
Identification of Myelin Basic Protein Proximity Interactome Using TurboID Labeling Proteomics
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Transfection
2.2. Proximity Labeling in Mammalian Cells with TurboID and Preparation of Proteomic Samples
2.3. Anti-FLAG Immunoprecipitation
2.4. Immunofluorescence, Image Acquisition, and Analysis in Cell Culture
2.5. Mass Spectrometry Analysis
2.6. Data Analysis
3. Results
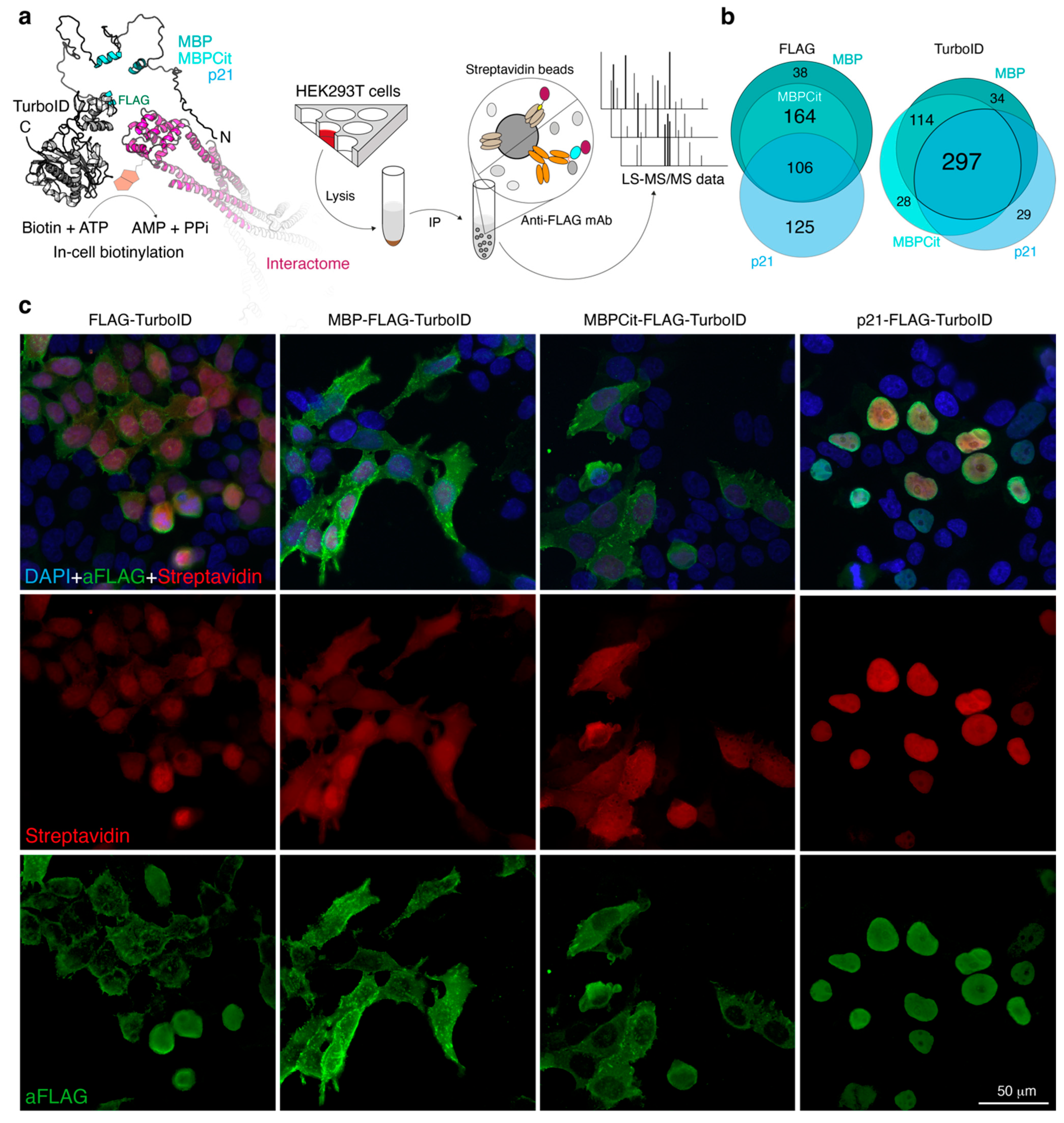
3.1. Study Design
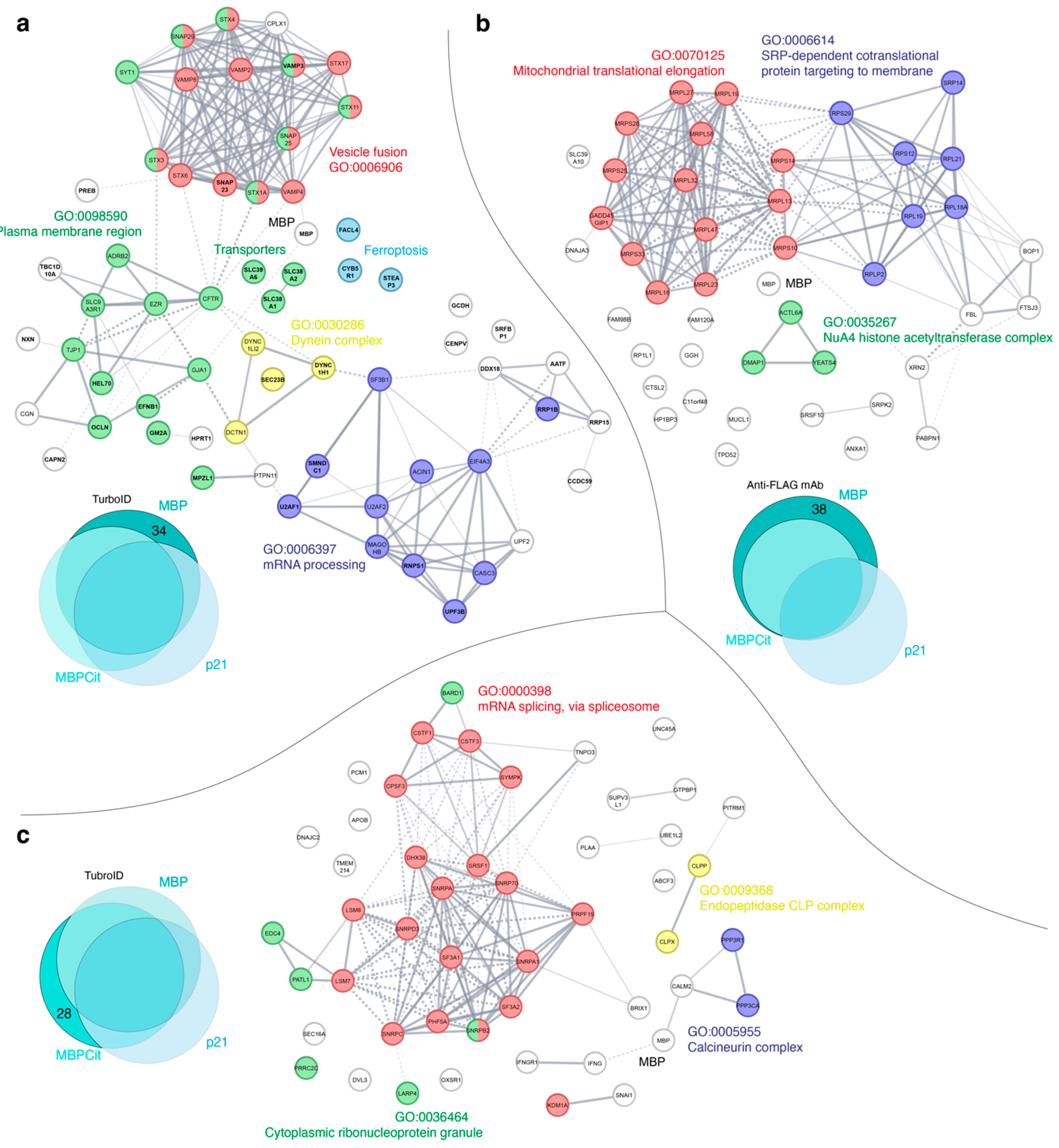
3.2. Protein-Protein Interaction Networks Functional Enrichment Analysis of MBP Interactome
4. Discussion
4.1. MBP Interactors Involved in the mRNA Processing and Maintenance
4.2. MBP Interactors—Members of Protein Synthesis Machinery
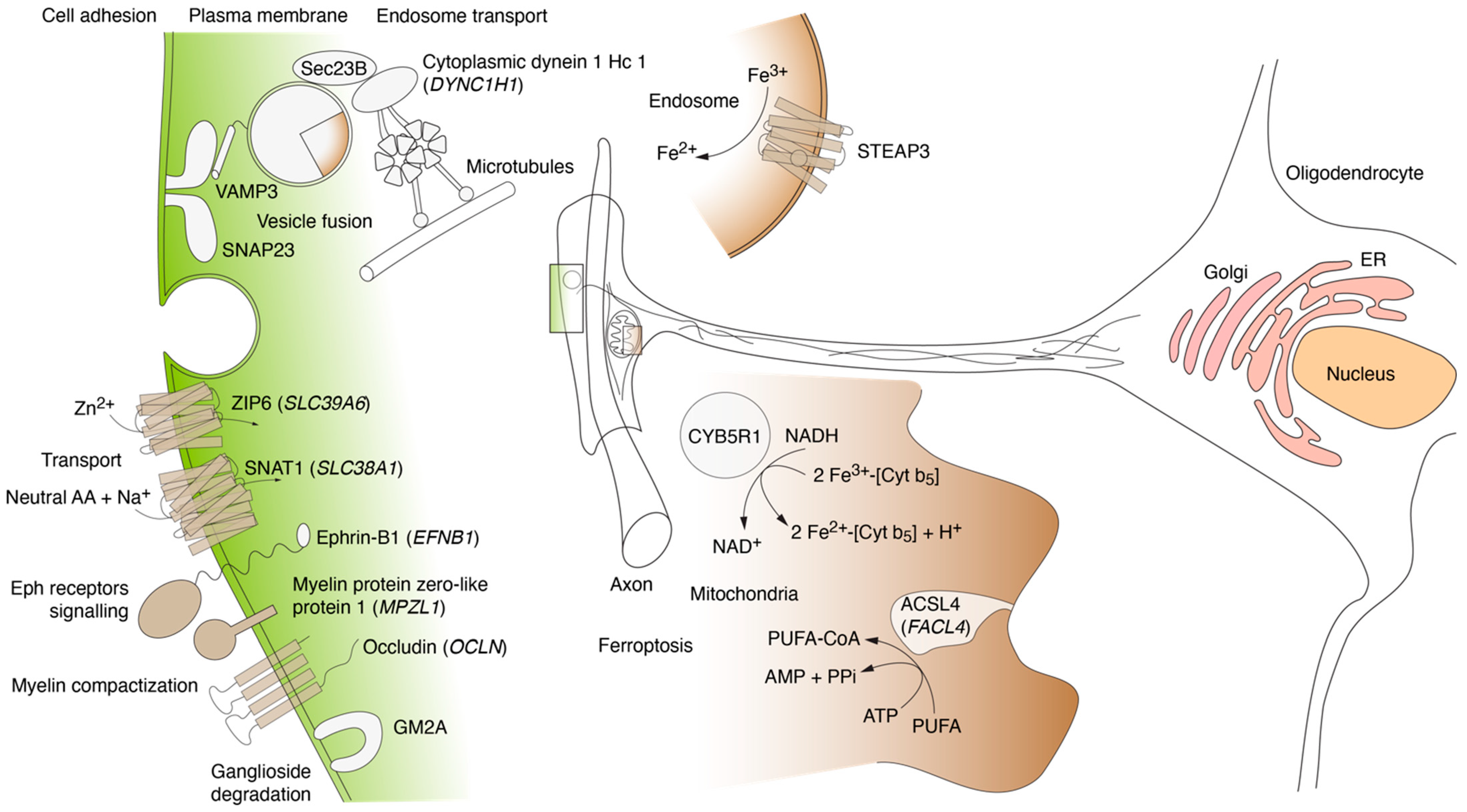
4.3. MBP Interactors Associated with Cellular Adhesion and Transmembrane Traffic
4.4. MBP Interactors Associated with Lipid Metabolism and Ferroptosis
4.5. MBP Interactors Involved in Vesicular Fusion and Trafficking to Plasma Membrane Region
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sorg, B.A.; Smith, M.M.; Campagnoni, A.T. Developmental Expression of the Myelin Proteolipid Protein and Basic Protein mRNAs in Normal and Dysmyelinating Mutant Mice. J. Neurochem. 1987, 49, 1146–1154. [Google Scholar] [CrossRef]
- Campagnoni, A.T.; Campagnoni, C.W. Myelin basic protein gene. In Myelin Biology and Disorders; Elsevier: Amsterdam, The Netherlands, 2004; pp. 387–400. [Google Scholar]
- Bagheri, H.; Friedman, H.; Siminovitch, K.A.; Peterson, A.C. Transcriptional regulators of the Golli/myelin basic protein locus integrate additive and stealth activities. PLoS Genet. 2020, 16, e1008752. [Google Scholar] [CrossRef] [PubMed]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Fujinami, R.S.; Oldstone, M.B.A. Amino Acid Homology Between the Encephalitogenic Site of Myelin Basic Protein and Virus: Mechanism for Autoimmunity. Science 1985, 230, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Einstein, E.R.; Robertson, D.M.; Dicaprio, J.M.; Moore, W. The isolation from bovine spinal cord of a homogeneous protein with encephalitogenic activity. J. Neurochem. 1962, 9, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Harauz, G.; Ladizhansky, V.; Boggs, J.M. Structural Polymorphism and Multifunctionality of Myelin Basic Protein. Biochemistry 2009, 48, 8094–8104. [Google Scholar] [CrossRef] [PubMed]
- Majava, V.; Wang, C.; Myllykoski, M.; Kangas, S.M.; Kang, S.U.; Hayashi, N.; Baumgärtel, P.; Heape, A.M.; Lubec, G.; Kursula, P. Structural analysis of the complex between calmodulin and full-length myelin basic protein, an intrinsically disordered molecule. Amino Acids 2010, 39, 59–71. [Google Scholar] [CrossRef]
- Muruganandam, G.; Bürck, J.; Ulrich, A.S.; Kursula, I.; Kursula, P. Lipid membrane association of myelin proteins and peptide segments studied by oriented and synchrotron radiation circular dichroism spectroscopy. J. Phys. Chem. B. 2013, 117, 14983–14993. [Google Scholar] [CrossRef] [Green Version]
- Raasakka, A.; Ruskamo, S.; Kowal, J.; Barker, R.; Baumann, A.; Martel, A.; Tuusa, J.; Myllykoski, M.; Bürck, J.; Ulrich, A.S.; et al. Membrane Association Landscape of Myelin Basic Protein Portrays Formation of the Myelin Major Dense Line. Sci. Rep. 2017, 7, 4974. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P.; Szász, C.; Buday, L. Structural disorder throws new light on moonlighting. Trends Biochem. Sci. 2005, 30, 484–489. [Google Scholar] [CrossRef]
- Libich, D.S.; Hill, C.M.D.; Bates, I.R.; Hallett, F.R.; Armstrong, S.; Siemiarczuk, A.; Harauz, G. Interaction of the 18.5-kD isoform of myelin basic protein with Ca 2+ -calmodulin: Effects of deimination assessed by intrinsic Trp fluorescence spectroscopy, dynamic light scattering, and circular dichroism. Protein Sci. 2003, 12, 1507–1521. [Google Scholar] [CrossRef] [Green Version]
