

The Role of Macrophages in the Pathogenesis of Atherosclerosis
 , ,
, ,  and
and 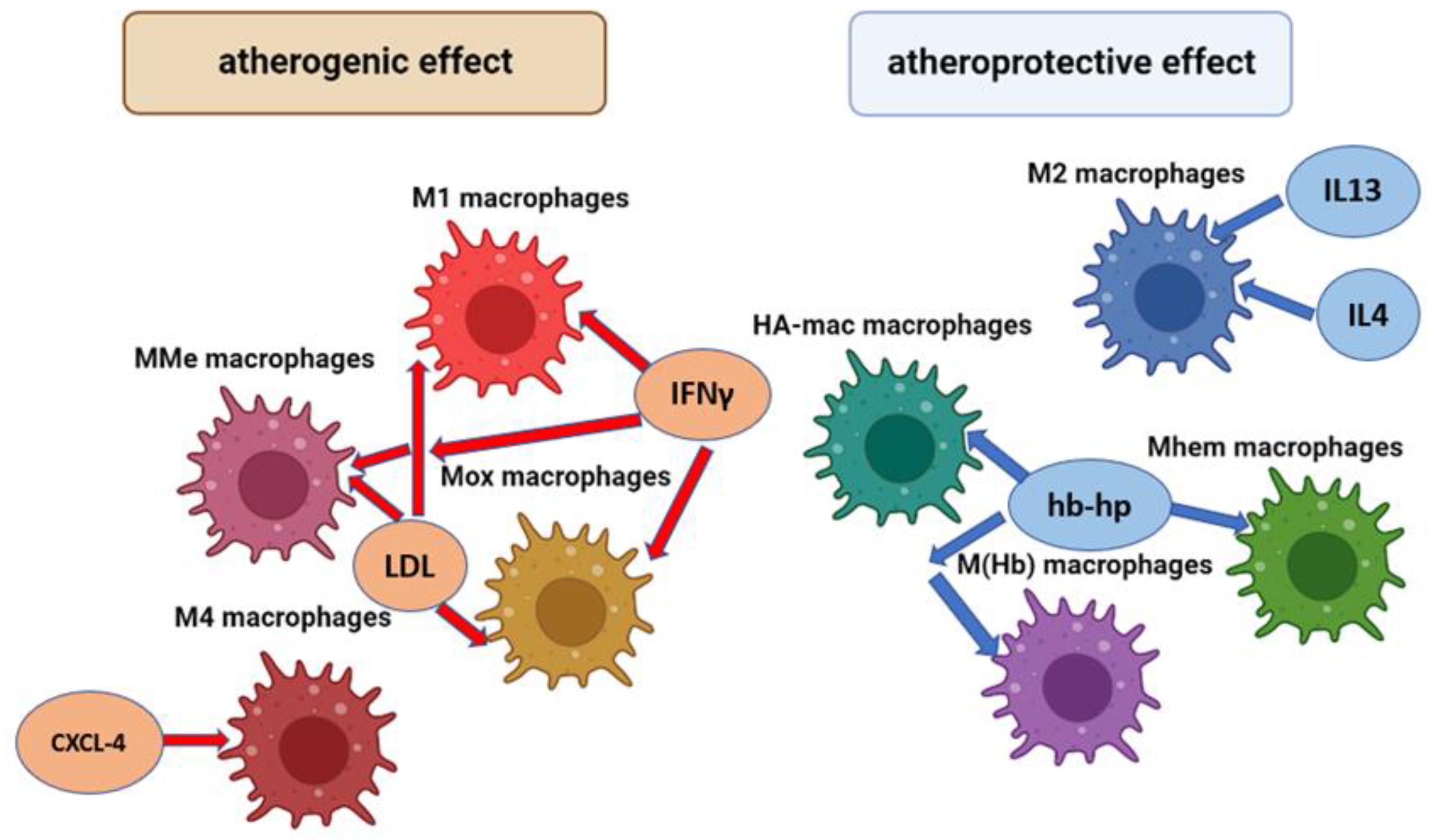{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Significance of Different Phenotypes of Macrophages in the Pathogenesis of Atherosclerosis
2.1. M1 Macrophages
2.2. M2 Macrophages
2.3. Other Macrophage Phenotypes
2.4. Molecular Mechanisms of Macrophage Phenotypic Shift
2.5. Distribution of Macrophages in Atherosclerosis
3. Lipid Activation of Macrophages in Atherosclerosis
4. Foam Cell Formation and Their Role in Disease Progression
5. Importance of Cytokines Produced by Macrophages in Atherosclerosis
5.1. Pro-Inflammatory Cytokines
5.2. Anti-Inflammatory Cytokines
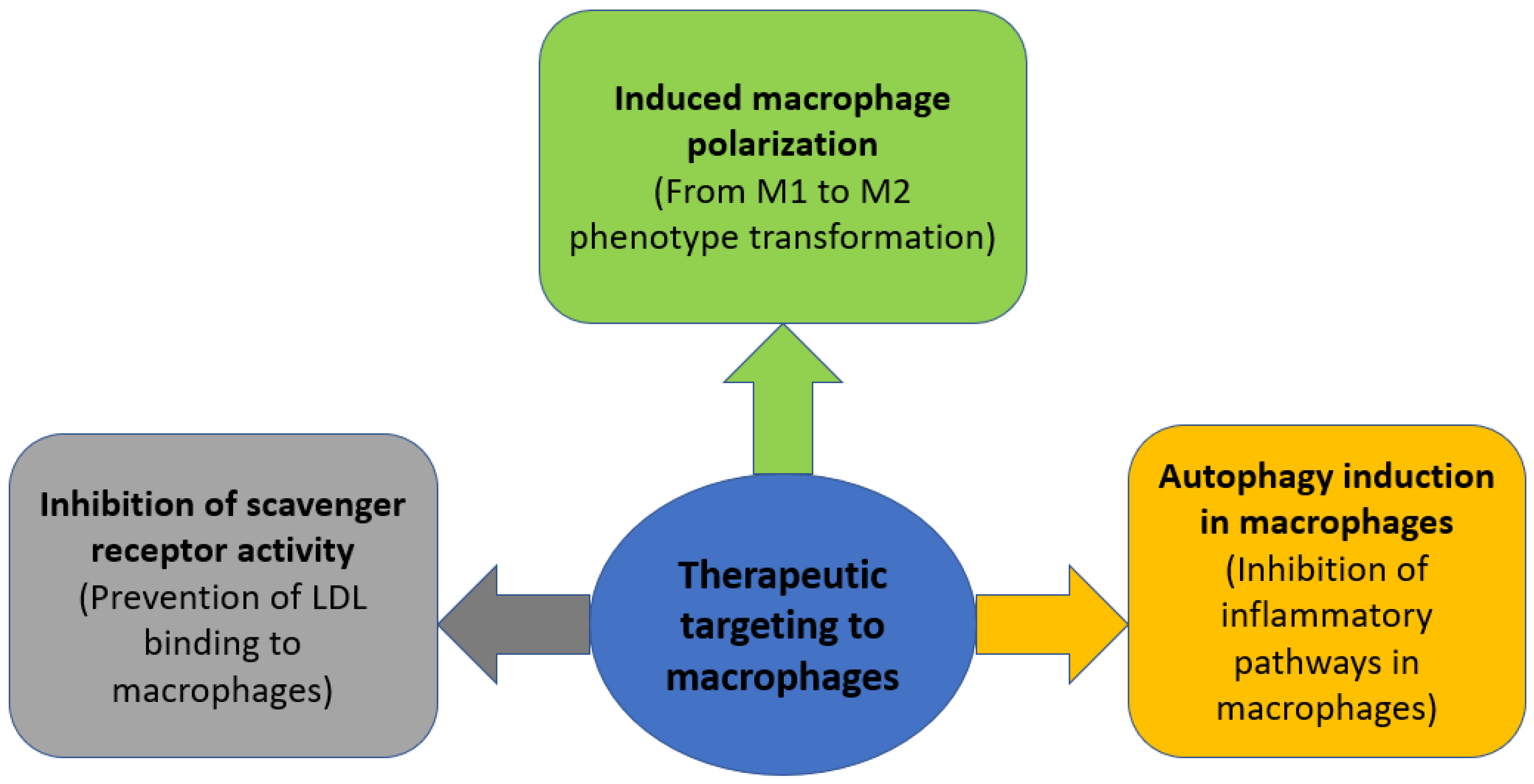
6. Therapeutic Strategies Targeting Macrophages in Atherosclerosis
6.1. Induced Macrophage Polarization
6.2. Inhibition of Scavenger Receptor Activity
6.3. Autophagy Induction in Macrophages
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gallino, A.; Aboyans, V.; Diehm, C.; Cosentino, F.; Stricker, H.; Falk, E.; Schouten, O.; Lekakis, J.; Amann-Vesti, B.; Siclari, F.; et al. Non-coronary atherosclerosis. Eur. Heart J. 2014, 35, 1112–1119. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in atherosclerosis. Circ. Res. 2019, 124, 315. [Google Scholar] [CrossRef]
- Kruk, M.E.; Gage, A.D.; Joseph, N.T.; Danaei, G.; García-Saisó, S.; Salomon, J.A. Mortality due to low-quality health systems in the universal health coverage era: A systematic analysis of amenable deaths in 137 countries. Lancet 2018, 392, 2203. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Melnichenko, A.A.; Grechko, A.V.; Myasoedova, V.A.; Orekhov, A.N. Potential of anti-inflammatory agents for treatment of atherosclerosis. Exp. Mol. Pathol. 2018, 104, 114–124. [Google Scholar] [CrossRef]
- Ranjit, N.; Diez-Roux, A.V.; Shea, S.; Cushman, M.; Seeman, T.; Jackson, S.A.; Ni, H. Psychosocial factors and inflammation in the multi-ethnic study of atherosclerosis. Arch Intern. Med. 2007, 167, 174–181. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Jinnouchi, H.; Guo, L.; Sakamoto, A.; Torii, S.; Sato, Y.; Cornelissen, A.; Kuntz, S.; Paek, K.H.; Fernandez, R.; Fuller, D.; et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 2019, 77, 1919–1932. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, A.; Khaitov, M.; Jackson, D.J.; Traub, S.; Trujillo-Torralbo, M.