
Cytological Diagnosis of Classic Myeloproliferative Neoplasms at the Age of Molecular Biology
 , , ,
, , ,
Abstract
:1. Introduction
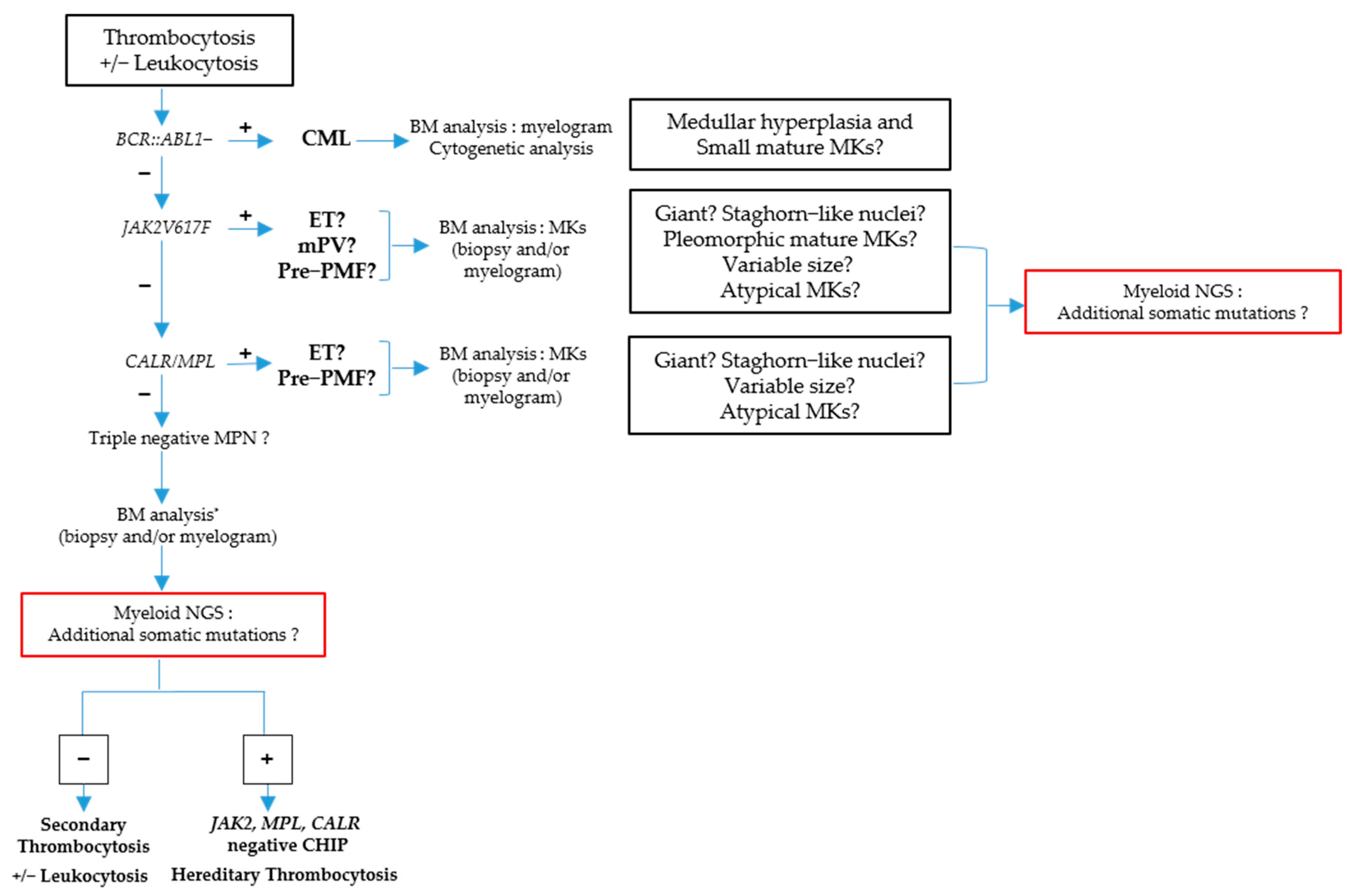
2. BCR::ABL1-Negative MPN
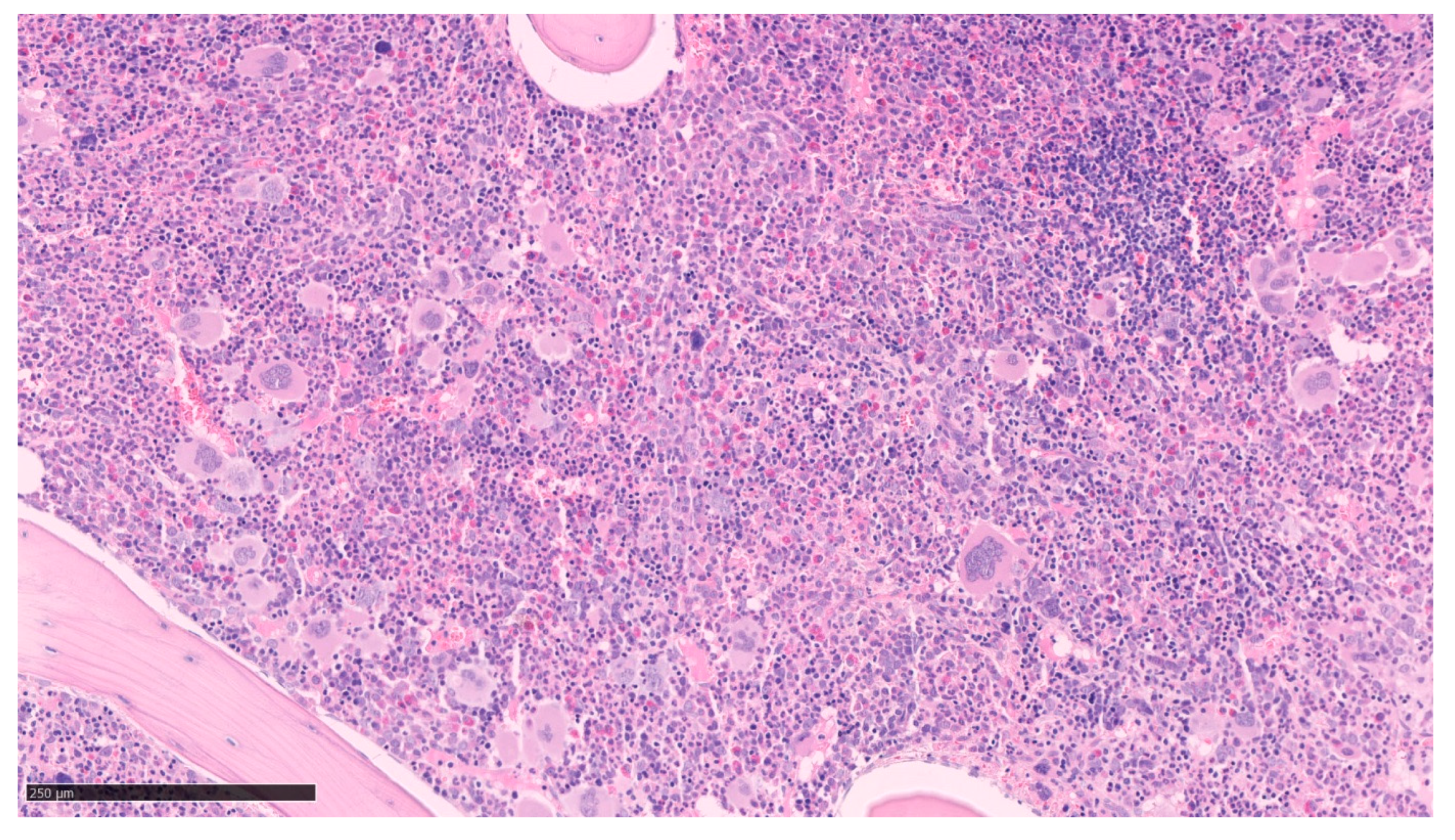
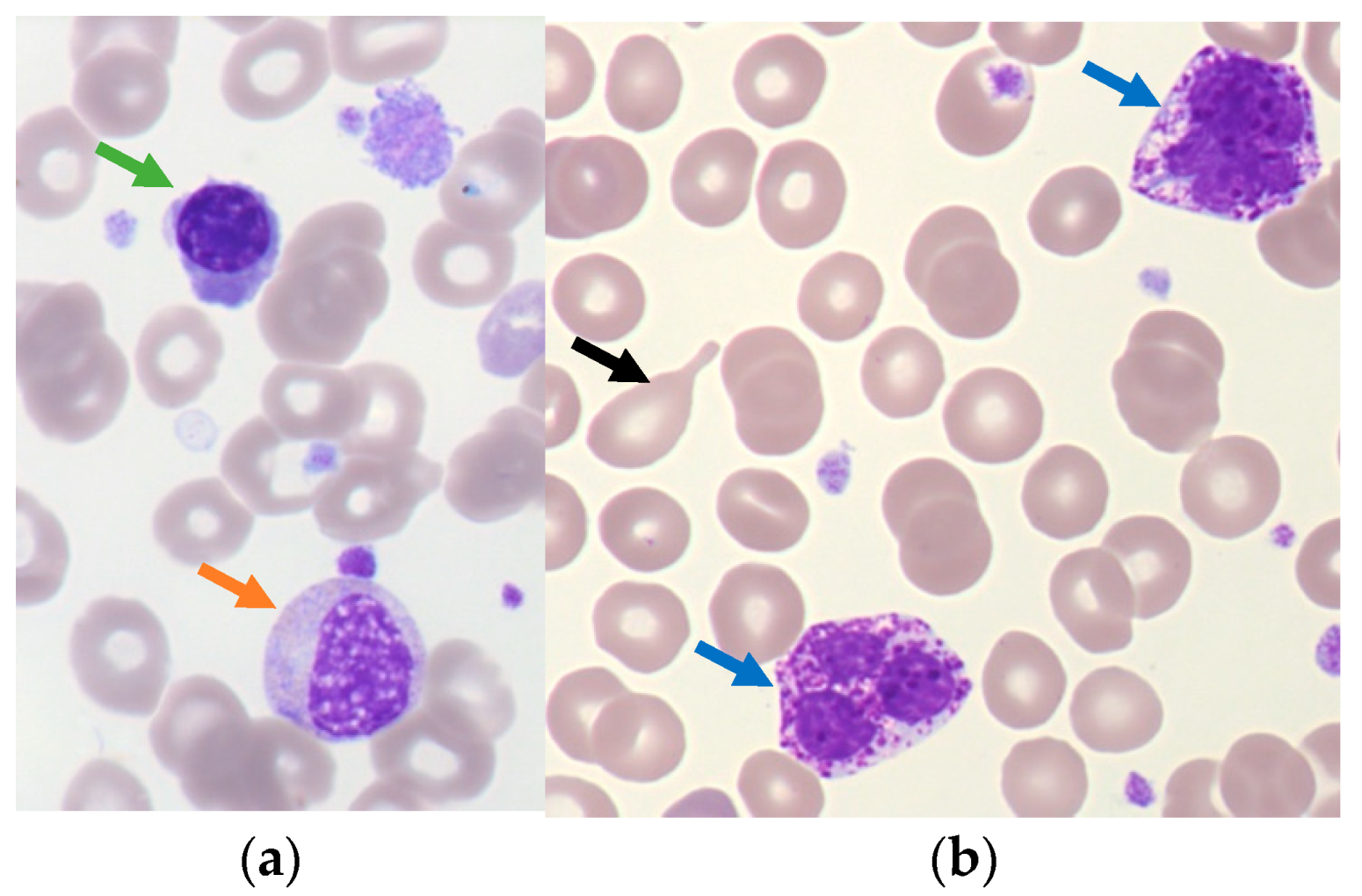
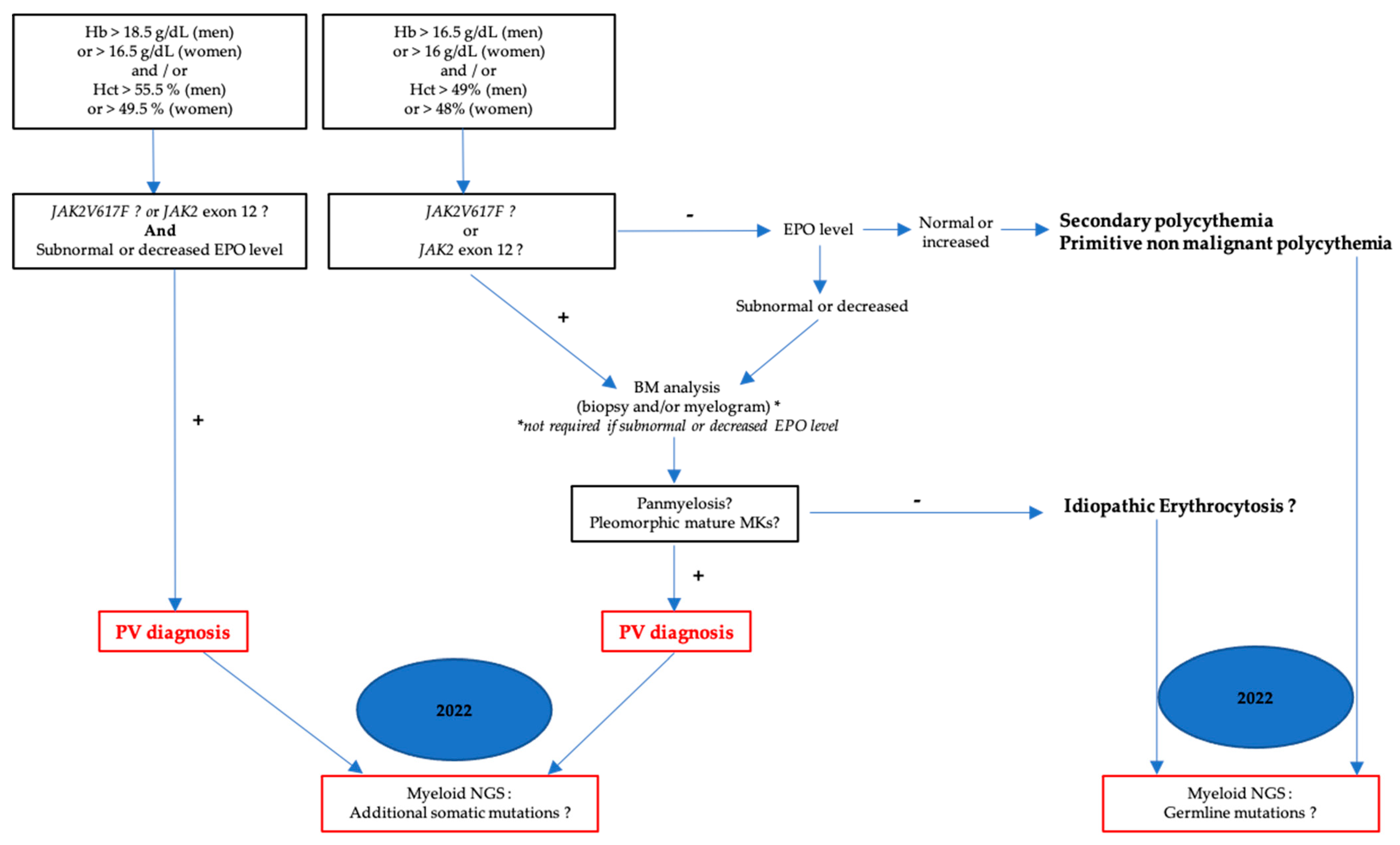
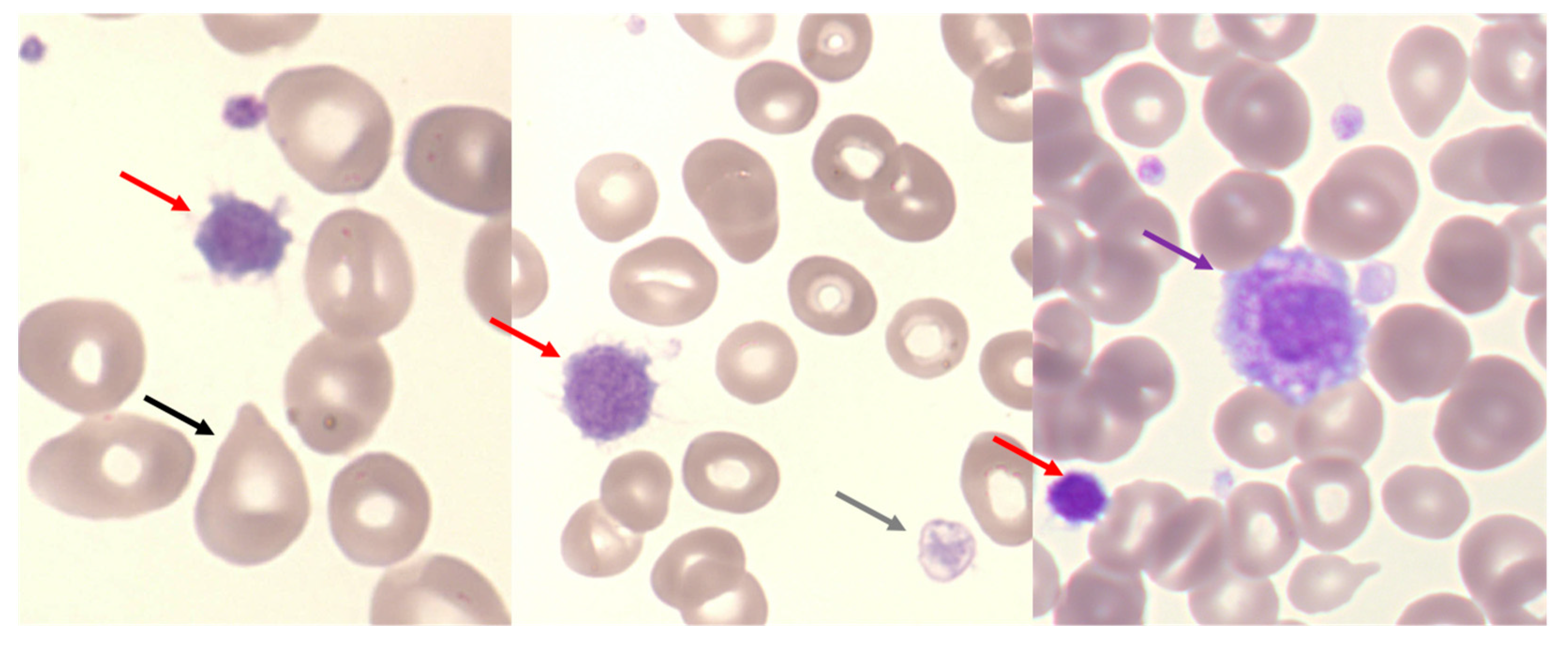
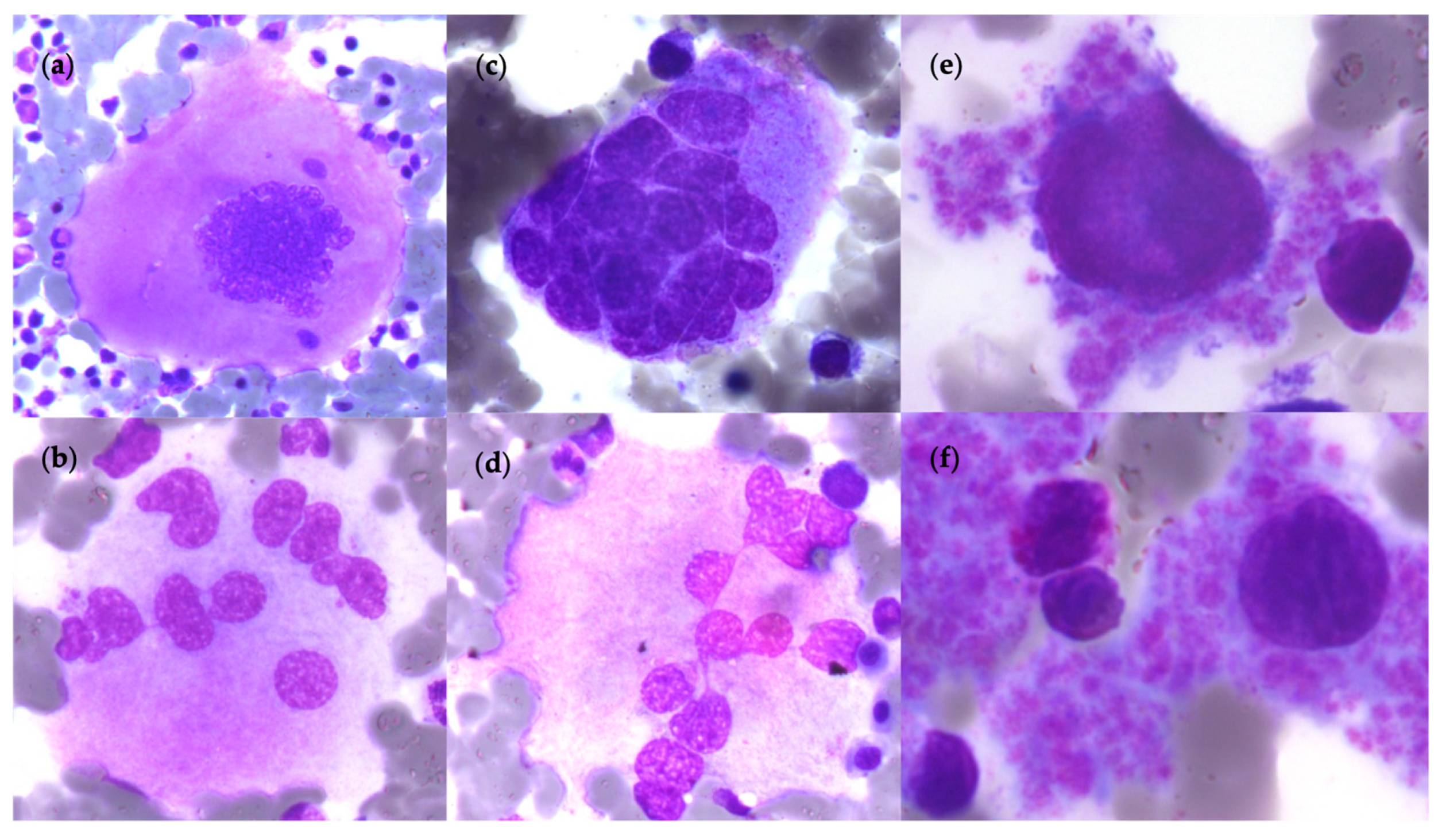
2.1. Polycythemia Vera (PV)
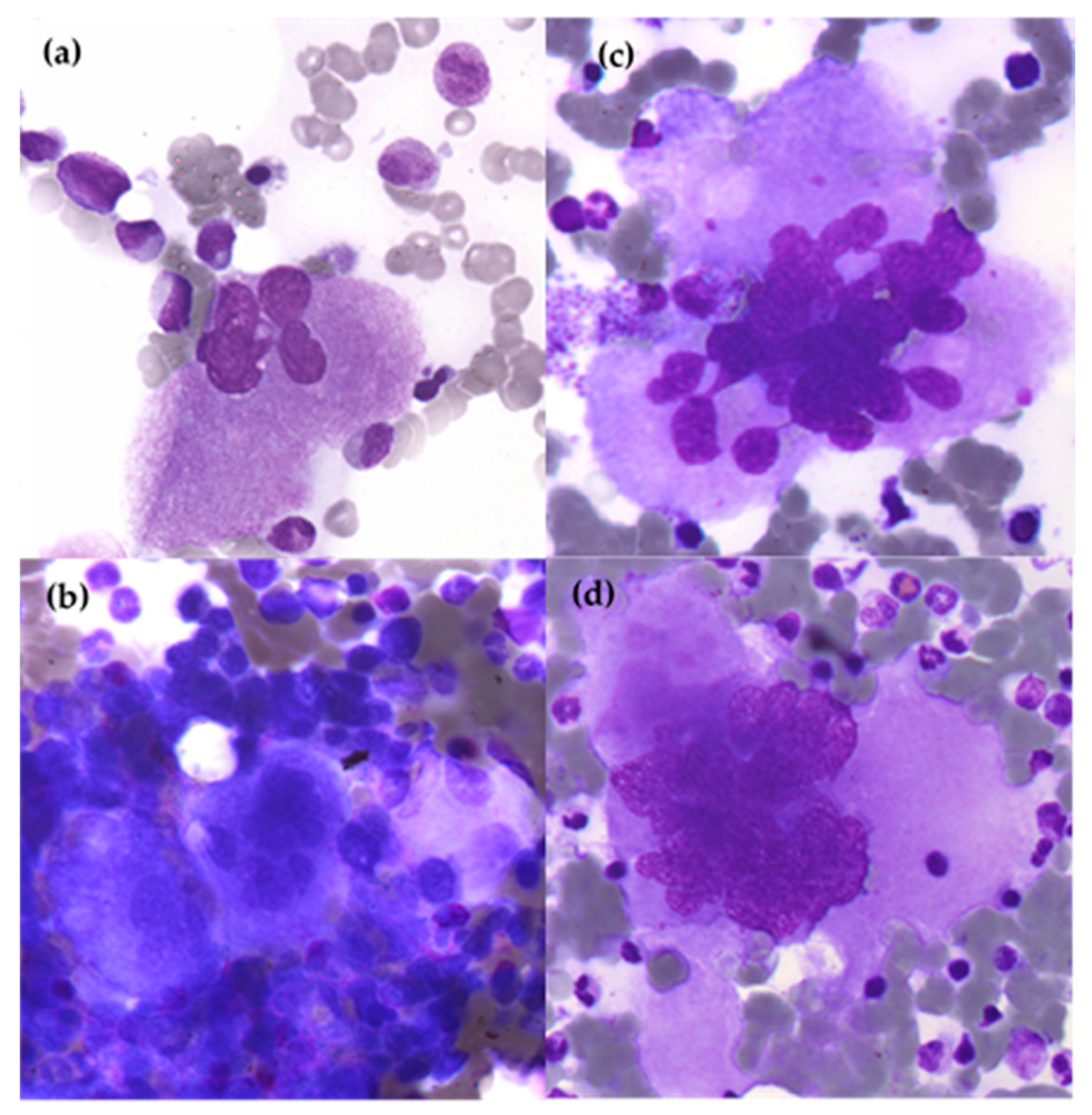
2.2. Essential Thrombocythemia (ET)
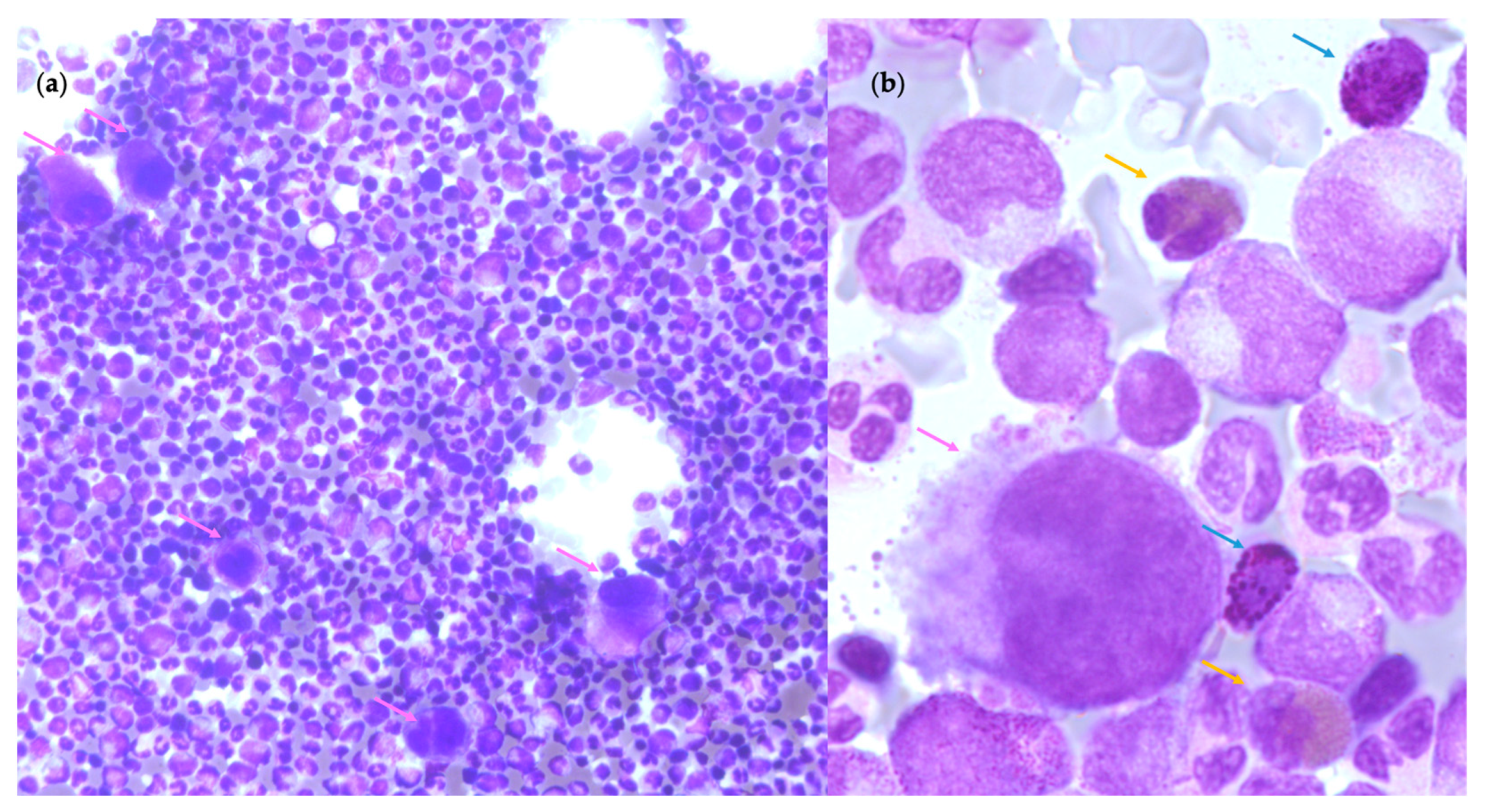
2.3. Primitive Myelofibrosis (PMF)
3. BCR::ABL1-Positive MPN: Chronic Myeloid Leukemia (CML)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Podoltsev, N.A.; Zeidan, A.M. Epidemiology of the classical myeloproliferative neoplasms: The four corners of an expansive and complex map. Blood Rev. 2020, 42, 100706. [Google Scholar] [CrossRef]
- Goldman, J.M. Chronic myeloid leukemia: A historical perspective. Semin. Hematol. 2010, 47, 302–311. [Google Scholar] [CrossRef]
- Cortes, J.; Pavlovsky, C.; Saußele, S. Chronic myeloid leukaemia. Lancet 2021, 398, 1914–1926. [Google Scholar] [CrossRef]
- Sawyers, C.L.; McLaughlin, J.; Witte, O.N. Genetic Requirement for Ras in the Transformation of Fibroblasts and Hematopoietic Cells by the Bcr-Abl Oncogene. J. Exp. Med. 1995, 181, 307–313. [Google Scholar] [CrossRef] [Green Version]
- Steelman, L.S.; Abrams, S.L.; Whelan, J.; Bertrand, F.E.; Ludwig, D.E.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008, 22, 686–707. [Google Scholar] [CrossRef] [Green Version]
- Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells 2021, 10, 117. [Google Scholar] [CrossRef]
- Tefferi, A.; Vardiman, J.W. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia 2008, 22, 14–22. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Lakey, M.A.; Pardanani, A.; Hoyer, J.D.; Nguyen, P.L.; Lasho, T.L.; Tefferi, A.; Hanson, C.A. Bone marrow morphologic features in polycythemia vera with JAK2 exon 12 mutations. Am. J. Clin. Pathol. 2010, 133, 942–948. [Google Scholar] [CrossRef] [Green Version]
