
Enrichment of Human Dermal Stem Cells from Primary Cell Cultures through the Elimination of Fibroblasts
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Cultivation of Cells
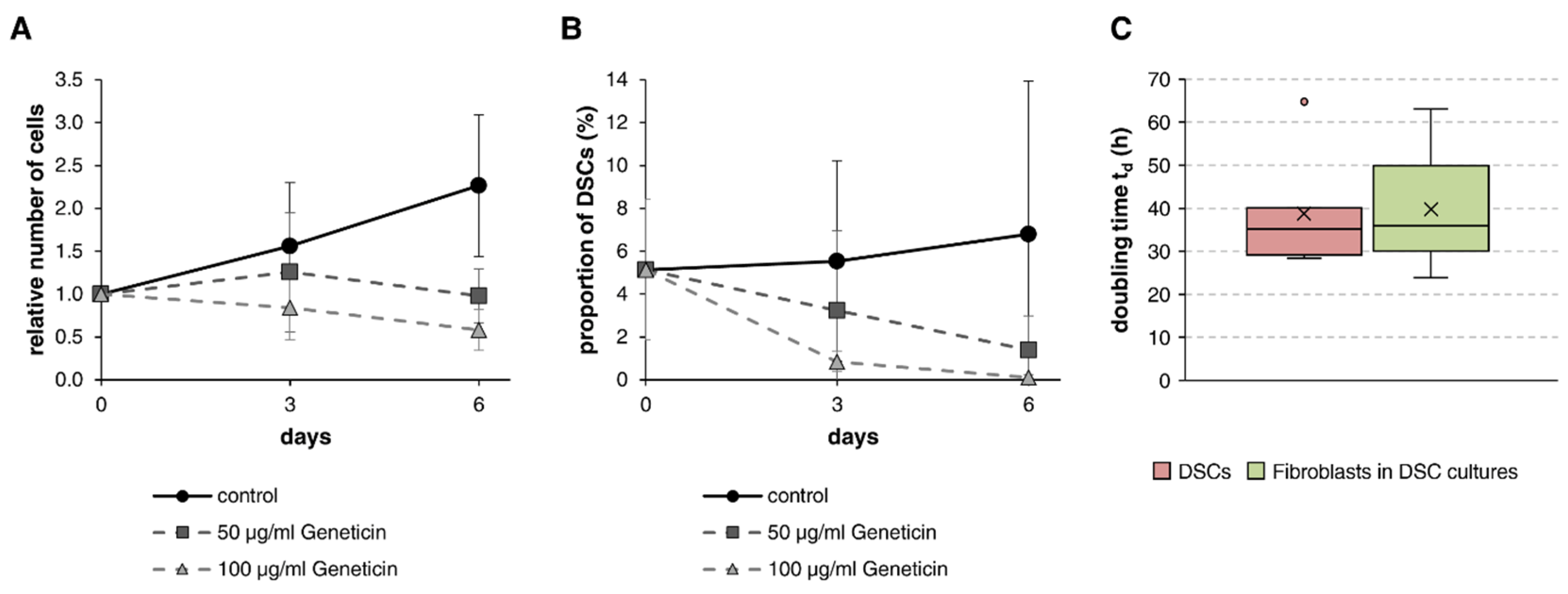
2.2. Doubling Time
2.3. Geneticin Treatment
2.4. Selective Detachment
2.5. Immunomagnetic Separation
2.5.1. EasySep™ Column-Free Separation
2.5.2. MACS® Automatic Column-Based Separation
2.6. Immunostaining
3. Results and Discussion
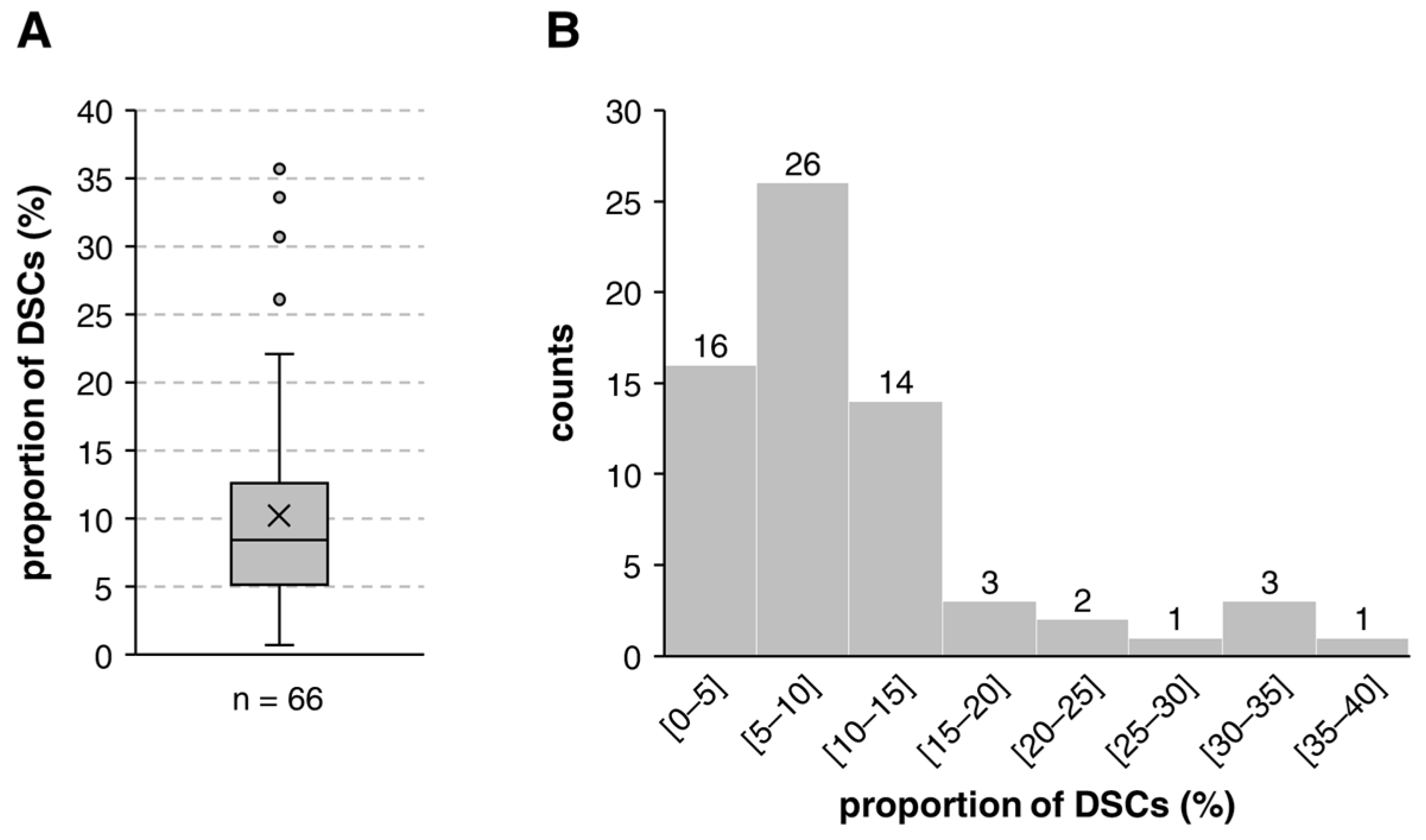
3.1. Stem Cell Frequency of DSC-Fibroblast Co-Cultures
3.2. Elimination of Fibroblasts with Geneticin
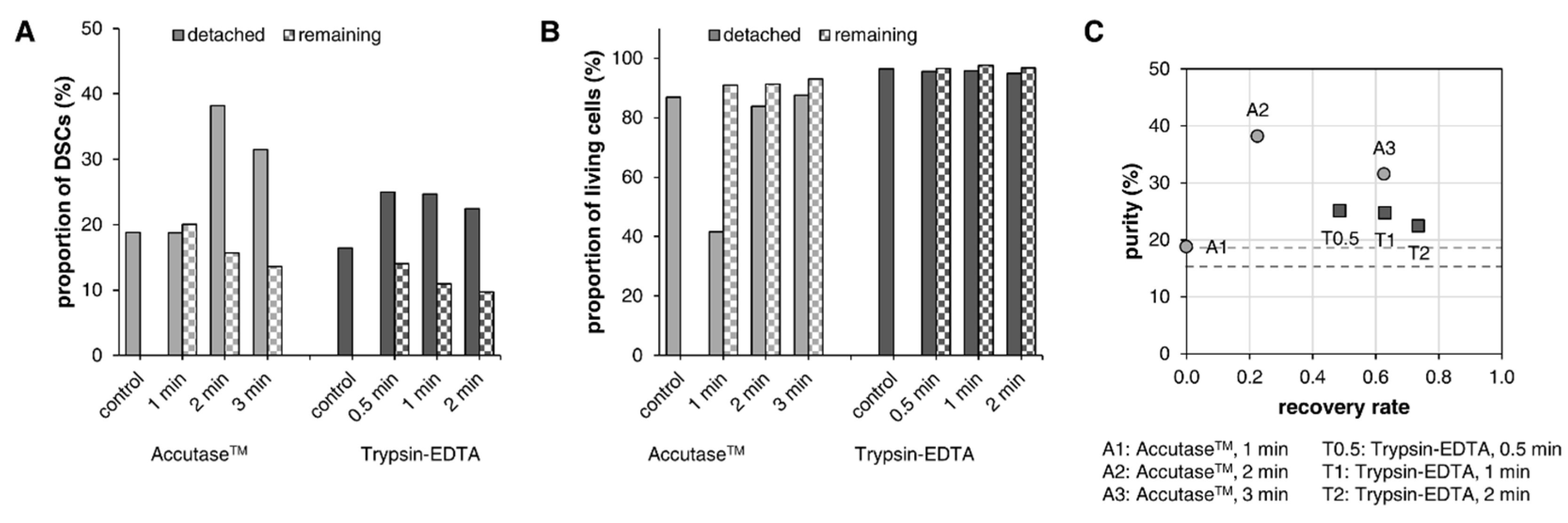
3.3. Segregation of DSCs and Fibroblasts via Selective Detachment
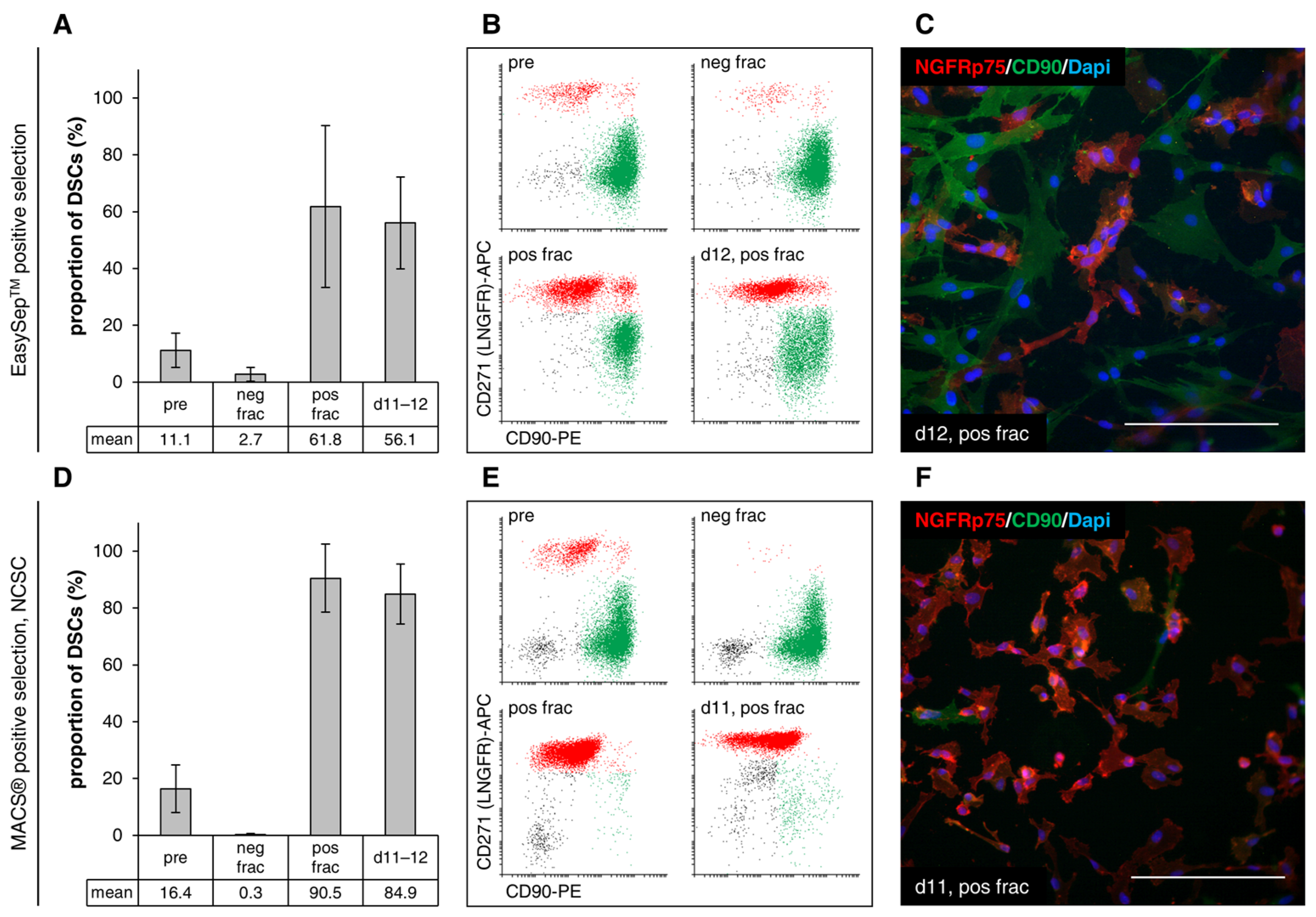
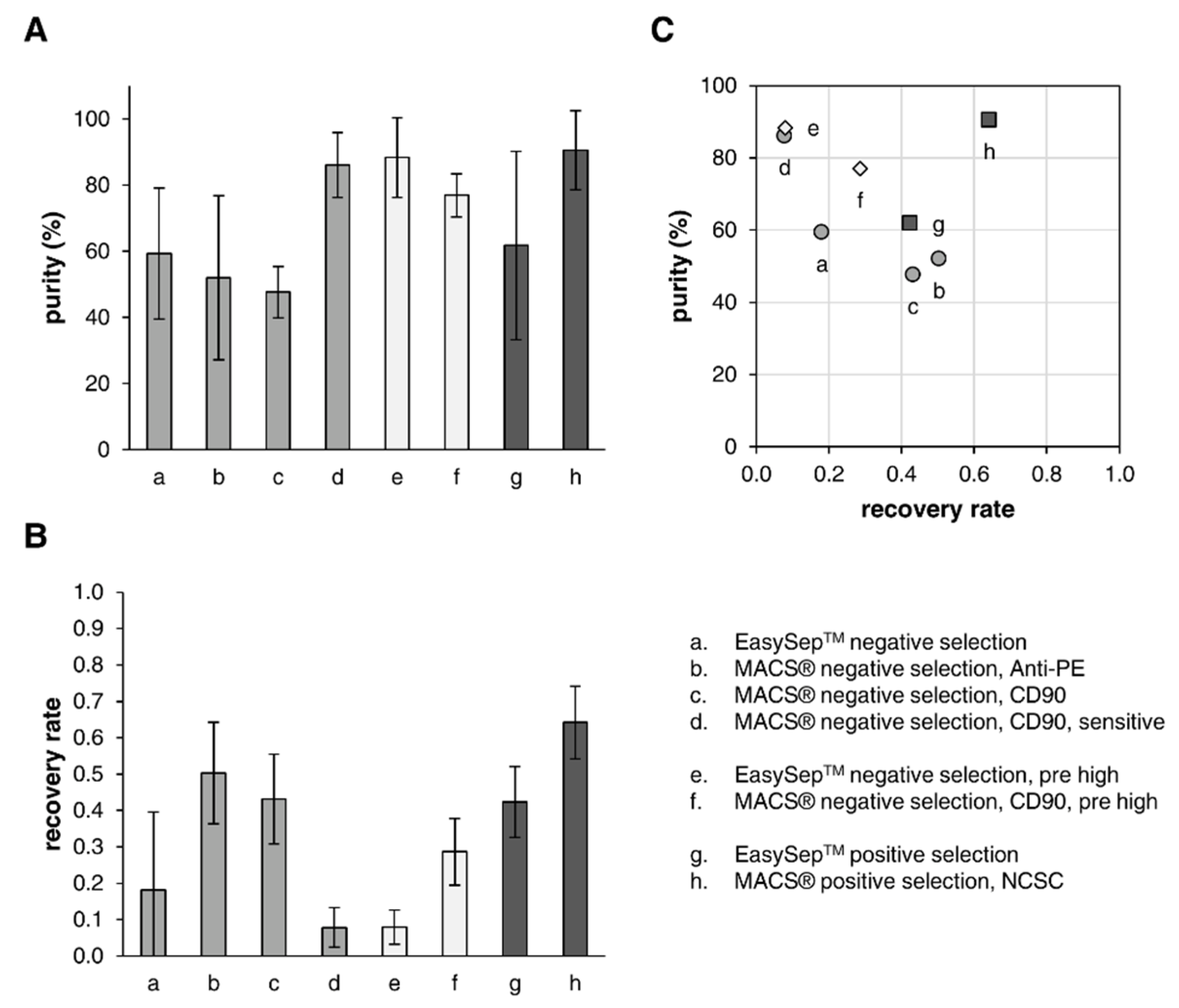
3.4. Separation of the Two Cell Types through Immunomagnetic Cell Sorting
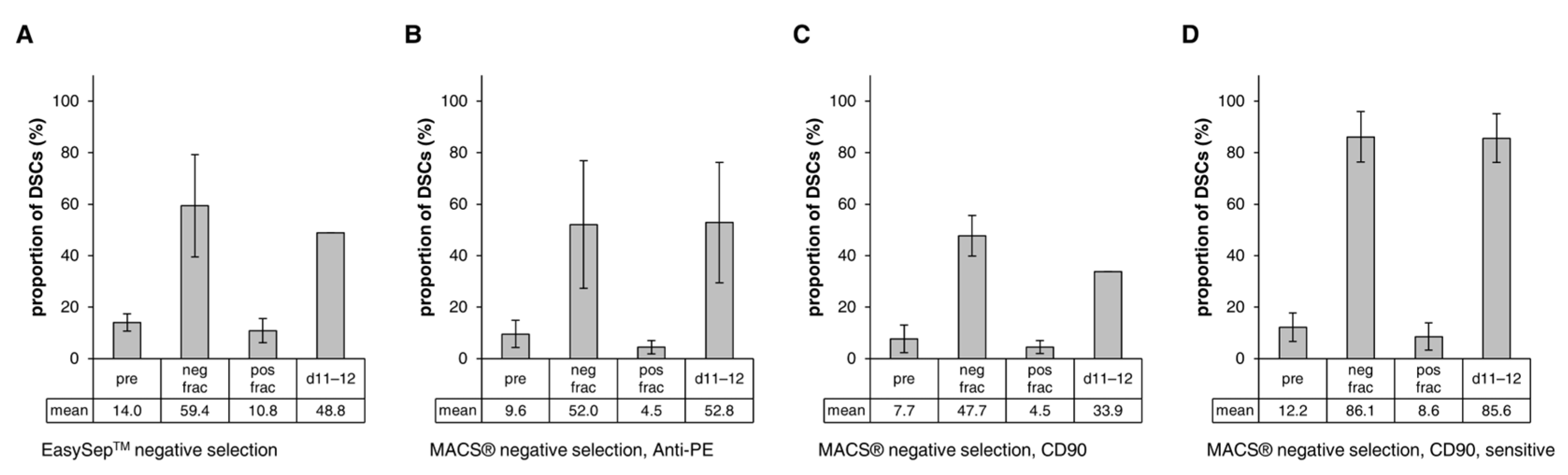
3.4.1. Negative Selection
3.4.2. Positive Selection
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hoerter, J.D.; Bradley, P.; Casillas, A.; Chambers, D.; Denholm, C.; Johnson, K.; Weiswasser, B. Extrafollicular dermal melanocyte stem cells and melanoma. Stem. Cells Int. 2012, 2012, 407079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabierowski, S.E.; Fukunaga-Kalabis, M.; Li, L.; Herlyn, M. Dermis-derived stem cells: A source of epidermal melanocytes and melanoma? Pigment. Cell Melanoma Res. 2011, 24, 422–429. [Google Scholar] [CrossRef]