- Boggs, J.M.; Rangaraj, G. Interaction of Lipid-Bound Myelin Basic Protein with Actin Filaments and Calmodulin. Biochemistry 2000, 39, 7799–7806. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolski, Z.; Osińska, H.; Mossakowska, M.; Baryłko, B. Ca2+-calmodulin-dependent polymerization of actin by myelin basic protein. Eur. J. Cell Biol. 1986, 42, 17–26. [Google Scholar] [PubMed]
- Hill, C.M.D.; Harauz, G. Charge effects modulate actin assembly by classic myelin basic protein isoforms. Biochem. Biophys. Res. Commun. 2005, 329, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Modesti, N.M.; Barra, H.S. The interaction of myelin basic protein with tubulin and the inhibition of tubulin carboxypeptidase activity. Biochem. Biophys. Res. Commun. 1986, 136, 482–489. [Google Scholar] [CrossRef]
- Hill, C.M.D.; Libich, D.S.; Harauz, G. Assembly of Tubulin by Classic Myelin Basic Protein Isoforms and Regulation by Post-Translational Modification. Biochemistry 2005, 44, 16672–16683. [Google Scholar] [CrossRef]
- Polverini, E.; Rangaraj, G.; Libich, D.S.; Boggs, J.M.; Harauz, G. Binding of the Proline-Rich Segment of Myelin Basic Protein to SH3 Domains: Spectroscopic, Microarray, and Modeling Studies of Ligand Conformation and Effects of Posttranslational Modifications. Biochemistry 2008, 47, 267–282. [Google Scholar] [CrossRef]
- Boggs, J.M.; Rangaraj, G.; Gao, W.; Heng, Y.-M. Effect of Phosphorylation of Myelin Basic Protein by MAPK on its Interactions with Actin and Actin Binding to a Lipid Membrane in Vitro. Biochemistry 2006, 45, 391–401. [Google Scholar] [CrossRef]
- Boggs, J.M.; Rangaraj, G.; Hill, C.M.D.; Bates, I.R.; Heng, Y.-M.; Harauz, G. Effect of Arginine Loss in Myelin Basic Protein, as Occurs in Its Deiminated Charge Isoform, on Mediation of Actin Polymerization and Actin Binding to a Lipid Membrane in Vitro. Biochemistry 2005, 44, 3524–3534. [Google Scholar] [CrossRef]
- Homchaudhuri, L.; Polverini, E.; Gao, W.; Harauz, G.; Boggs, J.M. Influence of membrane surface charge and post-translational modifications to myelin basic protein on its ability to tether the Fyn-SH3 domain to a membrane in vitro. Biochemistry 2009, 48, 2385–2393. [Google Scholar] [CrossRef]
- Boggs, J.M.; Rangaraj, G.; Heng, Y.-M.; Liu, Y.; Harauz, G. Myelin basic protein binds microtubules to a membrane surface and to actin filaments in vitro: Effect of phosphorylation and deimination. Biochim. Et. Biophys. Acta (BBA) Biomembr. 2011, 1808, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Boggs, J.M. Myelin basic protein: A multifunctional protein. Cell. Mol. Life Sci. 2006, 63, 1945–1961. [Google Scholar] [CrossRef]
- Kim, J.K.; Mastronardi, F.G.; Wood, D.D.; Lubman, D.M.; Zand, R.; Moscarello, M.A. Multiple sclerosis: An important role for post-translational modifications of myelin basic protein in pathogenesis. Mol. Cell. Proteom. 2003, 2, 453–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harauz, G.; Musse, A.A. A tale of two citrullines--structural and functional aspects of myelin basic protein deimination in health and disease. Neurochem. Res. 2007, 32, 137–158. [Google Scholar] [CrossRef]
- Smirnova, E.V.; Rakitina, T.V.; Ziganshin, R.H.; Arapidi, G.P.; Saratov, G.A.; Kudriaeva, A.A.; Belogurov, A.A. Comprehensive Atlas of the Myelin Basic Protein Interaction Landscape. Biomolecules 2021, 11, 1628. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.V.; Rakitina, T.V.; Saratov, G.A.; Kudriaeva, A.A.; Belogurov, A.A. Deconvolution of the MBP-Bri2 Interaction by a Yeast Two Hybrid System and Synergy of the AlphaFold2 and High Ambiguity Driven Protein-Protein Docking. Crystals 2022, 12, 197. [Google Scholar] [CrossRef]