- Coltro, G.; Loscocco, G.G.; Vannucchi, A.M. Classical Philadelphia-negative myeloproliferative neoplasms (MPNs): A continuum of different disease entities. Int. Rev. Cell Mol. Biol. 2021, 365, 1–69. [Google Scholar] [CrossRef]
- Yahmi, N.; Arkoun, B.; Raslova, H.; Vainchenker, W. Megakaryocyte differentiation: Cellular aspects and cytokine regulation. Hematologie 2022, 28, 192–219. [Google Scholar] [CrossRef]
- Constantinescu, S.N.; Vainchenker, W.; Levy, G.; Papadopoulos, N. Functional Consequences of Mutations in Myeloproliferative Neoplasms. HemaSphere 2021, 5, e578. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Spivak, J.L.; Silver, R.T. The revised World Health Organization diagnostic criteria for polycythemia vera, essential thrombocytosis, and primary myelofibrosis: An alternative proposal. Blood 2008, 112, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Skoda, R.; Vardiman, J.W. Myeloproliferative neoplasms: Contemporary diagnosis using histology and genetics. Nat Rev. Clin. Oncol. 2009, 6, 627–637. [Google Scholar] [CrossRef]
- Cattaneo, D.; Croci, G.A.; Bucelli, C.; Tabano, S.; Cannone, M.G.; Gaudioso, G.; Barbanti, M.C.; Barbullushi, K.; Bianchi, P.; Fermo, E.; et al. Triple-Negative Essential Thrombocythemia: Clinical-Pathological and Molecular Features. A Single-Center Cohort Study. Front. Oncol. 2021, 11, 637116. [Google Scholar] [CrossRef]
- Garmezy, B.; Schaefer, J.K.; Mercer, J.; Talpaz, M. A provider’s guide to primary myelofibrosis: Pathophysiology, diagnosis, and management. Blood Rev. 2021, 45, 100691. [Google Scholar] [CrossRef]
- Gleitz, H.F.E.; Benabid, A.; Schneider, R.K. Still a burning question: The interplay between inflammation and fibrosis in myeloproliferative neoplasms. Curr. Opin. Hematol. 2021, 28, 364–371. [Google Scholar] [CrossRef]
- Baumeister, J.; Chatain, N.; Sofias, A.M.; Lammers, T.; Koschmieder, S. Progression of Myeloproliferative Neoplasms (MPN): Diagnostic and Therapeutic Perspectives. Cells 2021, 10, 3551. [Google Scholar] [CrossRef]
- Siegel, F.P.; Petrides, P.E. Congenital and Acquired Polycythemias. Dtsch. Ärztebl. Int. 2008, 105, 62–68. [Google Scholar] [CrossRef]