-B.; Kudlay, D.A.; Dvornikov, A.S.; Del-Rosario, A.; Valenta, R.; Stanciu, L.A.; et al. M1-like macrophages are potent producers of anti-viral interferons and M1-associated marker-positive lung macrophages are decreased during rhinovirus-induced asthma exacerbations. Ebiomedicine 2020, 54, 102734. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Babaev, V.R.; Hebron, K.E.; Wiese, C.B.; Toth, C.L.; Ding, L.; Zhang, Y.; May, J.M.; Fazio, S.; Vickers, K.C.; Linton, M.F. Macrophage deficiency of Akt2 reduces atherosclerosis in Ldlr null mice. J. Lipid Res. 2014, 55, 2296–2308. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.-B.; Choi, M.J.; Ryu, D.; Yi, H.-S.; Lee, S.E.; Chang, J.Y.; Chung, H.K.; Kim, Y.K.; Kang, S.G.; Lee, J.H.; et al. Reduced oxidative capacity in macrophages results in systemic insulin resistance. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Xu, H.; Jiang, J.; Chen, W.; Li, W.; Chen, Z. Vascular macrophages in atherosclerosis. J. Immunol. Res. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J. Macrophages in atherosclerosis regression. Arter. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.J.; Donners, M.M.P.C. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2013, 17, 109–118. [Google Scholar] [CrossRef]
- Willemsen, L.; De Winther, M.P. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol. 2020, 250, 705–714. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Nikiforov, N.G.; Elizova, N.V.; Sobenin, I.A.; Orekhov, A.N. RETRACTED: Macrophage phenotypic plasticity in atherosclerosis: The associated features and the peculiarities of the expression of inflammatory genes. Int. J. Cardiol. 2015, 184, 436–445. [Google Scholar] [CrossRef]
- Cardilo-Reis, L.; Gruber, S.; Schreier, S.M.; Drechsler, M.; Papac-Milicevic, N.; Weber, C.; Wagner, O.; Stangl, H.; Soehnlein, O.; Binder, C.J. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol. Med. 2012, 4, 1072–1086. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2014, 12, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Haka, A.S.; Barbosa-Lorenzi, V.C.; Lee, H.J.; Falcone, D.J.; Hudis, C.A.; Dannenberg, A.J.; Maxfield, F.R. Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. J. Lipid Res. 2016, 57, 980–992. [Google Scholar] [CrossRef] [PubMed]