- Udeshi, N.D.; Pedram, K.; Svinkina, T.; Fereshetian, S.; Myers, S.A.; Aygun, O.; Krug, K.; Clauser, K.; Ryan, D.; Ast, T.; et al. Antibodies to biotin enable large-scale detection of biotinylation sites on proteins. Nat. Methods 2017, 14, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.A.; Wright, J.; Peckner, R.; Kalish, B.T.; Zhang, F.; Carr, S.A. Discovery of proteins associated with a predefined genomic locus via dCas9–APEX-mediated proximity labeling. Nat. Methods 2018, 15, 437–439. [Google Scholar] [CrossRef]
- Fazal, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 2019, 178, 473–490.e26. [Google Scholar] [CrossRef]
- Michalski, A.; Damoc, E.; Hauschild, J.-P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass Spectrometry-based Proteomics Using Q Exactive, a High-performance Benchtop Quadrupole Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2011, 10, M111.011015. [Google Scholar] [CrossRef] [Green Version]
- Eliuk, S.; Makarov, A. Evolution of Orbitrap Mass Spectrometry Instrumentation. Annu. Rev. Anal. Chem. 2015, 8, 61–80. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Choi-Rhee, E.; Schulman, H.; Cronan, J.E. Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci. 2004, 13, 3043–3050. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.I.; Birendra, K.C.; Zhu, W.; Motamedchaboki, K.; Doye, V.; Roux, K.J. Probing nuclear pore complex architecture with proximity-dependent biotinylation. Proc. Natl. Acad. Sci. USA 2014, 111, E2453–E2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, H.P. Size and Shape of Protein Molecules at the Nanometer Level Determined by Sedimentation, Gel Filtration, and Electron Microscopy. Biol. Proced. Online 2009, 11, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, M.; Majzoub, K.; Rao, D.S.; Neela, P.H.; Zarnegar, B.J.; Mondal, S.; Roth, J.G.; Gai, H.; Kovalski, J.R.; Siprashvili, Z.; et al. RNA–protein interaction detection in living cells. Nat. Methods 2018, 15, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havli, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef]
- Kovalchuk, S.I.; Jensen, O.N.; Rogowska-Wrzesinska, A. FlashPack: Fast and Simple Preparation of Ultrahigh-performance Capillary Columns for LC-MS. Mol. Cell. Proteom. 2019, 18, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Bandeira, N.; Sharma, V.; Perez-Riverol, Y.; Carver, J.J.; Kundu, D.J.; García-Seisdedos, D.; Jarnuczak, A.F.; Hewapathirana, S.; Pullman, B.S.; et al. The ProteomeXchange consortium in 2020: Enabling “big data” approaches in proteomics. Nucleic Acids Res. 2020, 48, D1145–D1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziganshin, R.H.; Ivanova, O.M.; Lomakin, Y.A.; Belogurov, A.A.; Kovalchuk, S.I.; Azarkin, I.V.; Arapidi, G.P.; Anikanov, N.A.; Shender, V.O.; Piradov, M.A.; et al. The Pathogenesis of the Demyelinating Form of Guillain-Barre Syndrome (GBS): Proteo-peptidomic and Immunological Profiling of Physiological Fluids. Mol. Cell. Proteom. 2016, 15, 2366–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic Cell-Derived Extracellular Vesicles Promote Malignancy of Glioblastoma Via Intercellular Transfer of Splicing Factors. Cancer Cell 2018, 34, 119–135.e10. [Google Scholar] [CrossRef] [Green Version]
- Musse, A.A.; Boggs, J.M.; Harauz, G. Deimination of membrane-bound myelin basic protein in multiple sclerosis exposes an immunodominant epitope. Proc. Natl. Acad. Sci. USA 2006, 103, 4422–4427. [Google Scholar] [CrossRef] [Green Version]