- Gianelli, U.; Iurlo, A.; Vener, C.; Moro, A.; Fermo, E.; Bianchi, P.; Graziani, D.; Radaelli, F.; Coggi, G.; Bosari, S.; et al. The Significance of Bone Marrow Biopsy and JAK2V617F Mutation in the Differential Diagnosis Between the “Early” Prepolycythemic Phase of Polycythemia Vera and Essential Thrombocythemia. Am. J. Clin. Pathol. 2008, 130, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Maynadié, M. Révision de la classification des syndromes myéloprolifératifs selon l’OMS en 2016. Rev. Francoph. Lab. 2017, 492, 25–28. [Google Scholar] [CrossRef]
- Zini, G.; Viscovo, M. Cytomorphology of normal, reactive, dysmorphic, and dysplastic megakaryocytes in bone marrow aspirates. Int. J. Lab. Hematol. 2021, 43, 23–28. [Google Scholar] [CrossRef]
- Maslah, N.; Soret, J.; Dosquet, C.; Vercellino, L.; Belkhodja, C.; Schlageter, M.-H.; Cassinat, B.; Kiladjian, J.-J.; Chomienne, C.; Giraudier, S. Masked polycythemia vera: Analysis of a single center cohort of 2480 red cell masses. Haematologica 2020, 105, e95–e97. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Polycythemia vera: Historical oversights, diagnostic details, and therapeutic views. Leukemia 2021, 35, 3339–3351. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Randi, M.L.; Bertozzi, I.; Marino, F.; Vannucchi, A.M.; Pieri, L.; et al. Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood 2012, 119, 2239–2241. [Google Scholar] [CrossRef]
- Nann, D.; Fend, F. Synoptic Diagnostics of Myeloproliferative Neoplasms: Morphology and Molecular Genetics. Cancers 2021, 13, 3528. [Google Scholar] [CrossRef]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef] [Green Version]
- Ronner, L.; Podoltsev, N.; Gotlib, J.; Heaney, M.L.; Kuykendall, A.T.; O’Connell, C.; Shammo, J.; Fleischman, A.G.; Scherber, R.M.; Mesa, R.; et al. Persistent leukocytosis in polycythemia vera is associated with disease evolution but not thrombosis. Blood 2020, 135, 1696–1703. [Google Scholar] [CrossRef]
- Boiocchi, L.; Gianelli, U.; Iurlo, A.; Fend, F.; Bonzheim, I.; Cattaneo, D.; Knowles, D.M.; Orazi, A. Neutrophilic leukocytosis in advanced stage polycythemia vera: Hematopathologic features and prognostic implications. Mod. Pathol. 2015, 28, 1448–1457. [Google Scholar] [CrossRef] [Green Version]