- Li, L.; Fukunaga-Kalabis, M.; Yu, H.; Xu, X.; Kong, J.; Lee, J.T.; Herlyn, M. Human dermal stem cells differentiate into functional epidermal melanocytes. J. Cell Sci. 2010, 123, 853–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mhamdi-Ghodbani, M.; Starzonek, C.; Degenhardt, S.; Bender, M.; Said, M.; Greinert, R.; Volkmer, B. UVB damage response of dermal stem cells as melanocyte precursors compared to keratinocytes, melanocytes, and fibroblasts from human foreskin. J. Photochem. Photobiol. B 2021, 220, 112216. [Google Scholar] [CrossRef] [PubMed]
- Saalbach, A.; Aust, G.; Haustein, U.F.; Herrmann, K.; Anderegg, U. The fibroblast-specific MAb AS02: A novel tool for detection and elimination of human fibroblasts. Cell Tissue Res. 1997, 290, 593–599. [Google Scholar] [CrossRef]
- Peng, K.; Sant, D.; Andersen, N.; Silvera, R.; Camarena, V.; Pinero, G.; Graham, R.; Khan, A.; Xu, X.M.; Wang, G.; et al. Magnetic separation of peripheral nerve-resident cells underscores key molecular features of human Schwann cells and fibroblasts: An immunochemical and transcriptomics approach. Sci. Rep. 2020, 10, 18433. [Google Scholar] [CrossRef]
- Calderón-Martínez, D.; Garavito, Z.; Spinel, C.; Hurtado, H. Schwann cell-enriched cultures from adult human peripheral nerve: A technique combining short enzymatic dissociation and treatment with cytosine arabinoside (Ara-C). J. Neurosci. Methods 2002, 114, 1–8. [Google Scholar] [CrossRef]
- Lehmann, H.C.; Chen, W.; Mi, R.; Wang, S.; Liu, Y.; Rao, M.; Höke, A. Human Schwann cells retain essential phenotype characteristics after immortalization. Stem. Cells Dev. 2012, 21, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Gledhill, K.; Guo, Z.; Umegaki-Arao, N.; Higgins, C.A.; Itoh, M.; Christiano, A.M. Melanin Transfer in Human 3D Skin Equivalents Generated Exclusively from Induced Pluripotent Stem Cells. PLoS ONE 2015, 10, e0136713. [Google Scholar] [CrossRef]
