
Pathogenesis and Mechanisms of SARS-CoV-2 Infection in the Intestine, Liver, and Pancreas
 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
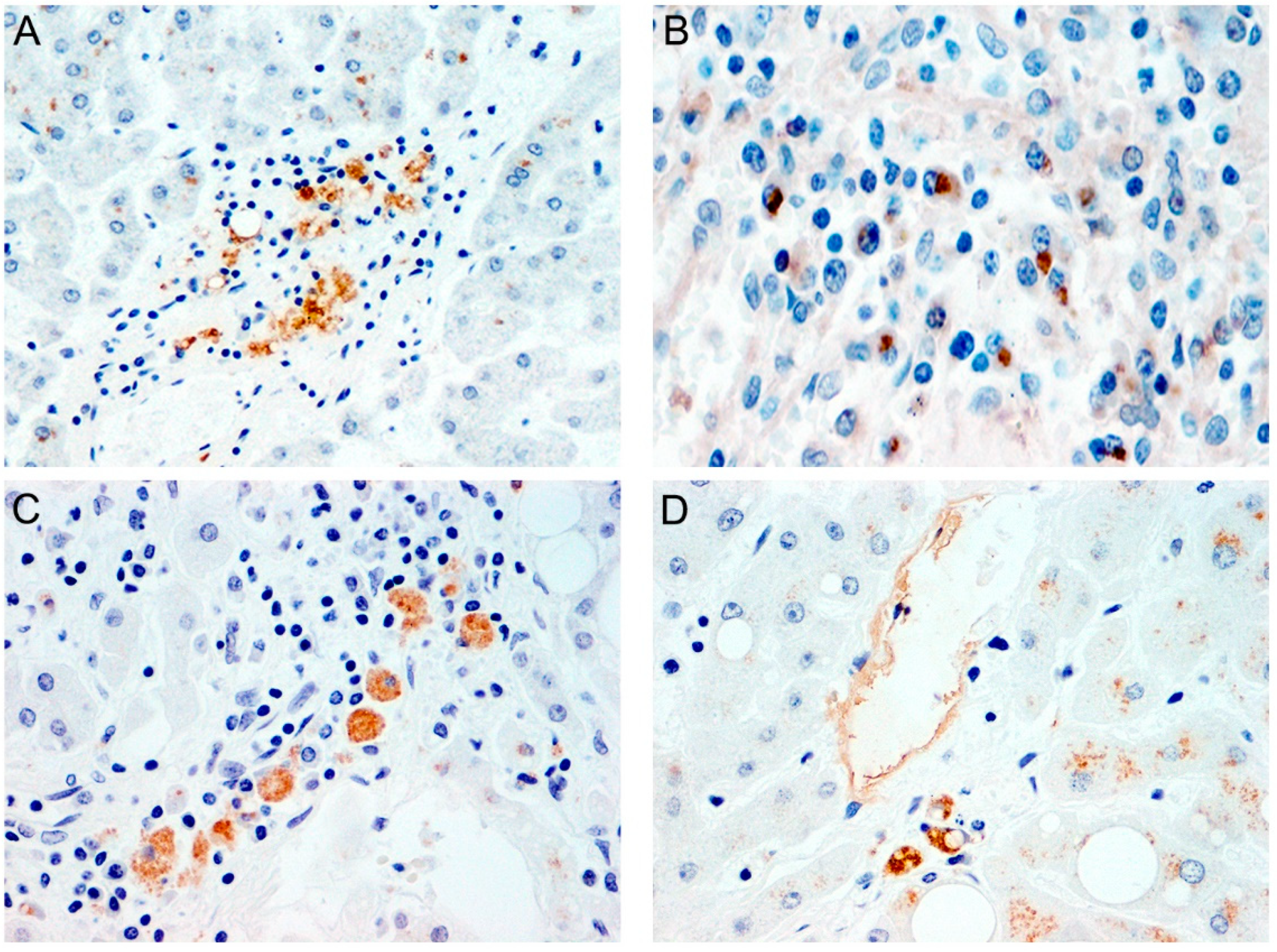{kind=link}
1. Introduction
2. Mechanisms of SARS-CoV-2 Infection in the Intestine
2.1. Viral Kinetics and Physiologic Luminal Barriers
2.2. Possible Viral Entry Mechanisms to the Intestinal Epithelial Cells
2.3. Autophagy and the Replication of SARS-CoV-2
2.4. The Role of Extracellular Vesicles/Exosomes in Viral Transmission and Spreading to the GI System
2.5. Proinflammatory Effects of COVID-19 Infection and Systemic Inflammatory Response Syndrome
2.6. Myeloid Derived Supressor Cells (MDSCs)
2.7. The Effects of COVID-19 Infection on the Coagulation System in Gastrointestinal Tract
2.8. COVID-19 and Microbiota Changes
3. Mechanisms of SARS-CoV-2 Infection in the Liver
4. Mechanisms of SARS-CoV-2 Infection in the Pancreas
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Who Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 17 November 2022).