- Szpurka, H.; Tiu, R.; Murugesan, G.; Aboudola, S.; Hsi, E.D.; Theil, K.S.; Sekeres, M.A.; Maciejewski, J.P. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood 2006, 108, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Palomo, L.; Meggendorfer, M.; Hutter, S.; Twardziok, S.; Ademà, V.; Fuhrmann, I.; Fuster-Tormo, F.; Xicoy, B.; Zamora, L.; Acha, P.; et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood 2020, 136, 1851–1862. [Google Scholar] [CrossRef]
- Hu, Z.; Ramos, C.E.B.; Medeiros, L.J.; Zhao, C.; Yin, C.C.; Li, S.; Hu, S.; Wang, W.; Thakral, B.; Xu, J.; et al. Utility of JAK2 V617F allelic burden in distinguishing chronic myelomonocytic Leukemia from Primary myelofibrosis with monocytosis. Hum. Pathol. 2019, 85, 290–298. [Google Scholar] [CrossRef]
- Hassan, A.; Dogara, L.G.; Babadoko, A.A.; Awwalu, S.; Mamman, A.I. Coexistence of JAK2 and BCR-ABL mutation in patient with myeloproliferative neoplasm. Niger. Med. J. 2015, 56, 74–76. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, N.; Qayum, S.; Jameel, A.; Ali, A.; Siraj, S.; Ali, J.; Yousafzai, Y.M. Clinical and laboratory relevance of JAK2 V617F and BCR-ABL coexistence in Philadelphia positive CML patients. Pak. J. Pharm. Sci. 2021, 34 (Suppl. 6), 2289–2295. [Google Scholar]
- Ali, E.A.H.; Al-Akiki, S.; Yassin, M.A. A Case Report of BCR-ABL-JAK2-Positive Chronic Myeloid Leukemia with Complete Hematological and Major Molecular Response to Dasatinib. Case Rep. Oncol. 2021, 14, 690–694. [Google Scholar] [CrossRef]
- Zhou, A.; Knoche, E.M.; Engle, E.K.; Fisher, D.A.C.; Oh, S.T. Concomitant JAK2 V617F-positive polycythemia vera and BCR-ABL-positive chronic myelogenous leukemia treated with ruxolitinib and dasatinib. Blood Cancer J. 2015, 5, e351. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Campbell, P.J.; Baxter, E.J.; Todd, T.; Stephens, P.; Edkins, S.; Wooster, R.; Stratton, M.R.; Futreal, P.A.; Green, A.R. The V617F JAK2 mutation is uncommon in cancers and in myeloid malignancies other than the classic myeloproliferative disorders. Blood 2005, 106, 2920–2921. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Scott, L.M.; Scott, M.A.; Campbell, P.J.; Green, A.R. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood 2006, 108, 2435–2437. [Google Scholar] [CrossRef]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 1599–1613. [Google Scholar] [CrossRef]