- Li, S.; Zenkel, M.; Kruse, F.E.; Giessl, A.; Schlotzer-Schrehardt, U. Identification, Isolation, and Characterization of Melanocyte Precursor Cells in the Human Limbal Stroma. Int. J. Mol. Sci. 2022, 23, 3756. [Google Scholar] [CrossRef]
- Hsiao, F.S.; Cheng, C.C.; Peng, S.Y.; Huang, H.Y.; Lian, W.S.; Jan, M.L.; Fang, Y.T.; Cheng, E.C.; Lee, K.H.; Cheng, W.T.; et al. Isolation of therapeutically functional mouse bone marrow mesenchymal stem cells within 3 h by an effective single-step plastic-adherent method. Cell Prolif. 2010, 43, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, M.J.; Tomlinson, S.; Yang, X.B.; Kirkham, J. Cell separation: Terminology and practical considerations. J. Tissue Eng. 2013, 4, 2041731412472690. [Google Scholar] [CrossRef] [PubMed]
- Kisselbach, L.; Merges, M.; Bossie, A.; Boyd, A. CD90 Expression on human primary cells and elimination of contaminating fibroblasts from cell cultures. Cytotechnology 2009, 59, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrot, P.; Laurent, P.; Levionnois, E.; Leleu, D.; Pain, C.; Truchetet, M.E.; Cario, M. A Method for Isolating and Culturing Skin Cells: Application to Endothelial Cells, Fibroblasts, Keratinocytes, and Melanocytes From Punch Biopsies in Systemic Sclerosis Skin. Front. Immunol. 2020, 11, 566607. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, T.; Nakao, Y.; Toyama, Y.; Asou, H.; Vacanti, C.A.; Vacanti, M.P. A novel technique to isolate adult Schwann cells for an artificial nerve conduit. J. Neurosci. Methods 2003, 122, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Bourland, J.; Mayrand, D.; Tremblay, N.; Moulin, V.J.; Fradette, J.; Auger, F.A. Isolation and Culture of Human Dermal Microvascular Endothelial Cells. Methods Mol. Biol. 2019, 1993, 79–90. [Google Scholar] [CrossRef]