- Kudriaeva, A.; Kuzina, E.S.; Zubenko, O.; Smirnov, I.V.; Belogurov, A. Charge-mediated proteasome targeting. FASEB J. 2019, 33, 6852–6866. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Du Plessis, L.; Skunca, N.; Dessimoz, C. The what, where, how and why of gene ontology--a primer for bioinformaticians. Brief. Bioinform. 2011, 12, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Kamholz, J.; Toffenetti, J.; Lazzarini, R.A. Organization and expression of the human myelin basic protein gene. J. Neurosci. Res. 1988, 21, 62–70. [Google Scholar] [CrossRef]
- Capello, E.; Voskuhl, R.R.; McFarland, H.F.; Raine, C.S. Multiple sclerosis: Re-expression of a developmental gene in chronic lesions correlates with remyelination. Ann. Neurol. 1997, 41, 797–805. [Google Scholar] [CrossRef]
- Nagasato, K.; Farris, R.W.; Dubois-Dalcq, M.; Voskuhl, R.R. Exon 2 containing myelin basic protein (MBP) transcripts are expressed in lesions of experimental allergic encephalomyelitis (EAE). J. Neuroimmunol. 1997, 72, 21–25. [Google Scholar] [CrossRef]
- Scarlato, M.; Beesley, J.; Pleasure, D. Analysis of oligodendroglial differentiation using cDNA arrays. J. Neurosci. Res. 2000, 59, 430–435. [Google Scholar] [CrossRef]
- Lu, Z.; Ku, L.; Chen, Y.; Feng, Y. Developmental abnormalities of myelin basic protein expression in fyn knock-out brain reveal a role of Fyn in posttranscriptional regulation. J. Biol. Chem. 2005, 280, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Müller, C.; Bauer, N.M.; Schäfer, I.; White, R. Making myelin basic protein -from mRNA transport to localized translation. Front. Cell. Neurosci. 2013, 7, 169. [Google Scholar] [CrossRef] [Green Version]
- Kinniburgh, A.J.; Martin, T.E. Detection of mRNA sequences in nuclear 30S ribonucleoprotein subcomplexes. Proc. Natl. Acad. Sci. USA 1976, 73, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matlin, A.J.; Clark, F.; Smith, C.W.J. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell. Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.-F.; Revil, T.; Chabot, B. hnRNP Proteins and Splicing Control. In Alternative Splicing in the Postgenomic Era; Advances in Experimental Medicine and Biology; Blencowe, B.J., Graveley, B.R., Eds.; Springer: New York, NY, USA, 2007; Volume 623, pp. 123–147. ISBN 978-0-387-77373-5. [Google Scholar]
- Wang, T.-H.; Wu, C.-C.; Huang, K.-Y.; Chuang, W.-Y.; Hsueh, C.; Li, H.-J.; Chen, C.-Y. Profiling of subcellular EGFR interactome reveals hnRNP A3 modulates nuclear EGFR localization. Oncogenesis 2020, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mourelatos, Z. SMN interacts with a novel family of hnRNP and spliceosomal proteins. EMBO J. 2001, 20, 5443–5452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, L.; Yao, X.; Williams, K.R.; Bassell, G.J. Negative regulation of RhoA translation and signaling by hnRNP-Q1 affects cellular morphogenesis. MBoC 2012, 23, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.R.; McAninch, D.S.; Stefanovic, S.; Xing, L.; Allen, M.; Li, W.; Feng, Y.; Mihailescu, M.R.; Bassell, G.J. hnRNP-Q1 represses nascent axon growth in cortical neurons by inhibiting Gap-43 mRNA translation. MBoC 2016, 27, 518–534. [Google Scholar] [CrossRef]
- Jankowsky, E. RNA helicases at work: Binding and rearranging. Trends Biochem. Sci. 2011, 36, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell. Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Gorbalenya, A.E. Autogenous translation regulation by Escherichia coli ATPase SecA may be mediated by an intrinsic RNA helicase activity of this protein. FEBS Lett. 1992, 298, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toone, W.M.; Rudd, K.E.; Friesen, J.D. deaD, a new Escherichia coli gene encoding a presumed ATP-dependent RNA helicase, can suppress a mutation in rpsB, the gene encoding ribosomal protein S2. J. Bacteriol. 1991, 173, 3291–3302. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.G.; Mitta, M.; Kim, Y.; Jiang, W.; Inouye, M. Cold shock induces a major ribosomal-associated protein that unwinds double-stranded RNA in Escherichia coli. Proc. Natl. Acad. Sci. USA 1996, 93, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charollais, J.; Dreyfus, M.; Iost, I. CsdA, a cold-shock RNA helicase from Escherichia coli, is involved in the biogenesis of 50S ribosomal subunit. Nucleic Acids Res. 2004, 32, 2751–2759. [Google Scholar] [CrossRef] [PubMed]