- Kilpivaara, O.; Levine, R.L. JAK2 and MPL mutations in myeloproliferative neoplasms: Discovery and science. Leukemia 2008, 22, 1813–1817. [Google Scholar] [CrossRef] [Green Version]
- Broséus, J.; Park, J.-H.; Carillo, S.; Hermouet, S.; Girodon, F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood 2014, 124, 3964–3966. [Google Scholar] [CrossRef] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Morsia, E.; Torre, E.; Poloni, A.; Olivieri, A.; Rupoli, S. Molecular Pathogenesis of Myeloproliferative Neoplasms: From Molecular Landscape to Therapeutic Implications. Int. J. Mol. Sci. 2022, 23, 4573. [Google Scholar] [CrossRef]
- Saeidi, K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit. Rev. Oncol./Hematol. 2016, 98, 375–389. [Google Scholar] [CrossRef]
- Murati, A. Nouvelles anomalies moléculaires pronostiques. Corresp. Onco-hématologie 2014, 9, 23–27. [Google Scholar]
- Loscocco, G.G.; Coltro, G.; Guglielmelli, P.; Vannucchi, A.M. Integration of Molecular Information in Risk Assessment of Patients with Myeloproliferative Neoplasms. Cells 2021, 10, 1962. [Google Scholar] [CrossRef]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [Green Version]
- Greenfield, G.; McMullin, M.F.; Mills, K. Molecular pathogenesis of the myeloproliferative neoplasms. J. Hematol. Oncol. 2021, 14, 103. [Google Scholar] [CrossRef]
- Rose, S.R.; Petersen, N.J.; Gardner, T.J.; Hamill, R.J.; Trautner, B.W. Etiology of Thrombocytosis in a General Medicine Population: Analysis of 801 Cases With Emphasis on Infectious Causes. J. Clin. Med. Res. 2012, 4, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H. Histological evaluation of myeloproliferative neoplasms. J. Clin. Exp. Hematop. 2018, 58, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Thiele, J.; Kvasnicka, H.M.; Müllauer, L.; Buxhofer-Ausch, V.; Gisslinger, B.; Gisslinger, H. Essential thrombocythemia versus early primary myelofibrosis: A multicenter study to validate the WHO classification. Blood 2011, 117, 5710–5718. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2, ISBN 978-92-832-4494-3.