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Aldaais, E.A.; Yegnaswamy, S.; Albahrani, F.; Alsowaiket, F.; Alramadan, S. Sequence and structural analysis of COVID-19 E and M proteins with MERS virus E and M proteins—A comparative study. Biochem. Biophys. Rep. 2021, 26, 101023. [Google Scholar] [CrossRef]
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 genome-encoded proteins. Sci. China Life Sci. 2022, 65, 280–294. [Google Scholar] [CrossRef]
- Rauti, R.; Shahoha, M.; Leichtmann-Bardoogo, Y.; Nasser, R.; Paz, E.; Tamir, R.; Miller, V.; Babich, T.; Shaked, K.; Ehrlich, A.; et al. Effect of SARS-CoV-2 proteins on vascular permeability. eLife 2021, 10, e69314. [Google Scholar] [CrossRef]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef]
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55, 2000607. [Google Scholar] [CrossRef] [Green Version]
- Tao, K.; Tzou, P.L.; Nouhin, J. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- He, X.; Lau, E.H.Y.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020, 26, 672–675. [Google Scholar] [CrossRef]
- Chavez, S.; Long, B.; Koyfman, A.; Liang, S.Y. Coronavirus Disease (COVID-19): A primer for emergency physicians. Am. J. Emerg. Med. 2021, 44, 220–229. [Google Scholar] [CrossRef]
- Long, B.; Carius, B.M.; Chavez, S.; Liang, S.Y.; Brady, W.J.; Koyfman, A.; Gottlieb, M. Clinical update on COVID-19 for the emergency clinician: Presentation and evaluation. Am. J. Emerg. Med. 2022, 54, 46–57. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lentz, S.; Roginski, M.A.; Montrief, T.; Ramzy, M.; Gottlieb, M.; Long, B. Initial emergency department mechanical ventilation strategies for COVID-19 hypoxemic respiratory failure and ARDS. Am. J. Emerg. Med. 2020, 38, 2194–2202. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardener, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Inf. Dis. 2022, 20, 533–534. [Google Scholar] [CrossRef]
- Mishchenko, E.L.; Va, V.A.I. Replication-transcription complex of coronaviruses: Functions of individual viral non-structural subunits, properties and architecture of their complexes. Vavilovskii Zhurnal Genet. Sel. 2022, 26, 121–127. [Google Scholar] [CrossRef]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [Green Version]
- Wolff, G.; Melia, C.E.; Snijder, E.J.; Barcena, M. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. 2020, 28, 1022–1033. [Google Scholar] [CrossRef]
- Mohamadian, M.; Chiti, H.; Shoghli, A.; Biglari, S.; Parsamanesh, N.; Esmaeilzadeh, A. COVID-19: Virology, biology and novel laboratory diagnosis. J. Gene Med. 2021, 23, e3303. [Google Scholar] [CrossRef]
- Woodby, B.; Arnold, M.M.; Valacchi, G. SARS-CoV-2 infection, COVID-19 pathogenesis, and exposure to air pollution: What is the connection? Ann. N. Y. Acad. Sci. 2021, 1486, 15–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Chen, R.; Ren, L.; Min, J.; Deng, F.; et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. 2021, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Ondruschka, B.; Heinrich, F.; Lindenmeyer, M.; Edler, C.; Möbius, D.; Czogalla, J.; Heinemann, A.; Braun, F.; Aepfelbacher, M.; Lütgehetmann, M.; et al. Multiorgan tropism of SARS-CoV-2 lineage B.1.1.7. Int. J. Leg. Med. 2021, 135, 2347–2349. [Google Scholar] [CrossRef] [PubMed]
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, S.; Beretta, P.; Morbini, P. Direct endothelial damage and vasculitis due to SARS-CoV-2 in small bowel submucosa of COVID-19 patient with diarrhea. J. Med. Virol. 2021, 93, 61–63. [Google Scholar] [CrossRef]
- Iba, T.; Connors, J.M.; Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm. Res. 2020, 69, 1181–1189. [Google Scholar] [CrossRef]
- Katneni, U.K.; Alexaki, A.; Hunt, R.C.; Schiller, T.; DiCuccio, M.; Buehler, P.W.; Ibla, J.C.; Kimchi-Sarfaty, C. Coagulopathy and Thrombosis as a Result of Severe COVID-19 Infection: A Microvascular Focus. Thromb. Haemost. 2020, 120, 1668–1679. [Google Scholar] [CrossRef]
- Xu, J.; Chu, M.; Zhong, F.; Tan, X.; Tang, G.; Mai, J.; Lai, N.; Guan, C.; Liang, Y.; Liao, G. Digestive symptoms of COVID-19 and expression of ACE2 in digestive tract organs. Cell Death Discov. 2020, 6, 76. [Google Scholar] [CrossRef]