- Gillian, A.L.; Svaren, J. The Ddx20/DP103 dead box protein represses transcriptional activation by Egr2/Krox-20. J. Biol. Chem. 2004, 279, 9056–9063. [Google Scholar] [CrossRef] [Green Version]
- Ueki, T.; Tsuruo, Y.; Yamamoto, Y.; Yoshimura, K.; Takanaga, H.; Seiwa, C.; Motojima, K.; Asou, H.; Yamamoto, M. A new monoclonal antibody, 4F2, specific for the oligodendroglial cell lineage, recognizes ATP-dependent RNA helicase Ddx54: Possible association with myelin basic protein. J. Neurosci. Res. 2012, 90, 48–59. [Google Scholar] [CrossRef]
- Zhan, R.; Yamamoto, M.; Ueki, T.; Yoshioka, N.; Tanaka, K.; Morisaki, H.; Seiwa, C.; Yamamoto, Y.; Kawano, H.; Tsuruo, Y.; et al. A DEAD-box RNA helicase Ddx54 protein in oligodendrocytes is indispensable for myelination in the central nervous system. J. Neurosci. Res. 2013, 91, 335–348. [Google Scholar] [CrossRef]
- Hoch-Kraft, P.; White, R.; Tenzer, S.; Krämer-Albers, E.-M.; Trotter, J.; Gonsior, C. Dual role of the RNA helicase DDX5 in post-transcriptional regulation of myelin basic protein in oligodendrocytes. J. Cell Sci. 2018, 131, jcs204750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolehmainen, E.; Sormunen, R. Myelin Basic Protein Induces Morphological Changes in the Endocrine Pancreas. Pancreas 1998, 16, 176–188. [Google Scholar] [CrossRef]
- Baburina, Y.L.; Gordeeva, A.E.; Moshkov, D.A.; Krestinina, O.V.; Azarashvili, A.A.; Odinokova, I.V.; Azarashvili, T.S. Interaction of myelin basic protein and 2′,3′-cyclic nucleotide phosphodiesterase with mitochondria. Biochem. Mosc. 2014, 79, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Remacle, A.G.; Hullugundi, S.K.; Dolkas, J.; Angert, M.; Cieplak, P.; Scott, D.; Chernov, A.V.; Shubayev, V.I.; Strongin, A.Y. Interaction of the cryptic fragment of myelin basic protein with mitochondrial voltage-dependent anion-selective channel-1 affects cell energy metabolism. Biochem. J. 2018, 475, 2355–2376. [Google Scholar] [CrossRef] [PubMed]
- Pool, M.R. Targeting of Proteins for Translocation at the Endoplasmic Reticulum. Int. J. Mol. Sci. 2022, 23, 3773. [Google Scholar] [CrossRef]
- Shao, S.; Hegde, R.S. A calmodulin-dependent translocation pathway for small secretory proteins. Cell 2011, 147, 1576–1588. [Google Scholar] [CrossRef] [Green Version]
- Toutenhoofd, S.L.; Strehler, E.E. The calmodulin multigene family as a unique case of genetic redundancy: Multiple levels of regulation to provide spatial and temporal control of calmodulin pools? Cell Calcium 2000, 28, 83–96. [Google Scholar] [CrossRef]
- Vogel, H.J. Calmodulin: A versatile calcium mediator protein. Biochem. Cell Biol. 1994, 72, 357–376. [Google Scholar] [CrossRef]
- Aramburu, J.; Rao, A.; Klee, C.B. Calcineurin: From structure to function. Curr. Top. Cell. Regul. 2000, 36, 237–295. [Google Scholar] [CrossRef]
- Crabtree, G.R.; Schreiber, S.L. SnapShot: Ca2+-calcineurin-NFAT signaling. Cell 2009, 138, 210–210.e1. [Google Scholar] [CrossRef] [Green Version]
- Weider, M.; Starost, L.J.; Groll, K.; Küspert, M.; Sock, E.; Wedel, M.; Fröb, F.; Schmitt, C.; Baroti, T.; Hartwig, A.C.; et al. Nfat/calcineurin signaling promotes oligodendrocyte differentiation and myelination by transcription factor network tuning. Nat. Commun. 2018, 9, 899. [Google Scholar] [CrossRef] [Green Version]