- Gianelli, U.; Vener, C.; Raviele, P.R.; Moro, A.; Savi, F.; Annaloro, C.; Somalvico, F.; Radaelli, F.; Franco, V.; Deliliers, G.L. Essential thrombocythemia or chronic idiopathic myelofibrosis? A single-center study based on hematopoieticbone marrow histology. Leuk. Lymphoma 2006, 47, 1774–1781. [Google Scholar] [CrossRef]
- Florena, A.M.; Tripodo, C.; Iannitto, E.; Porcasi, R.; Ingrao, S.; Franco, V. Value of bone marrow biopsy in the diagnosis of essential thrombocythemia. Haematologica 2004, 89, 911–919. [Google Scholar]
- Thiele, J.; Kvasnicka, H.M.; Schmitt-Graeff, A.; Zankovich, R.; Diehl, V. Follow-up examinations including sequential bone marrow biopsies in essential thrombocythemia (ET): A retrospective clinicopathological study of 120 patients. Am. J. Hematol. 2002, 70, 283–291. [Google Scholar] [CrossRef]
- Ryou, H.; Sirinukunwattana, K.; Aberdeen, A.; Grindstaff, G.; Stolz, B.J.; Byrne, H.; Harrington, H.A.; Sousos, N.; Godfrey, A.L.; Harrison, C.N.; et al. Continuous Indexing of Fibrosis (CIF): Improving the assessment and classification of MPN patients. Leukemia 2023, 37, 348–358. [Google Scholar] [CrossRef]
- Gerds, A.T.; Gotlib, J.; Ali, H.; Bose, P.; Dunbar, A.; Elshoury, A.; George, T.I.; Gundabolu, K.; Hexner, E.; Hobbs, G.S.; et al. Myeloproliferative Neoplasms, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 1033–1062. [Google Scholar] [CrossRef]
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [Green Version]
- Haider, M.; Gangat, N.; Lasho, T.; Abou Hussein, A.K.; Elala, Y.C.; Hanson, C.; Tefferi, A. Validation of the revised international prognostic score of thrombosis for essential thrombocythemia (IPSET-thrombosis) in 585 Mayo clinic patients. Am. J. Hematol. 2016, 91, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Beer, P.A.; Campbell, P.J.; Scott, L.M.; Bench, A.J.; Erber, W.N.; Bareford, D.; Wilkins, B.S.; Reilly, J.T.; Hasselbalch, H.C.; Bowman, R.; et al. MPL mutations in myeloproliferative disorders: Analysis of the PT-1 cohort. Blood 2008, 112, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Guerini, V.; Barosi, G.; Ruggeri, M.; Specchia, G.; Lo-Coco, F.; Delaini, F.; et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood 2008, 112, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Szuber, N.; Hanson, C.A.; Lasho, T.L.; Finke, C.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; Tefferi, A. MPL-mutated essential thrombocythemia: A morphologic reappraisal. Blood Cancer J. 2018, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Finazzi, G.; Falanga, A. Myeloproliferative neoplasms and thrombosis. Blood 2013, 122, 2176–2184. [Google Scholar] [CrossRef]
- Cottin, L.; Riou, J.; Orvain, C.; Ianotto, J.C.; Boyer, F.; Renard, M.; Truchan-Graczyk, M.; Murati, A.; Jouanneau-Courville, R.; Allangba, O.; et al. Sequential mutational evaluation of CALR-mutated myeloproliferative neoplasms with thrombocytosis reveals an association between CALR allele burden evolution and disease progression. Br. J. Haematol. 2020, 188, 935–944. [Google Scholar] [CrossRef]
- Marty, C.; Pecquet, C.; Nivarthi, H.; El-Khoury, M.; Chachoua, I.; Tulliez, M.; Villeval, J.-L.; Raslova, H.; Kralovics, R.; Constantinescu, S.N.; et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 2016, 127, 1317–1324. [Google Scholar] [CrossRef] [Green Version]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Courtier, F.; Carbuccia, N.; Garnier, S.; Guille, A.; Adélaïde, J.; Cervera, N.; Gelsi-Boyer, V.; Mozziconacci, M.-J.; Rey, J.; Vey, N.; et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica 2017, 102, e11–e14. [Google Scholar] [CrossRef]
- Abbou, N.; Piazzola, P.; Gabert, J.; Ernest, V.; Arcani, R.; Couderc, A.-L.; Tichadou, A.; Roche, P.; Farnault, L.; Colle, J.; et al. Impact of Molecular Biology in Diagnosis, Prognosis, and Therapeutic Management of BCR::ABL1-Negative Myeloproliferative Neoplasm. Cells 2022, 12, 105. [Google Scholar] [CrossRef]
- Luque Paz, D.; Jouanneau-Courville, R.; Riou, J.; Ianotto, J.-C.; Boyer, F.; Chauveau, A.; Renard, M.; Chomel, J.-C.; Cayssials, E.; Gallego-Hernanz, M.-P.; et al. Leukemic evolution of polycythemia vera and essential thrombocythemia: Genomic profiles predict time to transformation. Blood Adv. 2020, 4, 4887–4897. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Gobbi, G.; Merighi, S.; Gessi, S.; Vitale, M.; Carubbi, C. Cytokine Profiling in Myeloproliferative Neoplasms: Overview on Phenotype Correlation, Outcome Prediction, and Role of Genetic Variants. Cells 2020, 9, 2136. [Google Scholar] [CrossRef]
- Sirinukunwattana, K.; Aberdeen, A.; Theissen, H.; Sousos, N.; Psaila, B.; Mead, A.J.; Turner, G.D.H.; Rees, G.; Rittscher, J.; Royston, D. Artificial intelligence–based morphological fingerprinting of megakaryocytes: A new tool for assessing disease in MPN patients. Blood Adv. 2020, 4, 3284–3294. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Ruggeri, M.; Rodeghiero, F.; D’Amore, E.S.; Randi, M.L.; Bertozzi, I.; et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: A international study. J. Clin. 2011, 29, 3179–3184. [Google Scholar] [CrossRef]
- Costello, R.; Venton, G.; Baccini, V.; Lepidi, H. Myélofibrose primitive. EMC Hématologie 2018, 13, 12. [Google Scholar]
- Kvasnicka, H.M.; Beham-Schmid, C.; Bob, R.; Dirnhofer, S.; Hussein, K.; Kreipe, H.; Kremer, M.; Schmitt-Graeff, A.; Schwarz, S.; Thiele, J.; et al. Problems and pitfalls in grading of bone marrow fibrosis, collagen deposition and osteosclerosis–A consensus-based study. Histopathology 2016, 68, 905–915. [Google Scholar] [CrossRef]
- Barosi, G.; Viarengo, G.; Pecci, A.; Rosti, V.; Piaggio, G.; Marchetti, M.; Frassoni, F. Diagnostic and clinical relevance of the number of circulating CD34+ cells in myelofibrosis with myeloid metaplasia. Blood 2001, 98, 3249–3255. [Google Scholar] [CrossRef]
- Boiocchi, L.; Espinal-Witter, R.; Geyer, J.T.; Steinhilber, J.; Bonzheim, I.; Knowles, D.M.; Fend, F.; Orazi, A. Development of monocytosis in patients with primary myelofibrosis indicates an accelerated phase of the disease. Mod. Pathol. 2013, 26, 204–212. [Google Scholar] [CrossRef] [Green Version]