- Zhang, H.; Kang, Z.; Gong, H.; Xu, D.; Wang, J.; Li, Z.; Li, Z.; Cui, X.; Xiao, J.; Zhan, J.; et al. Digestive system is a potential route of COVID-19: An analysis of single-cell coexpression pattern of key proteins in viral entry process. Gut 2020, 69, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Aguila, E.J.T.; Cua, I.H.Y.; Dumagpi, J.E.L.; Francisco, C.P.D.; Raymundo, N.T.V.; Sy-Janairo, M.L.L.; Cabral-Prodigalidad, P.A.I.; Lontok, M.A.D. COVID-19 and its effects on the digestive system and endoscopy practice. JGH Open 2020, 4, 324–331. [Google Scholar] [CrossRef]
- Munne, K.; Bhanothu, V.; Bhor, V.; Patel, V.; Mahale, S.D.; Pande, S. Detection of SARS-CoV-2 infection by RT-PCR test: Factors influencing interpretation of results. Virusdisease 2021, 32, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; Paul van Schayck, J.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.H.; Ni, W.; Wu, Q.; Li, W.J.; Li, G.J.; Wang, W.D.; Tong, J.N.; Song, X.F.; Wing-Kin Wong, G.; Xing, Q.S. Prolonged viral shedding in feces of pediatric patients with coronavirus disease 2019. J. Microbiol. Immunol. Infect. 2020, 53, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, S.; Delgado Fuentes, A.; Fetterman, A.D. Recurrent Acute Pancreatitis in a Patient with COVID-19 Infection. Am. J. Case Rep. 2020, 21, e927076. [Google Scholar] [CrossRef] [PubMed]
- Kipkorir, V.; Cheruiyot, I.; Ngure, B.; Misiani, M.; Munguti, J. Prolonged SARS-CoV-2 RNA detection in anal/rectal swabs and stool specimens in COVID-19 patients after negative conversion in nasopharyngeal RT-PCR test. J. Med. Virol. 2020, 92, 2328–2331. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Hsu, M.T.; Lee, M.Y.; Chou, C.K. Gastrointestinal Involvement in SARS-CoV-2 Infection. Viruses 2022, 14, 1188. [Google Scholar] [CrossRef]
- Amirian, E.S. Potential fecal transmission of SARS-CoV-2: Current evidence and implications for public health. Int. J. Infect. Dis. 2020, 95, 363–370. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, L.; Deng, Q.; Zhang, G.; Wu, K.; Ni, L.; Yang, Y.; Liu, B.; Wang, W.; Wei, C.; et al. The presence of SARS-CoV-2 RNA in the feces of COVID-19 patients. J. Med. Virol. 2020, 92, 833–840. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.L.; Baluja, M.Q.; Graham, D.W.; Corbishley, A.; McDonald, J.E.; Malham, S.K.; Hillary, L.S.; Connor, T.R.; Gaze, W.H.; Moura, I.B.; et al. Shedding of SARS-CoV-2 in feces and urine and its potential role in person-to-person transmission and the environment-based spread of COVID-19. Sci. Total Environ. 2020, 749, 141364. [Google Scholar] [CrossRef]
- Peng, L.; Liu, J.; Xu, W.; Luo, Q.; Chen, D.; Lei, Z.; Huang, Z.; Li, X.; Deng, K.; Lin, B.; et al. SARS-CoV-2 can be detected in urine, blood, anal swabs, and oropharyngeal swabs specimens. J. Med. Virol. 2020, 92, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Rong, L.; Nian, W.; He, Y. Review article: Gastrointestinal features in COVID-19 and the possibility of faecal transmission. Aliment. Pharmacol. Ther. 2020, 51, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.W.X.; Tan, Y.K.; Chia, P.Y.; Lee, T.H.; Ng, O.T.; Wong, M.S.Y.; Marimuthu, K. Air, Surface Environmental, and Personal Protective Equipment Contamination by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) from a Symptomatic Patient. JAMA 2020, 323, 1610–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avgeris, M.; Adamopoulos, P.G.; Galani, A.; Xagorari, M.; Gourgiotis, D.; Trougakos, I.P.; Voulgaris, N.; Dimopoulos, M.A.; Thomaidis, N.S.; Scorilas, A. Novel Nested-Seq Approach for SARS-CoV-2 Real-Time Epidemiology and In-Depth Mutational Profiling in Wastewater. Int. J. Mol. Sci. 2021, 22, 8498. [Google Scholar] [CrossRef]
- Itarte, M.; Bofill-Mas, S.; Martínez-Puchol, S.; Torrell, H.; Ceretó, A.; Carrasco, M.; Forés, E.; Canela, N.; Girones, R.; Rusiñol, M. Looking for a needle in a haystack. SARS-CoV-2 variant characterization in sewage. Curr. Opin. Environ. Sci. Health 2021, 24, 100308. [Google Scholar] [CrossRef]
- Anderson, E.J.; Weber, S.G. Rotavirus infection in adults. Lancet Infect. Dis. 2004, 4, 91–99. [Google Scholar] [CrossRef]
- Atmar, R.L.; Opekun, A.R.; Gilger, M.A.; Estes, M.K.; Crawford, S.E.; Neill, F.H.; Graham, D.Y. Norwalk virus shedding after experimental human infection. Emerg. Infect. Dis. 2008, 14, 1553–1557. [Google Scholar] [CrossRef]