- Coats, B.R.; Schoenfelt, K.Q.; Barbosa-Lorenzi, V.C.; Peris, E.; Cui, C.; Hoffman, A.; Zhou, G.; Fernandez, S.; Zhai, L.; Hall, B.A.; et al. Metabolically activated adipose tissue macrophages perform detrimental and beneficial functions during diet-induced obesity. Cell Rep. 2017, 20, 3149–3161. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Changes in transcriptome of macrophages in atherosclerosis. J. Cell. Mol. Med. 2015, 19, 1163–1173. [Google Scholar] [CrossRef]
- Skuratovskaia, D.; Vulf, M.; Khaziakhmatova, O.; Malashchenko, V.; Komar, A.; Shunkin, E.; Shupletsova, V.; Goncharov, A.; Urazova, O.; Litvinova, L. Tissue-specific role of macrophages in noninfectious inflammatory disorders. Biomedicines 2020, 8, 400. [Google Scholar] [CrossRef]
- Martinez-Nunez, R.T.; Louafi, F.; Sanchez-Elsner, T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by MicroRNA-155 via Direct targeting of interleukin 13 receptor α1 (IL13Rα1). J. Biol. Chem. 2011, 286, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Amici, S.A.; Dong, J.; Guerau-De-Arellano, M. Molecular mechanisms modulating the phenotype of macrophages and microglia. Front. Immunol. 2017, 8, 1520. [Google Scholar] [CrossRef] [PubMed]
- De Gaetano, M.; Crean, D.; Barry, M.; Belton, O. M1-and M2-type macrophage responses are predictive of adverse outcomes in human atherosclerosis. Front. Immunol. 2016, 7, 275. [Google Scholar] [CrossRef]
- Lin, P.; Ji, H.-H.; Li, Y.-J.; Guo, S.-D. Macrophage plasticity and atherosclerosis therapy. Front. Mol. Biosci. 2021, 8, 679797. [Google Scholar] [CrossRef]
- Boyle, J.J.; Johns, M.; Lo, J.; Chiodini, A.; Ambrose, N.; Evans, P.C.; Mason, J.C.; Haskard, D.O. Heme induces heme oxygenase 1 via Nrf2. Arter. Thromb. Vasc. Biol. 2011, 31, 2685–2691. [Google Scholar] [CrossRef]
- Branchetti, E.; Bavaria, J.E.; Grau, J.B.; Shaw, R.E.; Poggio, P.; Lai, E.K.; Desai, N.D.; Gorman, J.H.; Gorman, R.C.; Ferrari, G. Circulating soluble receptor for advanced glycation end product identifies patients with bicuspid aortic valve and associated aortopathies. Arter. Thromb. Vasc. Biol. 2014, 34, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Sainger, R.; Grau, J.B.; Branchetti, E.; Poggio, P.; Lai, E.; Koka, E.; Vernick, W.J.; Gorman, R.C.; Bavaria, J.E.; Ferrari, G. Comparison of transesophageal echocardiographic analysis and circulating biomarker expression profile in calcific aortic valve disease. J. Hear. Valve Dis. 2013, 22, 156–165. [Google Scholar]