- Vaheri, A.; Carpén, O.; Heiska, L.; Helander, T.S.; Jääskeläinen, J.; Majander-Nordenswan, P.; Sainio, M.; Timonen, T.; Turunen, O. The ezrin protein family: Membrane-cytoskeleton interactions and disease associations. Curr. Opin. Cell Biol. 1997, 9, 659–666. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, L.; Xiao, H.; Yang, Y.; Shi, Y. Ezrin interacts with L-periaxin by the “head to head and tail to tail” mode and influences the location of L-periaxin in Schwann cell RSC96. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129520. [Google Scholar] [CrossRef]
- Melendez-Vasquez, C.V.; Rios, J.C.; Zanazzi, G.; Lambert, S.; Bretscher, A.; Salzer, J.L. Nodes of Ranvier form in association with ezrin-radixin-moesin (ERM)-positive Schwann cell processes. Proc. Natl. Acad. Sci. USA 2001, 98, 1235–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef]
- Saitou, M.; Ando-Akatsuka, Y.; Itoh, M.; Furuse, M.; Inazawa, J.; Fujimoto, K.; Tsukita, S. Mammalian occludin in epithelial cells: Its expression and subcellular distribution. Eur. J. Cell Biol. 1997, 73, 222–231. [Google Scholar] [PubMed]
- Beckmann, M.P.; Cerretti, D.P.; Baum, P.; Vanden Bos, T.; James, L.; Farrah, T.; Kozlosky, C.; Hollingsworth, T.; Shilling, H.; Maraskovsky, E. Molecular characterization of a family of ligands for eph-related tyrosine kinase receptors. EMBO J. 1994, 13, 3757–3762. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Gale, N.W.; Aldrich, T.H.; Maisonpierre, P.C.; Lhotak, V.; Pawson, T.; Goldfarb, M.; Yancopoulos, G.D. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science 1994, 266, 816–819. [Google Scholar] [CrossRef]
- Linneberg, C.; Harboe, M.; Laursen, L.S. Axo-Glia Interaction Preceding CNS Myelination Is Regulated by Bidirectional Eph-Ephrin Signaling. ASN Neuro 2015, 7, 1759091415602859. [Google Scholar] [CrossRef]
- Zhao, Z.J.; Zhao, R. Purification and Cloning of PZR, a Binding Protein and Putative Physiological Substrate of Tyrosine Phosphatase SHP-2. J. Biol. Chem. 1998, 273, 29367–29372. [Google Scholar] [CrossRef] [Green Version]
- Beigbeder, A.; Chartier, F.J.M.; Bisson, N. MPZL1 forms a signalling complex with GRB2 adaptor and PTPN11 phosphatase in HER2-positive breast cancer cells. Sci. Rep. 2017, 7, 11514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Guerrah, A.; Tang, H.; Zhao, Z.J. Cell Surface Glycoprotein PZR Is a Major Mediator of Concanavalin A-induced Cell Signaling. J. Biol. Chem. 2002, 277, 7882–7888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, K.M.; Nicholson, R.I. The LZT proteins; the LIV-1 subfamily of zinc transporters. Biochim. Biophys. Acta 2003, 1611, 16–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hediger, M.A.; Romero, M.F.; Peng, J.-B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteins. Pflügers Archiv. 2004, 447, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Perland, E.; Fredriksson, R. Classification Systems of Secondary Active Transporters. Trends Pharmacol. Sci. 2017, 38, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Roderick, H.L.; Camacho, P.; Jiang, J.X. Characterization of an N-system amino acid transporter expressed in retina and its involvement in glutamine transport. J. Biol. Chem. 2001, 276, 24137–24144. [Google Scholar] [CrossRef] [Green Version]
- Ohkuni, A.; Ohno, Y.; Kihara, A. Identification of acyl-CoA synthetases involved in the mammalian sphingosine 1-phosphate metabolic pathway. Biochem. Biophys. Res. Commun. 2013, 442, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Meloni, I.; Muscettola, M.; Raynaud, M.; Longo, I.; Bruttini, M.; Moizard, M.-P.; Gomot, M.; Chelly, J.; des Portes, V.; Fryns, J.-P.; et al. FACL4, encoding fatty acid-CoA ligase 4, is mutated in nonspecific X-linked mental retardation. Nat. Genet. 2002, 30, 436–440. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef]