- Liao, X.; Makris, M.; Luo, X.M. Fluorescence-activated Cell Sorting for Purification of Plasmacytoid Dendritic Cells from the Mouse Bone Marrow. J. Vis. Exp. 2016, 117, e54641. [Google Scholar] [CrossRef]
- Manohar, S.M.; Shah, P.; Nair, A. Flow cytometry: Principles, applications and recent advances. Bioanalysis 2021, 13, 181–198. [Google Scholar] [CrossRef]
- Polisetti, N.; Schlotzer-Schrehardt, U.; Reinhard, T.; Schlunck, G. Isolation and enrichment of melanocytes from human corneal limbus using CD117 (c-Kit) as selection marker. Sci. Rep. 2020, 10, 17588. [Google Scholar] [CrossRef]
- Willemsen, M.; Luiten, R.M.; Teunissen, M.B.M. Instant isolation of highly purified human melanocytes from freshly prepared epidermal cell suspensions. Pigment. Cell Melanoma Res. 2020, 33, 763–766. [Google Scholar] [CrossRef]
- Sobiepanek, A.; Kowalska, P.D.; Soszyńska, M.; Ścieżyńska, A. Implementation of Geneticin in the in vitro cell culture and in vivo studies. Rev. Res. Cancer Treat. 2020, 6, 79–87. [Google Scholar]
- Zborowski, M.; Chalmers, J.J. Magnetic cell sorting. Methods Mol. Biol. 2005, 295, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Ravelo, K.M.; Andersen, N.D.; Monje, P.V. Magnetic-Activated Cell Sorting for the Fast and Efficient Separation of Human and Rodent Schwann Cells from Mixed Cell Populations. Methods Mol. Biol. 2018, 1739, 87–109. [Google Scholar] [CrossRef] [PubMed]
