
Reduced Levels of Misfolded and Aggregated Mutant p53 by Proteostatic Activation
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Culture
2.3. Genotyping Cell Lines for p53 Status
2.4. In Silico Energy Calculations Using FoldX
2.5. SDS/Western Blot Analysis and Blue Native Page Analysis
2.6. Meso Scale Discovery MULTI-ARRAY Microplate Assay (USA)
2.7. SOD1 Transfection Experiments
2.8. Immunofluorescent Staining and High Content Imaging Screening
2.9. Gene Expression Analysis by Quantitative Real-Time PCR (qPCR)
2.10. Full Transcriptome RNA Sequencing
2.11. Calculation of the Inhibitory Concentration of Foldlin
2.12. Immunoprecipitation Experiments
2.13. Proteasome Activity Test (Proteasome-Glo) and Caspase Glo Test
2.14. Screening of Enzo Library
2.15. Statistical Analysis
3. Results
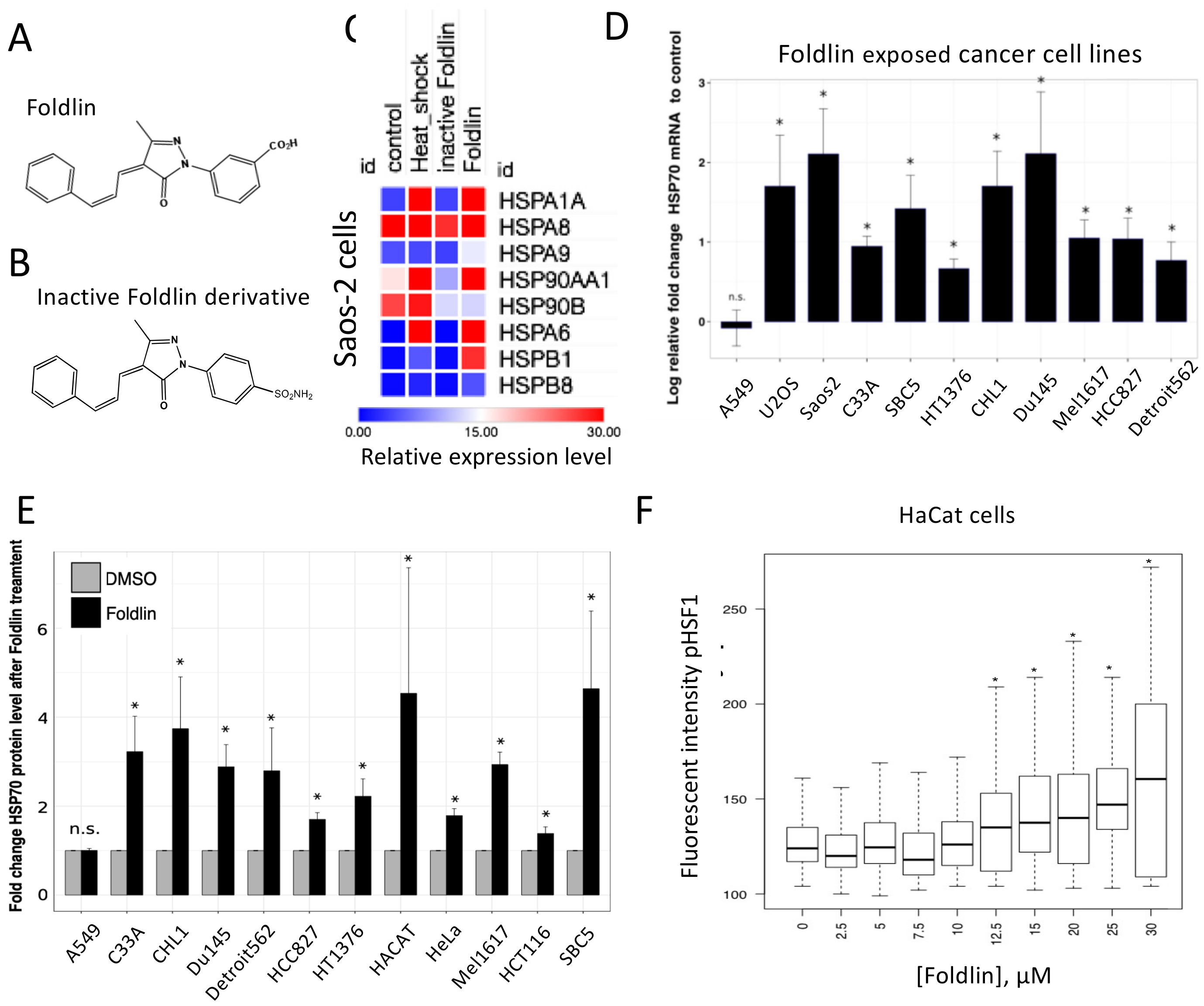
3.1. Pharmacological Induction of a Stress Response in Conventional and Primary Tumor Cell Lines
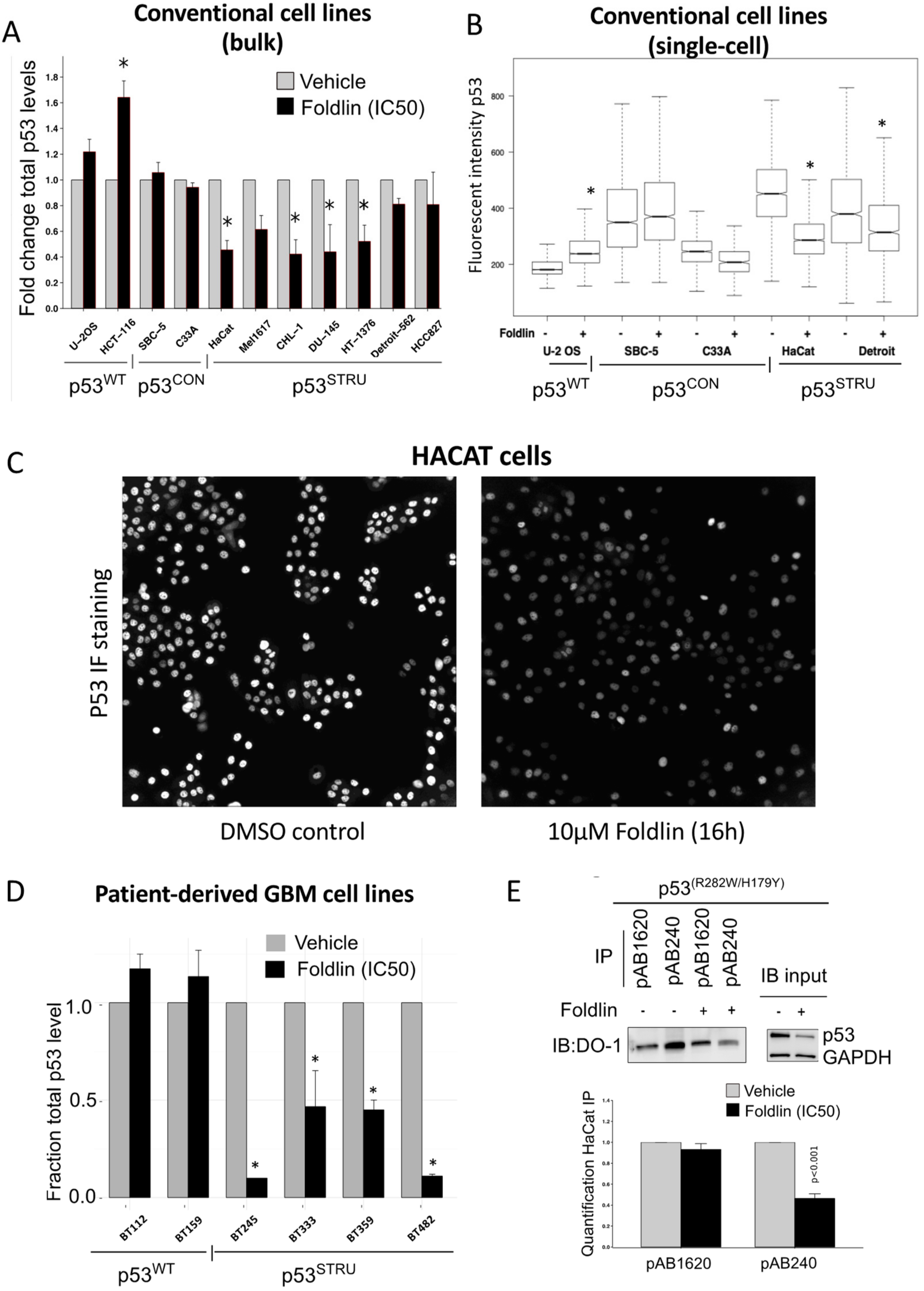
3.2. Pharmacological Activation of HSF1 Results in Reduced Levels of Misfolded and Aggregated mutp53
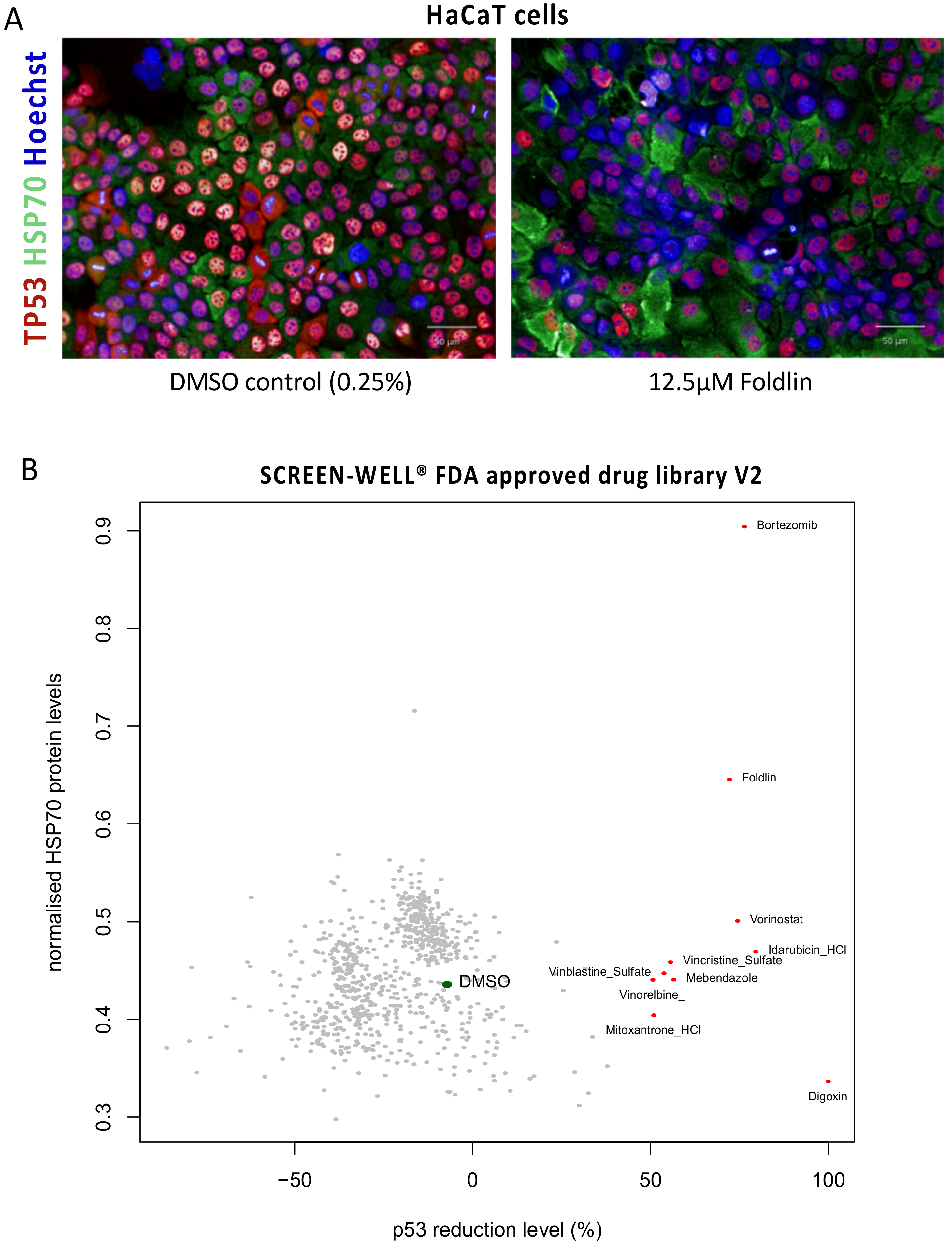
3.3. Pharmacological Activation of a Heat-Shock Response Mediated by Foldlin Prevents the Formation of Nuclear Inclusion Bodies of p53 upon Proteostatic Stress
3.4. Mapping the Proteostatic Network Induced by Foldlin
3.5. Mechanistic Analysis of Foldlin-Mediated Changes in p53 Protein Levels
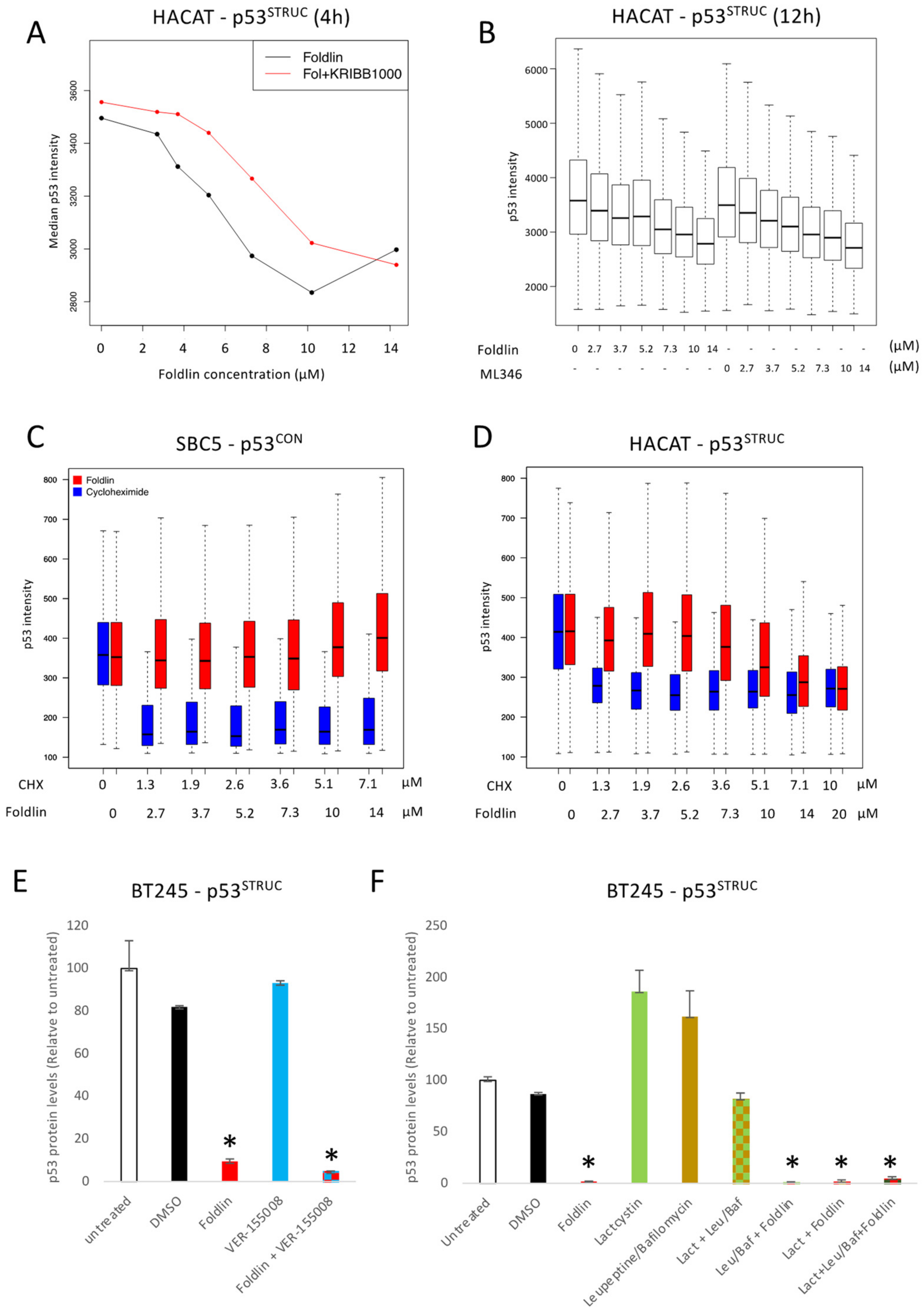
3.5.1. Foldlin Does Not Interfere with RNA Transcription or Protein Translation
3.5.2. Foldlin Does Not Depend on HSP70 Effector Functions
3.5.3. Foldlin Does Not Depend on HSP90 Effector Functions
3.5.4. Foldlin Activity Does Not Depend on Proteostatic Degradation, Autophagy or Shedding
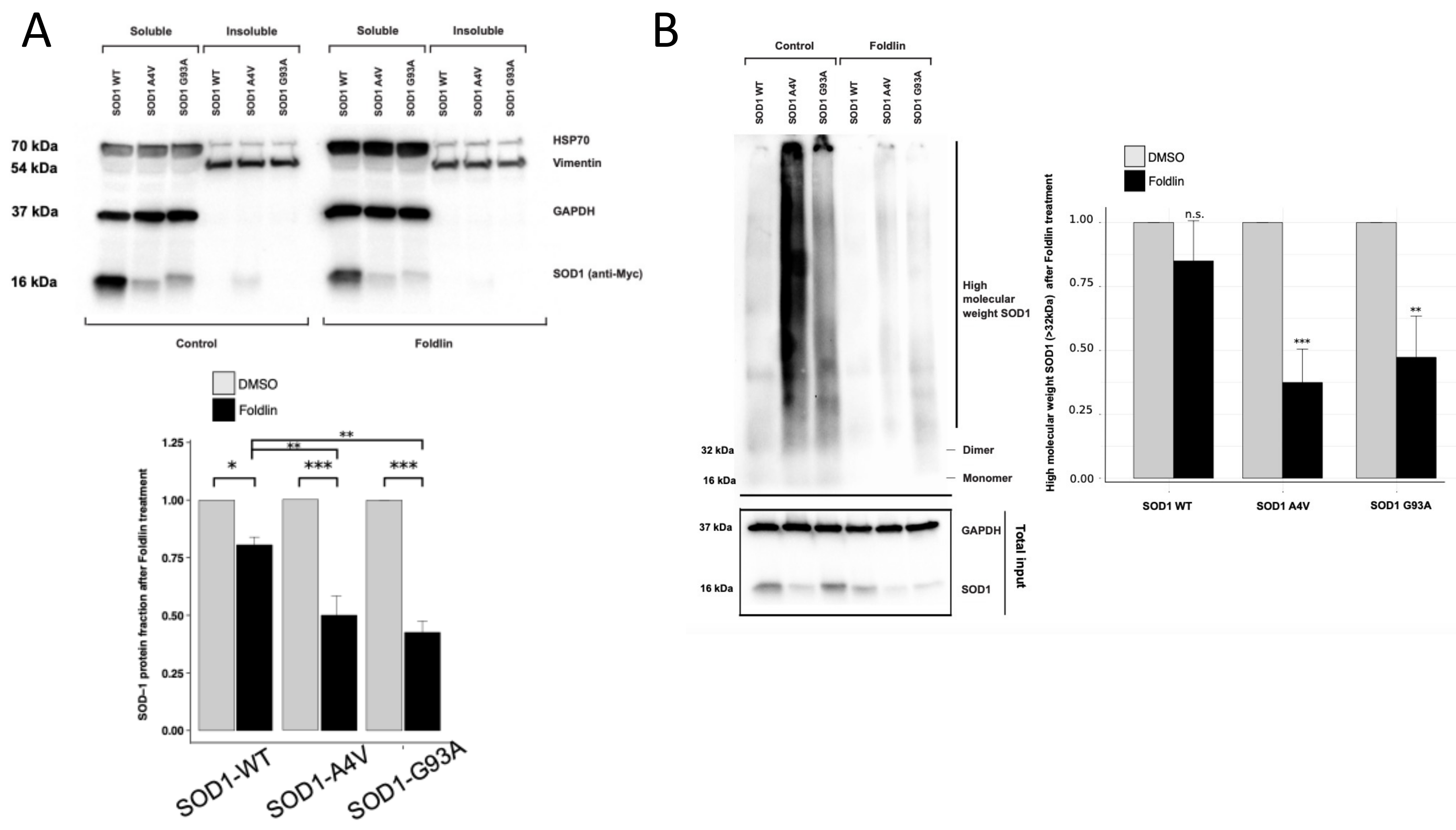
3.6. Reduced Levels of Misfolded and Aggregated SOD1 Variants upon the Pharmacological Activation of HSF1
3.7. Phenotypic Screen for p53 Level Modulators
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Hanel, W.; Marchenko, N.; Xu, S.; Xiaofeng Yu, S.; Weng, W.; Moll, U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [Green Version]
