto, H.; Nishida, E. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol. Cell. Biol. 2006, 26, 1679–1690. [Google Scholar] [CrossRef] [Green Version]
- Regan, C.P.; Li, W.; Boucher, D.M.; Spatz, S.; Su, M.S.; Kuida, K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. USA 2002, 99, 9248–9253. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Carr, J.; Ashby, P.R.; Murry-Tait, V.; Thompson, C.; Arthur, J.S. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev. Biol. 2003, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Kim, S.W.; Imanaka-Yoshida, K.; Yoshida, T.; Abel, E.D.; Eliceiri, B.; Yang, Y.; Ulevitch, R.J.; Lee, J.D. Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Investig. 2004, 113, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Lee, J.D. Role of the BMK1/ERK5 signaling pathway: Lessons from knockout mice. J. Mol. Med. 2004, 82, 800–808. [Google Scholar] [CrossRef]
- Roberts, O.L.; Holmes, K.; Muller, J.; Cross, D.A.; Cross, M.J. ERK5 is required for VEGF-mediated survival and tubular morphogenesis of primary human microvascular endothelial cells. J. Cell Sci. 2010, 123, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Nithianandarajah-Jones, G.N.; Cross, M.J. Analysis of VEGF-Mediated ERK5 Activity in Endothelial Cells. Methods Mol. Biol. 2015, 1332, 133–142. [Google Scholar] [CrossRef]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Muller, J.; Cross, M.J. The role of ERK5 in endothelial cell function. Biochem. Soc. Trans. 2014, 42, 1584–1589. [Google Scholar] [CrossRef] [Green Version]
- Mody, N.; Campbell, D.G.; Morrice, N.; Peggie, M.; Cohen, P. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem. J. 2003, 372, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, N.; Erazo, T.; Lizcano, J.M. ERK5 and Cell Proliferation: Nuclear Localization Is What Matters. Front. Cell Dev. Biol. 2016, 4, 105. [Google Scholar] [CrossRef] [Green Version]
- Duff, J.L.; Monia, B.P.; Berk, B.C. Mitogen-activated Protein (MAP) Kinase Is Regulated by the MAP Kinase Phosphatase (MKP-1) in Vascular Smooth Muscle Cells. J. Biol. Chem. 1995, 270, 7161–7166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, J.; Takahashi, M.; Ishida, M.; Lee, J.D.; Berk, B.C. c-Src Is Required for Oxidative Stress-mediated Activation of Big Mitogen-activated Protein Kinase 1 (BMK1). J. Biol. Chem. 1997, 272, 20389–20394. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Takiyama, K.; Koike, T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteom. 2006, 5, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Rodriguez, E.; Pandiella, A. Multisite phosphorylation of Erk5 in mitosis. J. Cell Sci. 2010, 123, 3146–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, E.L.; Sidaway, J.E.; Cross, M.J. Cardiotoxic drugs Herceptin and doxorubicin inhibit cardiac microvascular endothelial cell barrier formation resulting in increased drug permeability. Biol. Open 2016, 5, 1362–1370. [Google Scholar] [CrossRef] [Green Version]
- Tatake, R.J.; O’Neill, M.M.; Kennedy, C.A.; Wayne, A.L.; Jakes, S.; Wu, D.; Kugler, S.Z., Jr.; Kashem, M.A.; Kaplita, P.; Snow, R.J. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun. 2008, 377, 120–125. [Google Scholar] [CrossRef]
- Lin, E.C.; Amantea, C.M.; Nomanbhoy, T.K.; Weissig, H.; Ishiyama, J.; Hu, Y.; Sidique, S.; Li, B.; Kozarich, J.W.; Rosenblum, J.S. ERK5 kinase activity is dispensable for cellular immune response and proliferation. Proc. Natl. Acad. Sci. USA 2016, 113, 11865–11870. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Erazo, T.; Ferguson, F.M.; Buckley, D.L.; Gomez, N.; Munoz-Guardiola, P.; Dieguez-Martinez, N.; Deng, X.; Hao, M.; Massefski, W.; et al. Structural and Atropisomeric Factors Governing the Selectivity of Pyrimido-benzodiazipinones as Inhibitors of Kinases and Bromodomains. ACS Chem. Biol. 2018, 13, 2438–2448. [Google Scholar] [CrossRef] [Green Version]
- Kasler, H.G.; Victoria, J.; Duramad, O.; Winoto, A. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 2000, 20, 8382–8389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Jiang, Y.; Li, Z.; Kravchenko, V.V.; Ulevitch, R.J. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature 1997, 386, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Rouse, J.; Cohen, P.; Trigon, S.; Morange, M.; Alonso-Llamazares, A.; Zamanillo, D.; Hunt, T.; Nebreda, A.R. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 1994, 78, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Kuma, Y.; Sabio, G.; Bain, J.; Shpiro, N.; Marquez, R.; Cuenda, A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005, 280, 19472–19479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, O.L.; Holmes, K.; Muller, J.; Cross, D.A.; Cross, M.J. ERK5 and the regulation of endothelial cell function. Biochem. Soc. Trans. 2009, 37, 1254–1259. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Tapping, R.I.; Huang, S.; Watson, M.H.; Ulevitch, R.J.; Lee, J.D. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998, 395, 713–716. [Google Scholar] [CrossRef]