- Moore, X.-L.; Fang, L.; Sviridov, D.; Chin-Dusting, J.; Andrews, K.L.; Al-Sharea, A.; Lee, M.K.S.; Murphy, A.J. Native LDL promotes differentiation of human monocytes to macrophages with an inflammatory phenotype. Thromb. Haemost. 2016, 115, 762–772. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and their role in atherosclerosis: Pathophysiology and transcriptome analysis. BioMed Res. Int. 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Poggio, P.; Branchetti, E.; Grau, J.B.; Lai, E.K.; Gorman, R.C.; GormanIII, J.H.; Sacks, M.S.; Bavaria, J.E.; Ferrari, G. Osteopontin–CD44v6 interaction mediates calcium deposition via phospho-akt in valve interstitial cells from patients with noncalcified aortic valve sclerosis. Arter. Thromb. Vasc. Biol. 2014, 34, 2086–2094. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Nikiforov, N.G.; Starodubova, A.V.; Popkova, T.V.; Orekhov, A.N. Macrophages and foam cells: Brief overview of their role, linkage, and targeting potential in atherosclerosis. Biomedicines 2021, 9, 1221. [Google Scholar] [CrossRef] [PubMed]
- Parolari, A.; Poggio, P.; Myasoedova, V.; Songia, P.; Bonalumi, G.; Pilozzi, A.; Pacini, D.; Alamanni, F.; Tremoli, E. Biomarkers in coronary artery bypass surgery: Ready for prime time and outcome prediction? Front. Cardiovasc. Med. 2016, 2, 39. [Google Scholar] [CrossRef] [PubMed]
- Sanson, M.; Distel, E.; Fisher, E.A. HDL Induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS ONE 2013, 8, e74676. [Google Scholar] [CrossRef]
- Lee, M.K.S.; Moore, X.-L.; Fu, Y.; Al-Sharea, A.; Dragoljevic, D.; Fernandez-Rojo, M.A.; Parton, R.; Sviridov, D.; Murphy, A.J.; Chin-Dusting, J.P.F. High-density lipoprotein inhibits human M1 macrophage polarization through redistribution of caveolin-1. Br. J. Pharmacol. 2015, 173, 741–751. [Google Scholar] [CrossRef]
- Ivanova, E.A.; Bobryshev, Y.V.; Orekhov, A.N. LDL electronegativity index: A potential novel index for predicting cardiovascular disease. Vasc. Health Risk Manag. 2015, 11, 525–532. [Google Scholar] [CrossRef]
- Yu, X.-H.; Qian, K.; Jiang, N.; Zheng, X.-L.; Cayabyab, F.S.; Tang, C.-K. ABCG5/ABCG8 in cholesterol excretion and atherosclerosis. Clin. Chim. Acta 2014, 428, 82–88. [Google Scholar] [CrossRef]
- Moroni, F.; Ammirati, E.; Norata, G.D.; Magnoni, M.; Camici, P.G. The role of monocytes and macrophages in human atherosclerosis, plaque neoangiogenesis, and atherothrombosis. Mediat. Inflamm. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef]