- Lang, G.A.; Iwakuma, T.; Suh, Y.-A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Ano Bom, A.P.D.; Rangel, L.P.; Costa, D.C.F.; de Oliveira, G.A.P.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.I.; Cepeda, A.O.T.; Stumbo, A.C.; et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, C.B.; Stumbo, A.C.; Ano Bom, A.P.D.; Portari, E.A.; Cordeiro, Y.; Carneiro, Y.; Silva, J.L.; De Moura-Gallo, C. V Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 2011, 43, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang-Hartwich, Y.; Soteras, M.G.; Lin, Z.P.; Holmberg, J.; Sumi, N.; Craveiro, V.; Liang, M.; Romanoff, E.; Bingham, J.; Garofalo, F.; et al. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene 2015, 34, 3605–3616. [Google Scholar] [CrossRef]
- De Smet, F.; Saiz Rubio, M.; Hompes, D.; Naus, E.; De Baets, G.; Langenberg, T.; Hipp, M.S.; Houben, B.; Claes, F.; Charbonneau, S.; et al. Nuclear inclusion bodies of mutant and wild-type p53 in cancer: A hallmark of p53 inactivation and proteostasis remodelling by p53 aggregation. J. Pathol. 2017, 242, 24–38. [Google Scholar] [CrossRef]
- Iwahashi, N.; Ikezaki, M.; Komohara, Y.; Fujiwara, Y.; Noguchi, T.; Nishioka, K.; Sakai, K.; Nishio, K.; Ueda, M.; Ihara, Y.; et al. Cytoplasmic p53 aggregates accumulated in p53-mutated cancer correlate with poor prognosis. PNAS Nexus 2022, 1, pgac128. [Google Scholar] [CrossRef]
- Klus, P.; Cirillo, D.; Botta Orfila, T.; Gaetano Tartaglia, G. Neurodegeneration and Cancer: Where the Disorder Prevails. Sci. Rep. 2015, 5, 15390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.; Hrstka, R.; Coomber, D.; Lane, D.P.; Vojt\uesek, B. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 2008, 27, 3371–3383. [Google Scholar] [CrossRef] [Green Version]