- Sun, W.; Wei, X.; Kesavan, K.; Garrington, T.P.; Fan, R.; Mei, J.; Anderson, S.M.; Gelfand, E.W.; Johnson, G.L. MEK kinase 2 and the adaptor protein Lad regulate extracellular signal-regulated kinase 5 activation by epidermal growth factor via Src. Mol. Cell. Biol. 2003, 23, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Honda, T.; Obara, Y.; Yamauchi, A.; Couvillon, A.D.; Mason, J.J.; Ishii, K.; Nakahata, N. Phosphorylation of ERK5 on Thr732 is associated with ERK5 nuclear localization and ERK5-dependent transcription. PLoS ONE 2015, 10, e0117914. [Google Scholar] [CrossRef] [Green Version]
- Tubita, A.; Lombardi, Z.; Tusa, I.; Dello Sbarba, P.; Rovida, E. Beyond Kinase Activity: ERK5 Nucleo-Cytoplasmic Shuttling as a Novel Target for Anticancer Therapy. Int. J. Mol. Sci. 2020, 21, 938. [Google Scholar] [CrossRef] [Green Version]
- Erazo, T.; Moreno, A.; Ruiz-Babot, G.; Rodriguez-Asiain, A.; Morrice, N.A.; Espadamala, J.; Bayascas, J.R.; Gomez, N.; Lizcano, J.M. Canonical and kinase activity-independent mechanisms for extracellular signal-regulated kinase 5 (ERK5) nuclear translocation require dissociation of Hsp90 from the ERK5-Cdc37 complex. Mol. Cell. Biol. 2013, 33, 1671–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, D.; Xu, Z.; Duh, E.J. Vascular endothelial growth factor induces MEF2C and MEF2-dependent activity in endothelial cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3640–3648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Boerm, M.; McCarty, M.; Bucana, C.; Fidler, I.J.; Zhuang, Y.; Su, B. Mekk3 is essential for early embryonic cardiovascular development. Nat. Genet. 2000, 24, 309–313. [Google Scholar] [CrossRef]
- Wang, X.; Merritt, A.J.; Seyfried, J.; Guo, C.; Papadakis, E.S.; Finegan, K.G.; Kayahara, M.; Dixon, J.; Boot-Handford, R.P.; Cartwright, E.J.; et al. Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol. Cell. Biol. 2005, 25, 336–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesavan, K.; Lobel-Rice, K.; Sun, W.; Lapadat, R.; Webb, S.; Johnson, G.L.; Garrington, T.P. MEKK2 regulates the coordinate activation of ERK5 and JNK in response to FGF-2 in fibroblasts. J. Cell. Physiol. 2004, 199, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Clydesdale, G.; Cheng, J.; Kim, K.; Gan, L.; McConkey, D.J.; Ullrich, S.E.; Zhuang, Y.; Su, B. Disruption of Mekk2 in mice reveals an unexpected role for MEKK2 in modulating T-cell receptor signal transduction. Mol. Cell. Biol. 2002, 22, 5761–5768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrington, T.P.; Ishizuka, T.; Papst, P.J.; Chayama, K.; Webb, S.; Yujiri, T.; Sun, W.; Sather, S.; Russell, D.M.; Gibson, S.B.; et al. MEKK2 gene disruption causes loss of cytokine production in response to IgE and c-Kit ligand stimulation of ES cell-derived mast cells. EMBO J. 2000, 19, 5387–5395. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N. Undermining the endothelium by ablation of MAPK-MEF2 signaling. J. Clin. Investig. 2004, 113, 1110–1112. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Tschopp, O.; Di-Poi, N.; Bruder, E.; Baudry, A.; Dummler, B.; Wahli, W.; Hemmings, B.A. Dosage-dependent effects of Akt1/protein kinase Balpha (PKBalpha) and Akt3/PKBgamma on thymus, skin, and cardiovascular and nervous system development in mice. Mol. Cell. Biol. 2005, 25, 10407–10418. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mondru, A.K.; Aljasir, M.A.; Alrumayh, A.; Nithianandarajah, G.N.; Ahmed, K.; Muller, J.; Goldring, C.E.P.; Wilm, B.; Cross, M.J. VEGF Stimulates Activation of ERK5 in the Absence of C-Terminal Phosphorylation Preventing Nuclear Localization and Facilitating AKT Activation in Endothelial Cells. Cells 2023, 12, 967. https://doi.org/10.3390/cells12060967
Mondru AK, Aljasir MA, Alrumayh A, Nithianandarajah GN, Ahmed K, Muller J, Goldring CEP, Wilm B, Cross MJ. VEGF Stimulates Activation of ERK5 in the Absence of C-Terminal Phosphorylation Preventing Nuclear Localization and Facilitating AKT Activation in Endothelial Cells. Cells. 2023; 12(6):967. https://doi.org/10.3390/cells12060967
Chicago/Turabian StyleMondru, Anil Kumar, Mohammad A. Aljasir, Ahmed Alrumayh, Gopika N. Nithianandarajah, Katie Ahmed, Jurgen Muller, Christopher E. P. Goldring, Bettina Wilm, and Michael J. Cross. 2023. "VEGF Stimulates Activation of ERK5 in the Absence of C-Terminal Phosphorylation Preventing Nuclear Localization and Facilitating AKT Activation in Endothelial Cells" Cells 12, no. 6: 967. https://doi.org/10.3390/cells12060967