- De Graaf, M.; Beck, R.; Caccio, S.M.; Duim, B.; Fraaij, P.; Le Guyader, F.S.; Lecuit, M.; Le Pendu, J.; de Wit, E.; Schultsz, C. Sustained fecal-oral human-to-human transmission following a zoonotic event. Curr. Opin. Virol. 2017, 22, 1–6. [Google Scholar] [CrossRef]
- Ma, C.; Cong, Y.; Zhang, H. COVID-19 and the Digestive System. Am. J. Gastroenterol. 2020, 115, 1003–1006. [Google Scholar] [CrossRef]
- Yeo, C.; Kaushal, S.; Yeo, D. Enteric involvement of coronaviruses: Is faecal-oral transmission of SARS-CoV-2 possible? Lancet Gastroenterol. Hepatol. 2020, 5, 335–337. [Google Scholar] [CrossRef]
- Huang, N.; Pérez, P.; Kato, T.; Mikami, Y.; Okuda, K.; Gilmore, R.C.; Conde, C.D.; Gasmi, B.; Stein, S.; Beach, M.; et al. SARS-CoV-2 infection of the oral cavity and saliva. Nat. Med. 2021, 27, 892–903. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.; Tsang, O.T.; Yip, C.C.; Chan, K.H.; Wu, T.C.; Chan, J.M.; Leung, W.S.; Chik, T.S.; Choi, C.Y.; Kandamby, D.H.; et al. Consistent Detection of 2019 Novel Coronavirus in Saliva. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2020, 71, 841–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeang, K.-T. HIV-1: Molecular Biology and Pathogenesis Viral Mechanisms, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 55. [Google Scholar]
- Hariyanto, T.I.; Prasetya, I.B.; Kurniawan, A. Proton pump inhibitor use is associated with increased risk of severity and mortality from coronavirus disease 2019 (COVID-19) infection. Dig. Liver Dis. 2020, 52, 1410–1412. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Sloan, M.E.; Owings, A.H.; Figgins, E.; Gauthier, J.; Gharaibeh, R.; Robinson, T.; Williams, H.; Sindel, C.B.; Backus, F.; et al. Increased ACE2 Levels and Mortality Risk of Patients With COVID-19 on Proton Pump Inhibitor Therapy. Am. J. Gastroenterol. 2021, 116, 1638–1645. [Google Scholar] [CrossRef]
- Yozgat, A.; Kasapoğlu, B.; Can, G.; Tanoğlu, A.; Sakin, Y.S.; Yalçın, K.S.; Gürler, M.; Kaplan, M.; Kaban, M.G.; Kırsoy, M.; et al. Long-term proton pump inhibitor use is a risk factor for mortality in patients hospitalized for COVID-19. Turk. J. Med. Sci. 2021, 51, 1675–1681. [Google Scholar] [CrossRef]
- Martinsen, T.C.; Fossmark, R.; Waldum, H.L. The Phylogeny and Biological Function of Gastric Juice-Microbiological Consequences of Removing Gastric Acid. Int. J. Mol. Sci. 2019, 20, 6031. [Google Scholar] [CrossRef] [Green Version]
- Atlas, H.P. Tissue Expression of ace2—Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000130234-ACE2/tissue (accessed on 17 November 2022).
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef]
- Lee, J.J.; Kopetz, S.; Vilar, E.; Shen, J.P.; Chen, K.; Maitra, A. Relative Abundance of SARS-CoV-2 Entry Genes in the Enterocytes of the Lower Gastrointestinal Tract. Genes 2020, 11, 645. [Google Scholar] [CrossRef]
- Flint, S.J. (Ed.) Principles of Virology, 3rd ed.; ASM Press: Washington, DC, USA, 2009; Volume 2, p. 419. [Google Scholar]
- Siebers, A.; Finlay, B.B. M cells and the pathogenesis of mucosal and systemic infections. Trends Microbiol. 1996, 4, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Hörnich, B.F.; Großkopf, A.K.; Schlagowski, S.; Tenbusch, M.; Kleine-Weber, H.; Neipel, F.; Stahl-Hennig, C.; Hahn, A.S. SARS-CoV-2 and SARS-CoV Spike-Mediated Cell-Cell Fusion Differ in Their Requirements for Receptor Expression and Proteolytic Activation. J. Virol. 2021, 95, e00002-21. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Lin, E.; He, L.; Yu, J.; Tan, P.; Zhou, Y. Autophagy and Viral Infection. Adv. Exp. Med. Biol. 2019, 1209, 55–78. [Google Scholar] [CrossRef]
- Rautou, P.E.; Cazals-Hatem, D.; Feldmann, G.; Mansouri, A.; Grodet, A.; Barge, S.; Martinot-Peignoux, M.; Duces, A.; Bièche, I.; Lebrec, D.; et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am. J. Pathol. 2011, 178, 2708–2715. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Aydin, Y.; Koksal, A.R.; Reddy, V.; Lin, D.; Osman, H.; Heidari, Z.; Rhadhi, S.M.; Wimley, W.C.; Parsi, M.A.; Dash, S. Extracellular Vesicle Release Promotes Viral Replication during Persistent HCV Infection. Cells 2021, 10, 984. [Google Scholar] [CrossRef] [PubMed]