- Palanikumar, L.; Karpauskaite, L.; Al-Sayegh, M.; Chehade, I.; Alam, M.; Hassan, S.; Maity, D.; Ali, L.; Kalmouni, M.; Hunashal, Y.; et al. Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat. Commun. 2021, 12, 3962. [Google Scholar] [CrossRef]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Thai-Quynh Nguyen, A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.J.; Gaiddon, C.; Storr, T. A balancing act: Using small molecules for therapeutic intervention of the p53 pathway in cancer. Chem. Soc. Rev. 2020, 49, 6995–7014. [Google Scholar] [CrossRef]
- Haque, E.; Kamil, M.; Irfan, S.; Sheikh, S.; Hasan, A.; Nazir, A.; Mir, S.S. Blocking mutation independent p53 aggregation by emodin modulates autophagic cell death pathway in lung cancer. Int. J. Biochem. Cell Biol. 2018, 96, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Ferraz da Costa, D.C.; Campos, N.P.C.; Santos, R.A.; Guedes-da-Silva, F.H.; Martins-Dinis, M.M.D.C.; Zanphorlin, L.; Ramos, C.; Rangel, L.P.; Silva, J.L. Resveratrol prevents p53 aggregation in vitro and in breast cancer cells. Oncotarget 2018, 9, 29112–29122. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.R.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.A.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Chem. 2019, 11, 2491–2504. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 aggregation, interactions with tau, and impaired DNA damage response in Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 132. [Google Scholar] [CrossRef]
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting Hsp90/Hsp70-Based Protein Quality Control for Treatment of Adult Onset Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 353–371. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messiaen, J.; Claeys, A.; Shetty, A.; Spans, L.; Derweduwe, M.; Uyttebroeck, A.; Depreitere, B.; Vanden Bempt, I.; Sciot, R.; Ligon, K.L.; et al. Generation of patient-derived models from a metastatic pediatric diffuse leptomeningeal glioneuronal tumor with KIAA1549::BRAF fusion. Acta Neuropathol. 2022, 144, 793–797. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, C.; Seo, Y.H. Small Molecule Inhibitors of HSF1-Activated Pathways as Potential Next-Generation Anticancer Therapeutics. Molecules 2018, 23, 2757. [Google Scholar] [CrossRef] [Green Version]