- Federmann, B.; Abele, M.; Rosero Cuesta, D.S.; Vogel, W.; Boiocchi, L.; Kanz, L.; Quintanilla-Martinez, L.; Orazi, A.; Bonzheim, I.; Fend, F. The detection of SRSF2 mutations in routinely processed bone marrow biopsies is useful in the diagnosis of chronic myelomonocytic leukemia. Hum. Pathol. 2014, 45, 2471–2479. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M.; Facchetti, F.; Franco, V.; van der Walt, J.; Orazi, A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005, 90, 1128–1132. [Google Scholar]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Pancrazzi, A.; Bergamaschi, G.; Rosti, V.; Villani, L.; Antonioli, E.; Bosi, A.; Barosi, G.; Vannucchi, A.M.; for the GIMEMA-Italian Registry of Myelofibrosis and the MPD Research Consortium, Chicago, IL, USA. Anaemia characterises patients with myelofibrosis harbouring MplW515L/K mutation. Br. J. Haematol. 2007, 137, 244–247. [Google Scholar] [CrossRef]
- Cabagnols, X.; Defour, J.P.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.F.; et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2015, 29, 249–252. [Google Scholar] [CrossRef]
- Zhou, A.; Oh, S.T. Prognostication in MF: From CBC to cytogenetics to molecular markers. Best Pract. Res. Clin. Haematol. 2014, 27, 155–164. [Google Scholar] [CrossRef]
- Luque Paz, D.; Riou, J.; Verger, E.; Cassinat, B.; Chauveau, A.; Ianotto, J.-C.; Dupriez, B.; Boyer, F.; Renard, M.; Mansier, O.; et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: A FIM study. Blood Adv. 2021, 5, 1442–1451. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.P.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef]
- Hehlmann, R.; Hochhaus, A.; Baccarani, M. Chronic myeloid leukaemia. Lancet 2007, 370, 342–350. [Google Scholar] [CrossRef]
- Penot, A.; Preux, P.-M.; Le Guyader, S.; Collignon, A.; Herry, A.; Dufour, V.; Monnereau, A.; Woronoff, A.-S.; Troussard, X.; Pons, E.; et al. Incidence of chronic myeloid leukemia and patient survival: Results of five French population-based cancer registries 1980–2009. Leuk. Lymphoma 2015, 56, 1771–1777. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef] [Green Version]
- Vardiman, J.W.; Harris, N.L.; Brunning, R.D. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 2002, 100, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Turakhia, S.K.; Murugesan, G.; Cotta, C.V.; Theil, K.S. Thrombocytosis and STAT5 activation in chronic myelogenous leukaemia are not associated with JAK2 V617F or calreticulin mutations. J. Clin. Pathol. 2016, 69, 713–719. [Google Scholar] [CrossRef]
- Ng, T.F.; Wright, M.; De Kraa, R. Approximately 1% of chronic myeloid leukaemia cases present with isolated thrombocytosis and express common major breakpoints: A finding from a laboratory audit. Pathology 2019, 51, 98–99. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Jeong, S.Y.; Kim, C.; Lee, M.-Y.; Kim, J.; Kim, K.-H.; Lee, N.; Won, J.-H. Philadelphia+ Chronic Myeloid Leukemia with CALR Mutation: A Case Report and Literature Review. Cancer Res. Treat. 2020, 52, 987–991. [Google Scholar] [CrossRef]
- Liu, C.; Hu, R.; Du, Z.; Abecasis, M.; Wang, C. Atypical myeloproliferative neoplasm with concurrent BCR-ABL1 fusion and CALR mutation: A case report and literature review. Medicine 2020, 99, e18811. [Google Scholar] [CrossRef]
- Soderquist, C.R.; Ewalt, M.D.; Czuchlewski, D.R.; Geyer, J.T.; Rogers, H.J.; Hsi, E.D.; Wang, S.A.; Bueso-Ramos, C.E.; Orazi, A.; Arber, D.A.; et al. Myeloproliferative neoplasms with concurrent BCR–ABL1 translocation and JAK2 V617F mutation: A multi-institutional study from the bone marrow pathology group. Mod. Pathol. 2018, 31, 690–704. [Google Scholar] [CrossRef] [Green Version]
- Hasserjian, R.P.; Kelley, T.W.; Weinberg, O.K.; Morgan, E.A.; Fend, F. Genetic Testing in the Diagnosis and Biology of Myeloid Neoplasms (Excluding Acute Leukemias). Am. J. Clin. Pathol. 2019, 152, 302–321. [Google Scholar] [CrossRef]
- Bonzheim, I.; Mankel, B.; Klapthor, P.; Schmidt, J.; Hinrichsen, T.; Wachter, O.; Fend, F.; Quintanilla-Martinez, L. CALR-mutated essential thrombocythemia evolving to chronic myeloid leukemia with coexistent CALR mutation and BCR-ABL translocation. Blood 2015, 125, 2309–2311. [Google Scholar] [CrossRef] [Green Version]
- da Costa, V.E.F.; de Oliveira, R.D.; Traina, F.; Chahud, F.; Palma, L.C.; de Figueiredo-Pontes, L.L. Co-occurrence of BCR–ABL1-positive chronic myeloid leukaemia and CALR-mutated essential thrombocythaemia. Br. J. Haematol. 2020, 188, e21–e23. [Google Scholar] [CrossRef]
- Rieu, J.-B.; Tavitian, S.; Vergez, F.; Largeaud, L. Do not forget megakaryocytes morphology when you deal with chronic myeloid leukaemia. Ann. Biol. Clin. 2020, 78, 691–692. [Google Scholar] [CrossRef]
- Melo, J.V. 2 BCR-ABL gene variants. Baillière’s Clin. Haematol. 1997, 10, 203–222. [Google Scholar] [CrossRef]
- Sora, F.; Iurlo, A.; Sica, S.; Latagliata, R.; Annunziata, M.; Galimberti, S.; Castagnetti, F.; Pregno, P.; Sgherza, N.; Celesti, F.; et al. Chronic myeloid leukaemia with extreme thrombocytosis at presentation: Incidence, clinical findings and outcome. Br. J. Haematol. 2018, 181, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Dass, J.; Jain, S.; Tyagi, S.; Sazawal, S. Chronic myeloid leukemia with p210 BCR–ABL and monocytosis. Leuk. Lymphoma 2011, 52, 1380–1381. [Google Scholar] [CrossRef]
- Wang, X.; Wang, F.; Wang, Z.; Li, Y.; Wang, D.; Wu, H.; Zhang, B. p210BCR-ABL–Chronic myeloid leukemia presents with monocytosis. Clin. Case Rep. 2020, 8, 840–842. [Google Scholar] [CrossRef] [Green Version]
- Verma, D.; Kantarjian, H.M.; Jones, D.; Luthra, R.; Borthakur, G.; Verstovsek, S.; Rios, M.B.; Cortes, J. Chronic myeloid leukemia (CML) with P190BCR-ABL: Analysis of characteristics, outcomes, and prognostic significance. Blood 2009, 114, 2232–2235. [Google Scholar] [CrossRef] [Green Version]
- Bilhou-Nabéra, C.; Bidet, A.; Eclache, V.; Lippert, E.; Mozziconacci, M.-J. Cytogenetics in the management of Philadelphia-negative myeloproliferative neoplasms: An update by the Groupe francophone de cytogénétique hématologique (GFCH). Ann. Biol. Clin. 2016, 74, 517–523. [Google Scholar] [CrossRef]
- Johansson, B.; Fioretos, T.; Mitelman, F. Cytogenetic and Molecular Genetic Evolution of Chronic Myeloid Leukemia. Acta Haematol. 2002, 107, 76–94. [Google Scholar] [CrossRef]
- Wang, W.; Cortes, J.E.; Tang, G.; Khoury, J.D.; Wang, S.; Bueso-Ramos, C.E.; DiGiuseppe, J.A.; Chen, Z.; Kantarjian, H.M.; Medeiros, L.J.; et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood 2016, 127, 2742–2750. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-E.; Choi, S.Y.; Bang, J.-H.; Kim, S.-H.; Jang, E.-J.; Byeun, J.-Y.; Park, J.E.; Jeon, H.-R.; Oh, Y.J.; Kim, M.; et al. The long-term clinical implications of clonal chromosomal abnormalities in newly diagnosed chronic phase chronic myeloid leukemia patients treated with imatinib mesylate. Cancer Genet. 2012, 205, 563–571. [Google Scholar] [CrossRef]