- Shima Fakher, P.P.; Ghavami, S.; Mokarram, P. The Role of Autophagy in Respiratory Complications of COVID-19. Shiraz E-Med. J. 2020, 21, 3. [Google Scholar] [CrossRef] [Green Version]
- Foerster, E.G.; Mukherjee, T.; Cabral-Fernandes, L.; Rocha, J.D.B.; Girardin, S.E.; Philpott, D.J. How autophagy controls the intestinal epithelial barrier. Autophagy 2022, 18, 86–103. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.J.; Britton, P. Involvement of autophagy in coronavirus replication. Viruses 2012, 4, 3440–3451. [Google Scholar] [CrossRef]
- Shang, C.; Zhuang, X.; Zhang, H.; Li, Y.; Zhu, Y.; Lu, J.; Ge, C.; Cong, J.; Li, T.; Li, N.; et al. Inhibition of Autophagy Suppresses SARS-CoV-2 Replication and Ameliorates Pneumonia in hACE2 Transgenic Mice and Xenografted Human Lung Tissues. J. Virol. 2021, 95, e01537-21. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Saramago, M.; Costa, V.G.; Souza, C.S.; Bárria, C.; Domingues, S.; Viegas, S.C.; Lousa, D.; Soares, C.M.; Arraiano, C.M.; Matos, R.G. The nsp15 Nuclease as a Good Target to Combat SARS-CoV-2: Mechanism of Action and Its Inactivation with FDA-Approved Drugs. Microorganisms 2022, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Habibzadeh, P.; Dastsooz, H.; Eshraghi, M.; Łos, M.J.; Klionsky, D.J.; Ghavami, S. Autophagy: The Potential Link between SARS-CoV-2 and Cancer. Cancers 2021, 13, 5721. [Google Scholar] [CrossRef]
- Barbati, C.; Celia, A.I.; Colasanti, T.; Vomero, M.; Speziali, M.; Putro, E.; Buoncuore, G.; Savino, F.; Colafrancesco, S.; Ucci, F.M.; et al. Autophagy Hijacking in PBMC From COVID-19 Patients Results in Lymphopenia. Front. Immunol. 2022, 13, 903498. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Falzarano, D.; Gerdts, V.; Liu, Q. Construction of a Noninfectious SARS-CoV-2 Replicon for Antiviral-Drug Testing and Gene Function Studies. J. Virol. 2021, 95, e0068721. [Google Scholar] [CrossRef]
- Kumar, S.; Kashyap, P.; Chowdhury, S.; Kumar, S.; Panwar, A.; Kumar, A. Identification of phytochemicals as potential therapeutic agents that binds to Nsp15 protein target of coronavirus (SARS-CoV-2) that are capable of inhibiting virus replication. Phytomed. Int. J. Phytother. Phytopharm. 2021, 85, 153317. [Google Scholar] [CrossRef]
- Koepke, L.; Hirschenberger, M.; Hayn, M.; Kirchhoff, F.; Sparrer, K.M. Manipulation of autophagy by SARS-CoV-2 proteins. Autophagy 2021, 17, 2659–2661. [Google Scholar] [CrossRef]
- Su, W.Q.; Yu, X.J.; Zhou, C.M. SARS-CoV-2 ORF3a Induces Incomplete Autophagy via the Unfolded Protein Response. Viruses 2021, 13, 2467. [Google Scholar] [CrossRef]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2021, 56, 427–442.e425. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.; Wang, X.; Zhu, Y.; Wang, W.; Wang, Y.; Hu, G.; Liu, C.; Li, J.; Ren, S.; Xiao, M.Z.X.; et al. ORF3a-Mediated Incomplete Autophagy Facilitates Severe Acute Respiratory Syndrome Coronavirus-2 Replication. Front. Cell Dev. Biol. 2021, 9, 716208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Ι. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e1514. [Google Scholar] [CrossRef]
- Samimi, N.; Farjam, M.; Klionsky, D.J.; Rezaei, N. The role of autophagy in the pathogenesis of SARS-CoV-2 infection in different cell types. Autophagy 2022, 18, 1728–1731. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Shin, D.M.; Ramakrishna, L.; Goussetis, D.J.; Platanias, L.C.; Xiong, H.; Morse, H.C., III; Ozato, K. IRF8 directs stress-induced autophagy in macrophages and promotes clearance of Listeria monocytogenes. Nat. Commun. 2015, 6, 6379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorman, H.R.; Reategui, Y.; Poschel, D.B.; Liu, K. IRF8: Mechanism of Action and Health Implications. Cells 2022, 11, 2630. [Google Scholar] [CrossRef]
- Tomić, S.; Đokić, J.; Stevanović, D.; Ilić, N.; Gruden-Movsesijan, A.; Dinić, M.; Radojević, D.; Bekić, M.; Mitrović, N.; Tomašević, R.; et al. Reduced Expression of Autophagy Markers and Expansion of Myeloid-Derived Suppressor Cells Correlate with Poor T Cell Response in Severe COVID-19 Patients. Front. Immunol. 2021, 12, 614599. [Google Scholar] [CrossRef]