- Calamini, B.; Silva, M.C.; Madoux, F.; Hutt, D.M.; Khanna, S.; Chalfant, M.A.; Saldanha, S.A.; Hodder, P.; Tait, B.D.; Garza, D.; et al. small-molecule proteostasis regulators for protein conformational diseases. Nat. Chem. Biol. 2011, 8, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussiere, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattoo, R.U.H.; Sharma, S.K.; Priya, S.; Finka, A.; Goloubinoff, P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem. 2013, 288, 21399–21411. [Google Scholar] [CrossRef] [Green Version]
- Nillegoda, N.B.; Kirstein, J.; Szlachcic, A.; Berynskyy, M.; Stank, A.; Stengel, F.; Arnsburg, K.; Gao, X.; Scior, A.; Aebersold, R.; et al. Crucial HSP70 co-chaperone complex unlocks metazoan protein disaggregation. Nature 2015, 524, 247–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.J.; Kim, J.A.; Shin, K.D.; Shin, D.-S.; Han, Y.M.; Lee, Y.J.; Lee, J.S.; Kwon, B.-M.; Han, D.C. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J. Biol. Chem. 2011, 286, 1737–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calamini, B.; Silva, M.C.; Madoux, F.; Hutt, D.M.; Khanna, S.; Chalfant, M.A.; Allais, C.; Ouizem, S.; Saldanha, S.A.; Ferguson, J.; et al. ML346: A Novel Modulator of Proteostasis for Protein Conformational Diseases. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010; Volume 8. [Google Scholar]
- Islam, M.A.; Sooro, M.A.; Zhang, P. Autophagic Regulation of p62 is Critical for Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [Green Version]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef] [Green Version]
- Arlander, S.J.H.; Felts, S.J.; Wagner, J.M.; Stensgard, B.; Toft, D.O.; Karnitz, L.M. Chaperoning checkpoint kinase 1 (Chk1), an Hsp90 client, with purified chaperones. J. Biol. Chem. 2006, 281, 2989–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, K.T.; Goldberg, A.L.; Nigam, S.K. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J. Biol. Chem. 1997, 272, 9086–9092. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Dunner, K.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ōmura, S.; Crump, A. Lactacystin: First-in-class proteasome inhibitor still excelling and an exemplar for future antibiotic research. J. Antibiot. 2019, 72, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasil, A.d.A.; de Carvalho, M.D.C.; Gerhardt, E.; Queiroz, D.D.; Pereira, M.D.; Outeiro, T.F.; Eleutherio, E.C.A. Characterization of the activity, aggregation, and toxicity of heterodimers of WT and ALS-associated mutant Sod1. Proc. Natl. Acad. Sci. USA 2019, 116, 25991–26000. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zheng, M.; Li, Z.; Li, R.; Jia, L.; Xiong, X.; Southall, N.; Wang, S.; Xia, M.; Austin, C.P.; et al. Cardiac glycosides inhibit p53 synthesis by a mechanism relieved by Src or MAPK inhibition. Cancer Res. 2009, 69, 6556–6564. [Google Scholar] [CrossRef] [Green Version]
- Reece, D.; Imrie, K.; Stevens, A.; Smith, C.A.; Hematology Disease Site Groupof Cancer Care Ontario’s Program in Evidence-based Care. Bortezomib in multiple myeloma and lymphoma: A systematic review and clinical practice guideline. Curr. Oncol. 2006, 13, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Moxley, A.H.; Reisman, D. Context is key: Understanding the regulation, functional control, and activities of the p53 tumour suppressor. Cell Biochem. Funct. 2020, 39, 235–247. [Google Scholar] [CrossRef]
- Khoo, K.H.; Mayer, S.; Fersht, A.R. Effects of stability on the biological function of p53. J. Biol. Chem. 2009, 284, 30974–30980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, G.A.P.; Rangel, L.P.; Costa, D.C.; Silva, J.L. Misfolding, Aggregation, and Disordered Segments in c-Abl and p53 in Human Cancer. Front. Oncol. 2015, 5, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, P.J.; Klucken, J.; Shin, Y.; Hyman, B.T. Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem. Biophys. Res. Commun. 