- Moulin, V.J.; Mayrand, D.; Laforce-Lavoie, A.; Larochelle, S.; Genest, H. In Vitro Culture Methods of Skin Cells for Optimal Skin Reconstruction by Tissue Engineering. Regen. Med. Tissue Eng.-Cells Biomater. 2011, 10, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.Y.; Cao, J.; Ou-Yang, P.; Tsai, C.Y.; Lin, C.W.; Chen, C.C.; Tsai, M.K.; Lee, C.Y. Different methods of detaching adherent cells and their effects on the cell surface expression of Fas receptor and Fas ligand. Sci. Rep. 2022, 12, 5713. [Google Scholar] [CrossRef] [PubMed]
- Skog, M.; Sivler, P.; Steinvall, I.; Aili, D.; Sjoberg, F.; Elmasry, M. The Effect of Enzymatic Digestion on Cultured Epithelial Autografts. Cell Transpl. 2019, 28, 638–644. [Google Scholar] [CrossRef] [Green Version]
- Quan, Y.; Yan, Y.; Wang, X.; Fu, Q.; Wang, W.; Wu, J.; Yang, G.; Ren, J.; Wang, Y. Impact of cell dissociation on identification of breast cancer stem cells. Cancer Biomark. 2012, 12, 125–133. [Google Scholar] [CrossRef]
- Miltenyi, S.; Müller, W.; Weichel, W.; Radbruch, A. High gradient magnetic cell separation with MACS. Cytometry 1990, 11, 231–238. [Google Scholar] [CrossRef]
- Moore, D.K.; Motaung, B.; du Plessis, N.; Shabangu, A.N.; Loxton, A.G. Isolation of B-cells using Miltenyi MACS bead isolation kits. PLoS ONE 2019, 14, e0213832. [Google Scholar] [CrossRef]
- Peters, C.E.; Woodside, S.M.; Eaves, A.C. Isolation of subsets of immune cells. Methods Mol. Biol. 2005, 302, 95–116. [Google Scholar] [CrossRef]
- Frenea-Robin, M.; Marchalot, J. Basic Principles and Recent Advances in Magnetic Cell Separation. Magnetochemistry 2022, 8, 11. [Google Scholar] [CrossRef]
- Grützkau, A.; Radbruch, A. Small but mighty: How the MACS-technology based on nanosized superparamagnetic particles has helped to analyze the immune system within the last 20 years. Cytometry A 2010, 77, 643–647. [Google Scholar] [CrossRef]