- Livanos, A.E.; Jha, D.; Cossarini, F.; Gonzalez-Reiche, A.S.; Tokuyama, M.; Aydillo, T.; Parigi, T.L.; Ladinsky, M.S.; Ramos, I.; Dunleavy, K.; et al. Intestinal Host Response to SARS-CoV-2 Infection and COVID-19 Outcomes in Patients with Gastrointestinal Symptoms. Gastroenterology 2021, 160, 2435–2450.e2434. [Google Scholar] [CrossRef]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Li, F.; Li, J.; Wang, P.-H.; Yang, N.; Huang, J.; Ou, J.; Xu, T.; Zhao, X.; Liu, T.; Huang, X.; et al. SARS-CoV-2 spike promotes inflammation and apoptosis through autophagy by ROS-suppressed PI3K/AKT/mTOR signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166260. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juibari, A.D.; Rezadoost, M.H. The key role of Calpain in COVID-19 as a therapeutic strategy. Inflammopharmacology 2022, 30, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Boroumand-Noughabi, S.; Khoshnegah, Z.; Amel Jamehdar, S.; Ayatollahi, H.; Sheikhi, M.; Rostami, M.; Keramati, M.R. Deregulation of the Expression of Beclin1 and Light Chain 3(LC3), Autophagy-Related Genes, in COVID-19 Patients. Med. J. Islam. Repub. Iran 2022, 36, 99. [Google Scholar] [CrossRef]
- Li, K.; Hao, Z.; Zhao, X.; Du, J.; Zhou, Y. SARS-CoV-2 infection-induced immune responses: Friends or foes? Scand. J. Immunol. 2020, 92, e12895. [Google Scholar] [CrossRef]
- De Oliveira, A.P.; Lopes, A.L.F.; Pacheco, G.; de Sá Guimarães Nolêto, I.R.; Nicolau, L.A.D.; Medeiros, J.V.R. Premises among SARS-CoV-2, dysbiosis and diarrhea: Walking through the ACE2/mTOR/autophagy route. Med. Hypotheses 2020, 144, 110243. [Google Scholar] [CrossRef]
- Mutsafi, Y.; Altan-Bonnet, N. Enterovirus Transmission by Secretory Autophagy. Viruses 2018, 10, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustafa, A.; Khalel, R.S.; Aziz, R.K. Traces of SARS-CoV-2 RNA in Peripheral Blood Cells of Patients with COVID-19. OMICS J. Integr. Biol. 2021, 25, 475–483. [Google Scholar] [CrossRef]
- Richter, E.; Al Arashi, D.; Schulte, B.; Bode, C.; Marx, B.; Aldabbagh, S.; Schlüter, C.; Kümmerer, B.M.; Oldenburg, J.; Funk, M.B.; et al. Detectable SARS-CoV-2 RNAemia in Critically Ill Patients, but Not in Mild and Asymptomatic Infections. Transfus. Med. Hemother. 2021, 48, 154–160. [Google Scholar] [CrossRef]
- Sbarigia, C.; Vardanyan, D.; Buccini, L.; Tacconi, S.; Dini, L. SARS-CoV-2 and extracellular vesicles: An intricate interplay in pathogenesis, diagnosis and treatment. Front. Nanotechnol. 2022, 4, 987034. [Google Scholar] [CrossRef]
- Altan-Bonnet, N.; Perales, C.; Domingo, E. Extracellular vesicles: Vehicles of en bloc viral transmission. Virus Res. 2019, 265, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Santiana, M.; Ghosh, S.; Ho, B.A.; Rajasekaran, V.; Du, W.-L.; Mutsafi, Y.; De Jésus-Diaz, D.A.; Sosnovtsev, S.V.; Levenson, E.A.; Parra, G.I.; et al. Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-organismal Viral Transmission. Cell Host Microbe 2018, 24, 208–220.e208. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Sampey, G.; Chung, M.C.; Bailey, C.; van Hoek, M.L.; Kashanchi, F.; Hakami, R.M. The carrying pigeons of the cell: Exosomes and their role in infectious diseases caused by human pathogens. Pathog. Dis. 2014, 71, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, S.; Bihl, J. Exosome-Mediated Transfer of ACE2 (Angiotensin-Converting Enzyme 2) from Endothelial Progenitor Cells Promotes Survival and Function of Endothelial Cell. Oxid. Med. Cell Longev. 2020, 2020, 4213541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sur, S.; Khatun, M.; Steele, R.; Isbell, T.S.; Ray, R.; Ray, R.B. Exosomes from COVID-19 Patients Carry Tenascin-C and Fibrinogen-beta in Triggering Inflammatory Signals in Cells of Distant Organ. Int. J. Mol. Sci. 2021, 22, 3184. [Google Scholar] [CrossRef]
- Kongsomros, S.; Suksatu, A.; Kanjanasirirat, P.; Manopwisedjaroen, S.; Prasongtanakij, S.; Jearawuttanakul, K.; Borwornpinyo, S.; Hongeng, S.; Thitithanyanont, A.; Chutipongtanate, S. Anti-SARS-CoV-2 Activity of Extracellular Vesicle Inhibitors: Screening, Validation, and Combination with Remdesivir. Biomedicines 2021, 9, 1230. [Google Scholar] [CrossRef]