2004, 321, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.; Multhoff, G.; Mikhaylova, E.; Shibata, A.; Guzhova, I.; Margulis, B. Combination of Anti-Cancer Drugs with Molecular Chaperone Inhibitors. Int. J. Mol. Sci. 2019, 20, 5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santagata, S.; Xu, Y.-M.; Wijeratne, E.M.K.; Kontnik, R.; Rooney, C.; Perley, C.C.; Kwon, H.; Clardy, J.; Kesari, S.; Whitesell, L.; et al. Using the heat-shock response to discover anticancer compounds that target protein homeostasis. ACS Chem. Biol. 2012, 7, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chauve, L.; Phelps, G.; Brielmann, R.M.; Morimoto, R.I. E2F coregulates an essential HSF developmental program that is distinct from the heat-shock response. Genes Dev. 2016, 30, 2062–2075. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef] [PubMed]
- Krawczenko, A.; Bielawska-Pohl, A.; Wojtowicz, K.; Jura, R.; Paprocka, M.; Wojdat, E.; Kozłowska, U.; Klimczak, A.; Grillon, C.; Kieda, C.; et al. Expression and activity of multidrug resistance proteins in mature endothelial cells and their precursors: A challenging correlation. PLoS ONE 2017, 12, e0172371. [Google Scholar] [CrossRef] [Green Version]
- Kamel, M.M. Convenient synthesis, characterization, cytotoxicity and toxicity of pyrazole derivatives. Acta Chim. Slov. 2015, 62, 136–151. [Google Scholar] [CrossRef] [Green Version]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.; Barker, N.; Hoon, S.; He, P.; Thakur, T.; Abdeen, S.R.; Maruthappan, P.; Ghadessy, F.J.; Lane, D.P. Bortezomib Stabilizes and Activates p53 in Proliferative Compartments of Both Normal and Tumor Tissues In Vivo. Cancer Res. 2019, 79, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Dabiri, Y.; Kalman, S.; Gürth, C.-M.; Kim, J.Y.; Mayer, V.; Cheng, X. The essential role of TAp73 in bortezomib-induced apoptosis in p53-deficient colorectal cancer cells. Sci. Rep. 2017, 7, 5423. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Yang, S.; Li, Y.; Li, Y.; Devor, E.J.; Bi, J.; Wang, X.; Umesalma, S.; Quelle, D.E.; Thiel, W.H.; et al. Combination of Proteasome and Histone Deacetylase Inhibitors Overcomes the Impact of Gain-of-Function p53 Mutations. Dis. Markers 2018, 2018, 3810108. [Google Scholar] [CrossRef]