- Chalmers, J.J.; Xiong, Y.; Jin, X.; Shao, M.; Tong, X.; Farag, S.; Zborowski, M. Quantification of non-specific binding of magnetic micro- and nanoparticles using cell tracking velocimetry: Implication for magnetic cell separation and detection. Biotechnol. Bioeng. 2010, 105, 1078–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, G.S.; Silva, Z.; Videira, P.A. Antitumor Efficacy of Human Monocyte-Derived Dendritic Cells: Comparing Effects of two Monocyte Isolation Methods. Biol. Proced. Online 2018, 20, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Rinkevich, Y. Defining Skin Fibroblastic Cell Types Beyond CD90. Front. Cell Dev. Biol. 2018, 6, 133. [Google Scholar] [CrossRef] [Green Version]
- Farrell, E.; Wielopolski, P.; Pavljasevic, P.; van Tiel, S.; Jahr, H.; Verhaar, J.; Weinans, H.; Krestin, G.; O’Brien, F.J.; van Osch, G.; et al. Effects of iron oxide incorporation for long term cell tracking on MSC differentiation in vitro and in vivo. Biochem. Biophys. Res. Commun. 2008, 369, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, K.; Kroeger, I.; Abendroth, A.; Müller, S.; Mackensen, A.; Ullrich, E. Influence of NK cell magnetic bead isolation methods on phenotype and function of murine NK cells. J. Immunol. Methods 2012, 378, 1–10. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Azadmanesh, K.; Shokrgozar, M.A.; Journeay, W.S.; Laurent, S. Effect of nanoparticles on the cell life cycle. Chem. Rev. 2011, 111, 3407–3432. [Google Scholar] [CrossRef]
- Shen, M.J.; Olsthoorn, R.C.L.; Zeng, Y.; Bakkum, T.; Kros, A.; Boyle, A.L. Magnetic-Activated Cell Sorting Using Coiled-Coil Peptides: An Alternative Strategy for Isolating Cells with High Efficiency and Specificity. ACS Appl. Mater Interfaces 2021, 13, 11621–11630. [Google Scholar] [CrossRef]