- Feng, Y.; Ling, Y.; Bai, T.; Xie, Y.; Huang, J.; Li, J.; Xiong, W.; Yang, D.; Chen, R.; Lu, F.; et al. COVID-19 with Different Severities: A Multicenter Study of Clinical Features. Am. J. Respir. Crit. Care Med. 2020, 201, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.J.; Su, I.J.; Theron, M.; Wu, Y.C.; Lai, S.K.; Liu, C.C.; Lei, H.Y. An interferon-gamma-related cytokine storm in SARS patients. J. Med. Virol. 2005, 75, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e117. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Shao, B.; Dang, Q.; Chen, Z.; Zhou, Q.; Luo, H.; Yuan, W.; Sun, Z. Pathogenesis and Mechanism of Gastrointestinal Infection with COVID-19. Front. Immunol. 2021, 12, 674074. [Google Scholar] [CrossRef]
- Ntyonga-Pono, M.P. COVID-19 infection and oxidative stress: An under-explored approach for prevention and treatment? Pan Afr. Med. J. 2020, 35, 12. [Google Scholar] [CrossRef]
- Zhou, T.; Wu, J.; Zeng, Y.; Li, J.; Yan, J.; Meng, W.; Han, H.; Feng, F.; He, J.; Zhao, S.; et al. SARS-CoV-2 triggered oxidative stress and abnormal energy metabolism in gut microbiota. MedComm 2022, 3, e112. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, J.W.; Zhao, H.; Wang, G.Q. Cytokine release syndrome in severe COVID-19: Interleukin-6 receptor antagonist tocilizumab may be the key to reduce mortality. Int. J. Antimicrob. Agents 2020, 55, 105954. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef]
- Kuppalli, K.; Rasmussen, A.L. A glimpse into the eye of the COVID-19 cytokine storm. EBioMedicine 2020, 55, 102789. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Ichiki, T.; Yamakawa, T.; Tsuji, Y.; Kuronuma, K.; Takahashi, S.; Narimatsu, E.; Nakase, H. Impaired tryptophan metabolism in the gastrointestinal tract of patients with critical coronavirus disease 2019. Front. Med. 2022, 9, 941422. [Google Scholar] [CrossRef] [PubMed]
- Sorgdrager, F.J.H.; Naudé, P.J.W.; Kema, I.P.; Nollen, E.A.; Deyn, P.P. Tryptophan Metabolism in Inflammaging: From Biomarker to Therapeutic Target. Front. Immunol. 2019, 10, 2565. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Guo, C.; Doldan, P.; Boulant, S. Importance of Type I and III Interferons at Respiratory and Intestinal Barrier Surfaces. Front. Immunol. 2020, 11, 608645. [Google Scholar] [CrossRef]
- Lee, J.-H.; Koepke, L.; Kirchhoff, F.; Sparrer, K.M.J. Interferon antagonists encoded by SARS-CoV-2 at a glance. Med. Microbiol. Immunol. 2022, 1–7. [Google Scholar] [CrossRef]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Hsu, J.C.; Laurent-Rolle, M.; Pawlak, J.B.; Wilen, C.B.; Cresswell, P. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2101161118. [Google Scholar] [CrossRef] [PubMed]
- Low, Z.Y.; Zabidi, N.Z.; Yip, A.J.W.; Puniyamurti, A.; Chow, V.T.K.; Lal, S.K. SARS-CoV-2 Non-Structural Proteins and Their Roles in Host Immune Evasion. Viruses 2022, 14, 1991. [Google Scholar] [CrossRef]
- Oh, S.J.; Shin, O.S. SARS-CoV-2-mediated evasion strategies for antiviral interferon pathways. J. Microbiol. 2022, 60, 290–299. [Google Scholar] [CrossRef]
- Neurath, M.F. COVID-19 and immunomodulation in IBD. Gut 2020, 69, 1335–1342. [Google Scholar] [CrossRef]
- Chi, Y.; Ge, Y.; Wu, B.; Zhang, W.; Wu, T.; Wen, T.; Liu, J.; Guo, X.; Huang, C.; Jiao, Y.; et al. Serum Cytokine and Chemokine Profile in Relation to the Severity of Coronavirus Disease 2019 in China. J. Infect. Dis. 2020, 222, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, C.; Huang, F.; Yang, Y.; Wang, F.; Yuan, J.; Zhang, Z.; Qin, Y.; Li, X.; Zhao, D.; et al. Elevated plasma levels of selective cytokines in COVID-19 patients reflect viral load and lung injury. Natl. Sci. Rev. 2020, 7, 1003–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanella, G.; Capurso, G.; Burti, C.; Fanti, L.; Ricciardiello, L.; Souza Lino, A.; Boskoski, I.; Bronswijk, M.; Tyberg, A.; Krishna Kumar Nair, G.; et al. Gastrointestinal mucosal damage in patients with COVID-19 undergoing endoscopy: An international multicentre study. BMJ Open Gastroenterol. 2021, 8, e000578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.L.; Jacobsen, F.; Pepe-Mooney, B.J.; Mino-Kenudson, M.; Deshpande, V.; Shih, A.R.; Mattia, A.R.; Goessling, W.; Hwabejire, J.O.; Velmahos, G.C.; et al. Clinicopathological findings in patients with COVID-19-associated ischaemic enterocolitis. Histopathology 2021, 79, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Galanopoulos, M.; Gkeros, F.; Doukatas, A.; Karianakis, G.; Pontas, C.; Tsoukalas, N.; Viazis, N.; Liatsos, C.; Mantzaris, G.J. COVID-19 pandemic: Pathophysiology and manifestations from the gastrointestinal tract. World J. Gastroenterol. 2020, 26, 4579–4588. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Delano, M.J.; Scumpia, P.O.; Weinstein, J.S.; Coco, D.; Nagaraj, S.; Kelly-Scumpia, K.M.; O’Malley, K.A.; Wynn, J.L.; Antonenko, S.; Al-Quran, S.Z.; et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 2007, 204, 1463–1474. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Gong, W.; Song, J.; Shen, Z.; Cui, D. The paradoxical role of MDSCs in inflammatory bowel diseases: From bench to bedside. Front. Immunol. 2022, 13, 1021634. [Google Scholar] [CrossRef]