- Ling, X.; Calinski, D.; Chanan-Khan, A.A.; Zhou, M.; Li, F. Cancer cell sensitivity to bortezomib is associated with survivin expression and p53 status but not cancer cell types. J. Exp. Clin. Cancer Res. 2010, 29, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Forward Primer | Reversed Primer | Probe |
|---|---|---|---|
| HSPB8 | AAAGATGGATACGTGGAGGTG | GGGAAAGTGAGGCAAATACTG | /56-FAM/CTGGCAAAC/ZEN/ATGAAGAGAAACAGCAAGA/3IABkFQ/ |
| HSP90AA1 | GTCTGTGAAGGATCTGGTCATC | CAGCAGTAGGGTCATCTTCATC | /56-FAM/AGACAGGAG/ZEN/CGCAGTTTCATAAAGCA/3IABkFQ/ |
| HSP90B1 | AAACGGGCAAGGACATCTC | AAACCACAGCAAGATCCAAAAC | /56-FAM/TCAGCGGGT/ZEN/GTCTGGGATTAATTTCAA/3IABkFQ/ |
| HSPB1 | ATGTCAACCACTTCGCCC | GTGAAGCACCGGGAGATG | /56-FAM/AGATCACCG/ZEN/GCAAGCACGAGG/3IABkFQ/ |
| HSPA1A | AGGACATCAGCCAGAACAAG | CTGGTGATGGACGTGTAGAAG | /56-FAM/CTGCGAGAG/ZEN/GGCCAAGAGGAC/3IABkFQ/ |
| HSPA6 | AAGCAGACCCAGACTTTCAC | TCTCACCCTCATACACCTGG | /56-FAM/CACCTACTC/ZEN/GGACAACCAGCCT/3IABkFQ/ |
| HSPA8 | GGACAAGAGTACGGGAAAAGAG | GTCCCTCTGCTTCTCATCTTC | /56-FAM/TTCAATGTC/ZEN/TTCCTTGCTCAAACGGC/3IABkFQ/ |
| HSPA9 | GTATTCTCTACTGCCGCTGATG | TTCAATCTGAGGAACTCCACG | /56-FAM/TCTCCAGCC/ZEN/ATCTCTCTTTCACCCT/3IABkFQ/ |
| TP53 | AATACTCCACACGCAAATTTCC | CAAGCAGTCACAGCACATGA | /56-FAM/CCTCCTCAG/ZEN/CATCTTATCCGAGTGGA/3IABkFQ/ |
| ACTB | CCTTGCACATGCCGGAG | ACAGAGCCTCGCCTTTG | /56-FAM/TCATCCATG/ZEN/GTGAGCTGGCGG/3IABkFQ/ |
| GAPDH: | TGTAGTTGAGGTCAATGAAGGG | ACATCGCTCAGACACCATG | /5HEX/AAGGTCGGA/ZEN/GTCAACGGATTTGGTC/3IABkFQ/ |
| Cell Line | Tumor Type | P53 Genotype | Protein Classification | Aggregational Status(bNativePAGE) | Mean IC50 (±SEM)(μM) |
|---|---|---|---|---|---|
| U-2 OS | Osteosarcoma | WT | Wild type | No | 12.31 ± 1.8 |
| A549 * | Lung carcinoma | WT | Wild type | No | >50 |
| HCT-116 | Colorectal carcinoma | WT | Wild type | No | 10.87 ± 1.6 |
| SAOS-2 | Osteosarcoma | Null | Null | / | 13.07 ± 2.6 |