- Kappus, M.S.; Murphy, A.J.; Abramowicz, S.; Ntonga, V.; Welch, C.L.; Tall, A.R.; Westerterp, M. Activation of liver X receptor decreases atherosclerosis in Ldlr−/− mice in the absence of ATP-binding cassette transporters A1 and G1 in myeloid cells. Arter. Thromb. Vasc. Biol. 2014, 34, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Gomez, D.; Shankman, L.S.; Nguyen, A.T.; Owens, G.K. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat. Methods 2013, 10, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol loading reprograms the MicroRNA-143/145–myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arter. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Jansen, M.F.; Hollander, M.R.; van Royen, N.; Horrevoets, A.J.; Lutgens, E. CD40 in coronary artery disease: A matter of macrophages? Basic Res. Cardiol. 2016, 111, 1–16. [Google Scholar] [CrossRef]
- Fatkhullina, A.R.; Peshkova, I.O.; Koltsova, E.K. The role of cytokines in the development of atherosclerosis. Biochemistry 2016, 81, 1358–1370. [Google Scholar] [CrossRef]
- Duque, G.A.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [PubMed]
- Bhat, O.M.; Kumar, P.U.; Giridharan, N.; Kaul, D.; Kumar, M.M.; Dhawan, V. Interleukin-18-induced atherosclerosis involves CD36 and NF-κB crosstalk in Apo E−/− mice. J. Cardiol. 2015, 66, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, G.; Wiltshire, C.; Macaskill, S.; Tournu, H.; Budge, S.; Brown, A.J. Gcn4 co-ordinates morphogenetic and metabolic responses to amino acid star-vation in candida albicans. EMBO J. 2002, 21, 5448–5456. [Google Scholar] [CrossRef]
- Wainstein, M.V.; Mossmann, M.; Araujo, G.N.; Gonçalves, S.C.; Gravina, G.L.; Sangalli, M.; Veadrigo, F.; Matte, R.; Reich, R.; Costa, F.G.; et al. Elevated serum interleukin-6 is predictive of coronary artery disease in intermediate risk overweight patients referred for coronary angiography. Diabetol. Metab. Syndr. 2017, 9, 1–7. [Google Scholar] [CrossRef]
- Velásquez, I.M.; Gajulapuri, A.; Leander, K.; Berglund, A.; De Faire, U.; Gigante, B. Serum IL8 is not associated with cardiovascular events but with all-cause mortality. BMC Cardiovasc. Disord. 2019, 19, 1–8. [Google Scholar] [CrossRef]
- Bosmans, L.A.; Bosch, L.; Kusters, P.J.; Lutgens, E.; Seijkens, T.T. The CD40-CD40L dyad as immunotherapeutic target in cardiovascular disease. J. Cardiovasc. Transl. Res. 2020, 14, 13–22. [Google Scholar] [CrossRef]