- Van de Walle, A.; Perez, J.E.; Abou-Hassan, A.; Hémadi, M.; Luciani, N.; Wilhelm, C. Magnetic nanoparticles in regenerative medicine: What of their fate and impact in stem cells? Mater. Today Nano 2020, 11, 100084. [Google Scholar] [CrossRef]
- Müller, P.; Gaebel, R.; Lemcke, H.; Wiekhorst, F.; Hausburg, F.; Lang, C.; Zarniko, N.; Westphal, B.; Steinhoff, G.; David, R. Intramyocardial fate and effect of iron nanoparticles co-injected with MACS(®) purified stem cell products. Biomaterials 2017, 135, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Gaebel, R.; Lemcke, H.; Steinhoff, G.; David, R. Data on the fate of MACS® MicroBeads intramyocardially co-injected with stem cell products. Data Brief. 2017, 13, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Laghmouchi, A.; Hoogstraten, C.; Falkenburg, J.H.F.; Jedema, I. Long-term in vitro persistence of magnetic properties after magnetic bead-based cell separation of T cells. Scand J. Immunol. 2020, 92, e12924. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, M.; Rieger, M.; Reinert, S.; Alexander, D. Isolation of osteoprogenitors from human jaw periosteal cells: A comparison of two magnetic separation methods. PLoS ONE 2012, 7, e47176. [Google Scholar] [CrossRef] [Green Version]
- Meyers, C.A.; Xu, J.; Zhang, L.; Chang, L.; Wang, Y.; Asatrian, G.; Ding, C.; Yan, N.; Zou, E.; Broderick, K.; et al. Skeletogenic Capacity of Human Perivascular Stem Cells Obtained Via Magnetic-Activated Cell Sorting. Tissue Eng. Part A 2019, 25, 1658–1666. [Google Scholar] [CrossRef]