- Vanisree, A.J.; Thamizhoviya, G. Enriched Environment Minimizes Anxiety/Depressive-Like Behavior in Rats Exposed to Immobilization Stress and Augments Hippocampal Neurogenesis (In Vitro). J. Mol. Neurosci. 2021, 71, 2071–2084. [Google Scholar] [CrossRef]
- Zarruk, J.G.; Berard, J.L.; Passos dos Santos, R.; Kroner, A.; Lee, J.; Arosio, P.; David, S. Expression of iron homeostasis proteins in the spinal cord in experimental autoimmune encephalomyelitis and their implications for iron accumulation. Neurobiol. Dis. 2015, 81, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Klima, H.; Tanaka, A.; Schnabel, D.; Nakano, T.; Schröder, M.; Suzuki, K.; Sandhoff, K. Characterization of full-length cDNAs and the gene coding for the human GM2 activator protein. FEBS Lett. 1991, 289, 260–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonny, B.; Madden, D.; Hamamoto, S.; Orci, L.; Schekman, R. Dynamics of the COPII coat with GTP and stable analogues. Nat. Cell Biol. 2001, 3, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Khoriaty, R.; Hesketh, G.G.; Bernard, A.; Weyand, A.C.; Mellacheruvu, D.; Zhu, G.; Hoenerhoff, M.J.; McGee, B.; Everett, L.; Adams, E.J.; et al. Functions of the COPII gene paralogs SEC23A and SEC23B are interchangeable in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E7748–E7757. [Google Scholar] [CrossRef] [Green Version]
- Yu, I.-M.; Hughson, F.M. Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol. 2010, 26, 137–156. [Google Scholar] [CrossRef]
- Tamura, N.; Mima, J. Membrane-anchored human Rab GTPases directly mediate membrane tethering in vitro. Biol. Open. 2014, 3, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Ayala, J.; Touchot, N.; Zahraoui, A.; Tavitian, A.; Prochiantz, A. The product of rab2, a small GTP binding protein, increases neuronal adhesion, and neurite growth in vitro. Neuron 1990, 4, 797–805. [Google Scholar] [CrossRef]
- Itoh, T.; Fukuda, M. Identification of EPI64 as a GTPase-activating protein specific for Rab27A. J. Biol. Chem. 2006, 281, 31823–31831. [Google Scholar] [CrossRef]
- Rothman, J.E. Mechanisms of Intracellular Protein Transport. Nature 1994, 372, 55–63. [Google Scholar] [CrossRef]
- Bennett, M.K.; Scheller, R.H. A Molecular Description of Synaptic Vesicle Membrane Trafficking. Annu. Rev. Biochem. 1994, 63, 63–100. [Google Scholar] [CrossRef]
- Jahn, R.; Südhof, T.C. Synaptic Vesicles and Exocytosis. Annu. Rev. Neurosci. 1994, 17, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Söllner, T.; Whiteheart, S.W.; Brunner, M.; Erdjument-Bromage, H.; Geromanos, S.; Tempst, P.; Rothman, J.E. SNAP Receptors Implicated in Vesicle Targeting and Fusion. Nature 1993, 362, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.K.; García-Arrarás, J.E.; Elferink, L.A.; Peterson, K.; Fleming, A.M.; Hazuka, C.D.; Scheller, R.H. The Syntaxin Family of Vesicular Transport Receptors. Cell 1993, 74, 863–873. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Ushkaryov, Y.A.; Edelmann, L.; Link, E.; Binz, T.; Niemann, H.; Jahn, R.; Südhof, T.C. Cellubrevin Is a Ubiquitous Tetanus-Toxin Substrate Homologous to a Putative Synaptic Vesicle Fusion Protein. Nature 1993, 364, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Gorza, L.; Schiavo, G.; Schiavo, N.; Scheller, R.H.; Montecucco, C. VAMP/Synaptobrevin Isoforms 1 and 2 Are Widely and Differentially Expressed in Nonneuronal Tissues. J. Cell Biol. 1996, 132, 167–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calakos, N.; Bennett, M.K.; Peterson, K.E.; Scheller, R.H. Protein-Protein Interactions Contributing to the Specificity of Intracellular Vesicular Trafficking. Science 1994, 263, 1146–1149. [Google Scholar] [CrossRef]