- Chi, Z.; Melendez, A.J. Role of cell adhesion molecules and immune-cell migration in the initiation, onset and development of atherosclerosis. Cell Adhes. Migr. 2007, 1, 171–175. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting early atherosclerosis: A focus on oxidative stress and inflammation. Oxid. Med. Cell Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef]
- Ailuno, G.; Baldassari, S.; Zuccari, G.; Schlich, M.; Caviglioli, G. Peptide-based nanosystems for vascular cell adhesion molecule-1 targeting: A real opportunity for therapeutic and diagnostic agents in inflammation associated disorders. J. Drug Deliv. Sci. Technol. 2019, 55, 101461. [Google Scholar] [CrossRef]
- Subedi, L.; Lee, S.E.; Madiha, S.; Gaire, B.P.; Jin, M.; Yumnam, S.; Kim, S.Y. Phytochemicals against TNFα-mediated neuroinflammatory diseases. Int. J. Mol. Sci. 2020, 21, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douna, H.; Amersfoort, J.; Schaftenaar, F.H.; Kroon, S.; van Puijvelde, G.H.; Kuiper, J.; Foks, A.C. Bidirectional effects of IL-10+ regulatory B cells in Ldlr mice. Atherosclerosis 2018, 280, 118–125. [Google Scholar] [CrossRef]
- Jiang, Y.; Gao, Q.; Wang, L.; Guo, C.; Zhu, F.; Wang, B.; Wang, Q.; Gao, F.; Chen, Y.; Zhang, L. Deficiency of programmed cell death 4 results in increased IL-10 expression by macrophages and thereby attenuates atherosclerosis in hyperlipidemic mice. Cell. Mol. Immunol. 2015, 13, 524–534. [Google Scholar] [CrossRef]
- Toma, I.; McCaffrey, T.A. Transforming growth factor-β and atherosclerosis: Interwoven atherogenic and atheroprotective aspects. Cell Tissue Res. 2011, 347, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.R.; Petersen, L.K.; York, A.W.; Zablocki, K.R.; Joseph, L.B.; Kholodovych, V.; Prud’Homme, R.K.; Uhrich, K.E.; Moghe, P.V. Sugar-based amphiphilic nanoparticles arrest atherosclerosis in vivo. Proc. Natl. Acad. Sci. USA 2015, 112, 2693–2698. [Google Scholar] [CrossRef] [PubMed]
- Hehir, S.; Plourde, N.M.; Gu, L.; Poree, D.E.; Welsh, W.J.; Moghe, P.V.; Uhrich, K.E. Carbohydrate composition of amphiphilic macromolecules influences physicochemical properties and binding to atherogenic scavenger receptor A. Acta Biomater. 2012, 8, 3956–3962. [Google Scholar] [CrossRef]
- Marleau, S.; Harb, D.; Bujold, K.; Avallone, R.; Iken, K.; Wang, Y.; Demers, A.; Sirois, M.G.; Febbraio, M.; Silverstein, R.L.; et al. EP 80317, a ligand of the CD36 scavenger receptor, protects apolipoprotein E-deficient mice from developing atherosclerotic lesions. FASEB J. 2005, 19, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Ji, H.; Jin, L.; Lin, S.; Huang, Y.; Xu, K.; Yang, Q.; Sun, D.; Wu, W. Macrophage-based therapies for atherosclerosis management. J. Immunol. Res. 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef]