- Øren, A.; Husebø, C.; Iversen, A.C.; Austgulen, R. A comparative study of immunomagnetic methods used for separation of human natural killer cells from peripheral blood. J. Immunol. Methods 2005, 303, 1–10. [Google Scholar] [CrossRef]
- Miltenyi Biotec. Magnetic Cell Separation. Available online: https://www.miltenyibiotec.com/DE-en/resources/macs-handbook/macs-technologies/cell-separation/magnetic-cell-separation.html (accessed on 9 May 2022).
- Miltenyi Biotec. MACS® Cell Separation, Select the Best Brochure. Available online: https://www.miltenyibiotec.com/_Resources/Persistent/b5349effdd595b72195e588aff033be3e24706bd/IM0020021.pdf (accessed on 9 May 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EasySep™ Column-Free | MACS® Automatic Column-Based |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starzonek, C.; Mhamdi-Ghodbani, M.; Henning, S.; Bender, M.; Degenhardt, S.; Chen, I.-P.; Said, M.; Greinert, R.; Volkmer, B. Enrichment of Human Dermal Stem Cells from Primary Cell Cultures through the Elimination of Fibroblasts. Cells 2023, 12, 949. https://doi.org/10.3390/cells12060949
Starzonek C, Mhamdi-Ghodbani M, Henning S, Bender M, Degenhardt S, Chen I-P, Said M, Greinert R, Volkmer B. Enrichment of Human Dermal Stem Cells from Primary Cell Cultures through the Elimination of Fibroblasts. Cells. 2023; 12(6):949. https://doi.org/10.3390/cells12060949
Chicago/Turabian StyleStarzonek, Christin, Mouna Mhamdi-Ghodbani, Stefan Henning, Marc Bender, Sarah Degenhardt, I-Peng Chen, Mohamed Said, Rüdiger Greinert, and Beate Volkmer. 2023. "Enrichment of Human Dermal Stem Cells from Primary Cell Cultures through the Elimination of Fibroblasts" Cells 12, no. 6: 949. https://doi.org/10.3390/cells12060949