- Ferro-Novick, S.; Jahn, R. Vesicle Fusion from Yeast to Man. Nature 1994, 370, 191–193. [Google Scholar] [CrossRef]
- Oyler, G.A.; Higgins, G.A.; Hart, R.A.; Battenberg, E.; Billingsley, M.; Bloom, F.E.; Wilson, M.C. The Identification of a Novel Synaptosomal-Associated Protein, SNAP-25, Differentially Expressed by Neuronal Subpopulations. J. Cell Biol. 1989, 109, 3039–3052. [Google Scholar] [CrossRef] [Green Version]
- Bark, I.C.; Hahn, K.M.; Ryabinin, A.E.; Wilson, M.C. Differential Expression of SNAP-25 Protein Isoforms during Divergent Vesicle Fusion Events of Neural Development. Proc. Natl. Acad. Sci. USA 1995, 92, 1510–1514. [Google Scholar] [CrossRef] [Green Version]
- Brumell, J.H.; Volchuk, A.; Sengelov, H.; Borregaard, N.; Cieutat, A.M.; Bainton, D.F.; Grinstein, S.; Klip, A. Subcellular Distribution of Docking/Fusion Proteins in Neutrophils, Secretory Cells with Multiple Exocytic Compartments. J. Immunol. Baltim. Md 1950 1995, 155, 5750–5759. [Google Scholar] [CrossRef]
- McMahon, H.T.; Südhof, T.C. Synaptic Core Complex of Synaptobrevin, Syntaxin, and SNAP25 Forms High Affinity Alpha-SNAP Binding Site. J. Biol. Chem. 1995, 270, 2213–2217. [Google Scholar] [CrossRef] [Green Version]
- Pevsner, J.; Hsu, S.C.; Braun, J.E.; Calakos, N.; Ting, A.E.; Bennett, M.K.; Scheller, R.H. Specificity and Regulation of a Synaptic Vesicle Docking Complex. Neuron 1994, 13, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; McMahon, H.; Yamasaki, S.; Binz, T.; Hata, Y.; Südhof, T.C.; Niemann, H. Synaptic Vesicle Membrane Fusion Complex: Action of Clostridial Neurotoxins on Assembly. EMBO J. 1994, 13, 5051–5061. [Google Scholar] [CrossRef]
- Ravichandran, V.; Chawla, A.; Roche, P.A. Identification of a Novel Syntaxin- and Synaptobrevin/VAMP-Binding Protein, SNAP-23, Expressed in Non-Neuronal Tissues. J. Biol. Chem. 1996, 271, 13300–13303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganley, I.G.; Espinosa, E.; Pfeffer, S.R. A Syntaxin 10-SNARE Complex Distinguishes Two Distinct Transport Routes from Endosomes to the Trans-Golgi in Human Cells. J. Cell Biol. 2008, 180, 159–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijlard, M.; Klunder, B.; de Jonge, J.C.; Nomden, A.; Tyagi, S.; de Vries, H.; Hoekstra, D.; Baron, W. Transcriptional Expression of Myelin Basic Protein in Oligodendrocytes Depends on Functional Syntaxin 4: A Potential Correlation with Autocrine Signaling. Mol. Cell. Biol. 2015, 35, 675–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, M.; Takeo, K.; Almeida, R.G.; Cooper, M.H.; Wu, K.; Iyer, M.; Kantarci, H.; Zuchero, J.B. CNS Myelination Requires VAMP2/3-Mediated Membrane Expansion in Oligodendrocytes. Nat. Commun. 2022, 13, 5583. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smirnova, E.V.; Rakitina, T.V.; Ziganshin, R.H.; Saratov, G.A.; Arapidi, G.P.; Belogurov, A.A., Jr.; Kudriaeva, A.A. Identification of Myelin Basic Protein Proximity Interactome Using TurboID Labeling Proteomics. Cells 2023, 12, 944. https://doi.org/10.3390/cells12060944
Smirnova EV, Rakitina TV, Ziganshin RH, Saratov GA, Arapidi GP, Belogurov AA Jr., Kudriaeva AA. Identification of Myelin Basic Protein Proximity Interactome Using TurboID Labeling Proteomics. Cells. 2023; 12(6):944. https://doi.org/10.3390/cells12060944
Chicago/Turabian StyleSmirnova, Evgeniya V., Tatiana V. Rakitina, Rustam H. Ziganshin, George A. Saratov, Georgij P. Arapidi, Alexey A. Belogurov, Jr., and Anna A. Kudriaeva. 2023. "Identification of Myelin Basic Protein Proximity Interactome Using TurboID Labeling Proteomics" Cells 12, no. 6: 944. https://doi.org/10.3390/cells12060944