| HeLa | Epithelial cervix adenocarcinoma | null | Null | No | 11.73 ± 1.5 |
| SBC-5 | Lung carcinoma | R248L | Contact mutant | No | 15.88 ± 1.3 |
| C-33A * | Retinoblastoma | R273C | Mixed conformation | Yes | 13.94 ± 1.4 |
| HT-1376 * | Bladder carcinoma | P250L | Structural mutant | Yes | >50 |
| CHL-1 * | Melanoma | H193R | Structural mutant | Yes | 6.67 ± 1.4 |
| DU 145 * | Prostate carcinoma | P223L/V274F | Structural mutant | Yes | 17.24 ± 1.4 |
| MEL1617 * | Melanoma | Y220C | Structural mutant | Yes | 8.28 ± 1.7 |
| HaCat * | Immortalized keratinocyte | R282W/H197Y | Structural mutant | Yes | 11.15 ± 1.4 |
| HCC827 * | Lung carcinoma | ^V218 | Structural mutant | Yes | 8.17 ± 1.5 |
| Detroit-562 | Pharyngeal carcinoma | R175H | Structural mutant | Yes | 23.5 ± 1.9 |
| GBM Cell Line | p53 Mutation Status | IC50 Foldlin (μM) |
|---|---|---|
| BT112 | WT | 5.02 |
| BT569 | WT | / |
| BT164 | Q164 * | 10.84 |
| BT271 | WT | / |
| BT239 | WT | 5.71 |
| BT333 | V173M | 6.68 |
| BT359 | C275Y | 14.09 |
| BT482 | Fs152 | 8.93 |
| BT245 | R249S | / |
| BT607 | WT | 7.22 |
| BT159 | WT | 1.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naus, E.; Derweduwe, M.; Lampi, Y.; Claeys, A.; Pauwels, J.; Langenberg, T.; Claes, F.; Xu, J.; Haemels, V.; Atak, Z.K.; et al. Reduced Levels of Misfolded and Aggregated Mutant p53 by Proteostatic Activation. Cells 2023, 12, 960. https://doi.org/10.3390/cells12060960
Naus E, Derweduwe M, Lampi Y, Claeys A, Pauwels J, Langenberg T, Claes F, Xu J, Haemels V, Atak ZK, et al. Reduced Levels of Misfolded and Aggregated Mutant p53 by Proteostatic Activation. Cells. 2023; 12(6):960. https://doi.org/10.3390/cells12060960
Chicago/Turabian StyleNaus, Evelyne, Marleen Derweduwe, Youlia Lampi, Annelies Claeys, Jarne Pauwels, Tobias Langenberg, Filip Claes, Jie Xu, Veerle Haemels, Zeynep Kalender Atak, and et al. 2023. "Reduced Levels of Misfolded and Aggregated Mutant p53 by Proteostatic Activation" Cells 12, no. 6: 960. https://doi.org/10.3390/cells12060960