- Togasaki, E.; Takeda, J.; Yoshida, K.; Shiozawa, Y.; Takeuchi, M.; Oshima, M.; Saraya, A.; Iwama, A.; Yokote, K.; Sakaida, E.; et al. Frequent somatic mutations in epigenetic regulators in newly diagnosed chronic myeloid leukemia. Blood Cancer J. 2017, 7, e559. [Google Scholar] [CrossRef] [Green Version]
- Romzova, M.; Smitalova, D.; Hynst, J.; Tom, N.; Loja, T.; Herudkova, Z.; Jurcek, T.; Stejskal, L.; Zackova, D.; Mayer, J.; et al. Hierarchical distribution of somatic variants in newly diagnosed chronic myeloid leukaemia at diagnosis and early follow-up. Br. J. Haematol. 2021, 194, 604–612. [Google Scholar] [CrossRef]
- Branford, S.; Kim, D.D.H.; Apperley, J.F.; Eide, C.A.; Mustjoki, S.; Ong, S.T.; Nteliopoulos, G.; Ernst, T.; Chuah, C.; Gambacorti-Passerini, C.; et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia 2019, 33, 1835–1850. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO 2016 classification [18] | Major criteria: 1-Hb > 16.5 g/dL (men), >16.0 g/dL (women) or, Hct > 49% (men), >48% (women) or, increased red cell mass. 2-BM biopsy showing hypercellularity for age with trilineage growth (panmyelosis) including prominent erythroid, granulocytic and megakaryocytic proliferation with pleomorphic, mature megakaryocytes. 3-JAK2V617F or JAK2 exon 12 mutation. Minor criterion: Subnormal serum EPO level |
| PV | ET | PMF | |
|---|---|---|---|
| Driver mutations (Frequencies %) | |||
| Signaling MPN driver | JAK2V617F (95%) | JAK2V617F (50–70%) | JAK2V617F (40–50%) |
| [33,48,54,55] | JAK2 exon 12 (3%) | ||
| MPL (<1%) | MPL (3–15%) | MPL (3–15%) | |
| CALR (<1%) | CALR (25%) | CALR (25–30%) | |
| Additional mutations (Frequencies %) | |||
| DNA methylation | TET2 (9–16%) | TET2 (4–5%) | TET2 (7–17%) |
| [10,55] | DNMT3A (1–7%) | DNMT3A (5–10%) | DNMT3A (5–10%) |
| IDH 1/2 (2%) | IDH1/IDH2 (3–6%) | IDH1/IDH2 (3–6%) | |
| Histone modification | ASXL1 (7%) | ASXL1 (4%) | ASXL1 (18–35%) |
| [10,56] | EZH2 (0–3%) | EZH2 (0–3%) | EZH2 (6–13%) |
| mRNA Splicing | SF3B1 (3–5%) | SF3B1 (3–5%) | SF3B1 (5–7%) |
| [10,33] | SRSF2 (0–3%) | SRSF2 (0–3%) | SRSF2 (8–17%) |
| U2AF1 (1–2%) | U2AF1 (1–2%) | U2AF1 (5–15%) | |
| ZRSR2 (0–2%) | ZRSR2 (0–2%) | ZRSR2 (0–2%) | |
| Transcription factors | TP53 (1–3%) | TP53 (1–3%) | TP53 (4%) |
| [10,54] | IKZF1 (<3%) | IKZF1 (<3%) | IKZF1 (<3%) |
| RUNX1 (<3%) | RUNX1 (<3%) | RUNX1 (<3%) | |
| Other signaling | LNK/ SH2B3 (2–10%) | LNK/ SH2B3 (3–6%) | LNK/ SH2B3 (11%) |
| pathways | CBL (0–2%) | CBL (0–2%) | CBL (0–6%) |
| [10,33] | NRAS, KRAS (0–1%) | NRAS, KRAS (0–1%) | NRAS, KRAS (3–15%) |
| WHO 2016 classification [18] | Major criteria: 1-Platelet count ≥ 450 × 109/L. 2-BM biopsy showing proliferation mainly of the megakaryocytic lineage with increased number of enlarged, mature megakaryocytes with hyperlobulated nuclei. No significant increase or left shift in neutrophil granulopoiesis or erythropoiesis and very rarely minor increase in reticulin fibers (≤grade 1). 3-No WHO criteria for other myeloid neoplasms. 4-JAK2, CALR or MPL mutation. Minor criterion: Presence of clonal marker or absence of evidence for reactive thrombocytosis |
| Pre-PMF | Overt PMF | |
|---|---|---|
| WHO 2016 classification [18] | Major criteria: 1-Megakaryocytic proliferation and atypia, without reticulinic fibrosis > grade 1, accompanied by increased age-adjusted BM cellularity, granulocytic proliferation and often decreased erythropoiesis. 2-No WHO criteria for other myeloid neoplasms. 3-JAK2, CALR or MPL mutation or, in the absence of these mutations, presence of another clonal marker or absence of minor reactive BM reticulin fibrosis. | Major criteria: 1-Megakaryocyte proliferation and atypia accompanied by either reticulin and/or collagen fibrosis. 2-No WHO criteria for other myeloid neoplasms. 3-JAK2, CALR or MPL mutation or other clonal marker or, absence of reactive bone marrow fibrosis. |
| Minor criteria: 1-Anemia not attributed to a comorbid condition. 2-Palpable splenomegaly. 3-Leukocytosis ≥ 11 × 109/L. 4-Elevated LDH (confirmed in two consecutive determinations). 5-Leukoerythroblastosis. * | ||
| Pre-PMF | Overt PMF | |
|---|---|---|
| CBC and blood smear | Thrombocytosis | Leukoerythroblastosis, thrombopenia/anemia |
| Myelogram/ BM biopsy | Megakaryocytic proliferation with atypia and large spectrum of shapes (abnormal maturation, hyperchromatic, hypo or hyperlobulated, irregular nuclei (bulbous, “cloudlike”), tight clusters Reticulin fibrosis ≤ grade 1 (pre-PMF) Reticulin and/or collagen fibrosis grade 2 or 3 (overt PMF) [15] | |
| Accelerated Phase (AP) | Blast Phase (BP) | |
|---|---|---|
| WHO 2016 classification [18] | Any 1 or more of the following: Hematological/Cytogenetic criteria or response to TKI criteria:
Provisional response-to-tyrosine kinase inhibitor (TKI) criteria:
| Myeloid blast cells ≥ 20% of total in blood or BM. Extramedullary proliferation of blasts. |
| WHO 2022 classification [19] | No criteria AP is omitted | Myeloid blast cells ≥ 20% of the total in the blood or BM. Extramedullary proliferation of blasts. Presence of increased lymphoblasts in PB or BM. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Combaluzier, S.; Quessada, J.; Abbou, N.; Arcani, R.; Tichadou, A.; Gabert, J.; Costello, R.; Loosveld, M.; Venton, G.; Berda-Haddad, Y. Cytological Diagnosis of Classic Myeloproliferative Neoplasms at the Age of Molecular Biology. Cells 2023, 12, 946. https://doi.org/10.3390/cells12060946
Combaluzier S, Quessada J, Abbou N, Arcani R, Tichadou A, Gabert J, Costello R, Loosveld M, Venton G, Berda-Haddad Y. Cytological Diagnosis of Classic Myeloproliferative Neoplasms at the Age of Molecular Biology. Cells. 2023; 12(6):946. https://doi.org/10.3390/cells12060946
Chicago/Turabian StyleCombaluzier, Sophie, Julie Quessada, Norman Abbou, Robin Arcani, Antoine Tichadou, Jean Gabert, Régis Costello, Marie Loosveld, Geoffroy Venton, and Yaël Berda-Haddad. 2023. "Cytological Diagnosis of Classic Myeloproliferative Neoplasms at the Age of Molecular Biology" Cells 12, no. 6: 946. https://doi.org/10.3390/cells12060946