- Cantero-Navarro, E.; Rayego-Mateos, S.; Orejudo, M.; Tejedor-Santamaria, L.; Tejera-Muñoz, A.; Sanz, A.B.; Marquez-Exposito, L.; Marchant, V.; Santos-Sanchez, L.; Egido, J.; et al. Role of macrophages and related cytokines in kidney disease. Front. Med. 2021, 8, 688060. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, D.; Wang, R.; Wu, M.; Zhu, L.; Peng, W.; Tu, H.; Deng, X.; Zhu, H.; Zhang, Z.; et al. Targeted therapy of rheumatoid arthritis via macrophage repolarization. Drug Deliv. 2021, 28, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Çetin, M.; Erdoğan, T.; Kırış, T.; Özer, S.; Çinier, G.; Emlek, N.; Durak, H.; Şatıroğlu. Elevated serum YKL40 level is a predictor of MACE during the long-term follow up in hypertensive patients. Clin. Exp. Hypertens. 2019, 42, 271–274. [Google Scholar] [CrossRef]
- Li, M.; Xin, S.; Gu, R.; Zheng, L.; Hu, J.; Zhang, R.; Dong, H. Novel diagnostic biomarkers related to oxidative stress and macrophage ferroptosis in atherosclerosis. Oxidative Med. Cell. Longev. 2022, 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wang, X.-K.; Lv, Y.-H.; Wang, X.; Cui, Y.-C. The integrated analysis identifies three critical genes as novel diagnostic biomarkers involved in immune infiltration in atherosclerosis. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, F. Atherosclerosis and the role of immune cells. World J. Clin. Cases 2015, 3, 345–352. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blagov, A.V.; Markin, A.M.; Bogatyreva, A.I.; Tolstik, T.V.; Sukhorukov, V.N.; Orekhov, A.N. The Role of Macrophages in the Pathogenesis of Atherosclerosis. Cells 2023, 12, 522. https://doi.org/10.3390/cells12040522
Blagov AV, Markin AM, Bogatyreva AI, Tolstik TV, Sukhorukov VN, Orekhov AN. The Role of Macrophages in the Pathogenesis of Atherosclerosis. Cells. 2023; 12(4):522. https://doi.org/10.3390/cells12040522
Chicago/Turabian StyleBlagov, Alexander V., Alexander M. Markin, Anastasia I. Bogatyreva, Taisiya V. Tolstik, Vasily N. Sukhorukov, and Alexander N. Orekhov. 2023. "The Role of Macrophages in the Pathogenesis of Atherosclerosis" Cells 12, no. 4: 522. https://doi.org/10.3390/cells12040522