- Dean, M.J.; Ochoa, J.B.; Sanchez-Pino, M.D.; Zabaleta, J.; Garai, J.; Del Valle, L.; Wyczechowska, D.; Baiamonte, L.B.; Philbrook, P.; Majumder, R.; et al. Severe COVID-19 Is Characterized by an Impaired Type I Interferon Response and Elevated Levels of Arginase Producing Granulocytic Myeloid Derived Suppressor Cells. Front. Immunol. 2021, 12, 695972. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, M.; Segal, F.; Hartl, D. Myeloid-Derived Suppressor Cells as a Potential Biomarker and Therapeutic Target in COVID-19. Front. Immunol. 2021, 12, 697405. [Google Scholar] [CrossRef] [PubMed]
- Bedoya, S.A.; Ye, F.; Best, V.; Ramilo, O.; Ramilo, O.; Mejias, A. 2783. Expansion of Monocytic Myeloid-Derived Suppressor Cells in Infants with Severe Respiratory Syncytial Virus (RSV) Infection. Open Forum Infect. Dis. 2019, 6, S983. [Google Scholar] [CrossRef] [Green Version]
- Beliakova-Bethell, N.; Maruthai, K.; Xu, R.; Salvador, L.C.M.; Garg, A. Monocytic-Myeloid Derived Suppressor Cells Suppress T-Cell Responses in Recovered SARS-CoV-2-Infected Individuals. Front. Immunol. 2022, 13, 894543. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, M.M.; Kottyan, L.C.; Singh, H.; Pasare, C. Suppression of Inflammasome Activation by IRF8 and IRF4 in cDCs Is Critical for T Cell Priming. Cell Rep. 2020, 31, 107604. [Google Scholar] [CrossRef] [PubMed]
- Vatansever, H.S.; Becer, E. Relationship between IL-6 and COVID-19: To be considered during treatment. Future Virol. 2020, 15, 817–822. [Google Scholar] [CrossRef]
- Waight, J.D.; Netherby, C.; Hensen, M.L.; Miller, A.; Hu, Q.; Liu, S.; Bogner, P.N.; Farren, M.R.; Lee, K.P.; Liu, K.; et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J. Clin. Investig. 2013, 123, 4464–4478. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Cortegana, C.; Liró, J.; Palazón-Carrión, N.; Salamanca, E.; Sojo-Dorado, J.; de la Cruz-Merino, L.; Pascual, Á.; Rodríguez-Baño, J.; Sánchez-Margalet, V. Increased Blood Monocytic Myeloid Derived Suppressor Cells but Low Regulatory T Lymphocytes in Patients with Mild COVID-19. Viral Immunol. 2021, 34, 639–645. [Google Scholar] [CrossRef]
- Reizine, F.; Lesouhaitier, M.; Gregoire, M.; Pinceaux, K.; Gacouin, A.; Maamar, A.; Painvin, B.; Camus, C.; Le Tulzo, Y.; Tattevin, P.; et al. SARS-CoV-2-Induced ARDS Associates with MDSC Expansion, Lymphocyte Dysfunction, and Arginine Shortage. J. Clin. Immunol. 2021, 41, 515–525. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Hernandez, C.P.; Morrow, K.; Sierra, R.; Zabaleta, J.; Wyczechowska, D.D.; Ochoa, A.C. L-arginine deprivation regulates cyclin D3 mRNA stability in human T cells by controlling HuR expression. J. Immunol. 2010, 185, 5198–5204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Pribis, J.P.; Rodriguez, P.C.; Morris, S.M., Jr.; Vodovotz, Y.; Billiar, T.R.; Ochoa, J.B. The central role of arginine catabolism in T-cell dysfunction and increased susceptibility to infection after physical injury. Ann. Surg. 2014, 259, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Agrati, C.; Sacchi, A.; Bordoni, V.; Cimini, E.; Notari, S.; Grassi, G.; Casetti, R.; Tartaglia, E.; Lalle, E.; D’Abramo, A.; et al. Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ. 2020, 27, 3196–3207. [Google Scholar] [CrossRef]
- Adebayo, A.; Varzideh, F.; Wilson, S.; Gambardella, J.; Eacobacci, M.; Jankauskas, S.; Donkor, K.; Kansakar, U.; Trimarco, V.; Mone, P.; et al. L-arginine and COVID-19: An update. Nutrients 2021, 13, 3951. [Google Scholar] [CrossRef] [PubMed]
- Grassi, G.; Notari, S.; Gili, S.; Bordoni, V.; Casetti, R.; Cimini, E.; Tartaglia, E.; Mariotti, D.; Agrati, C.; Sacchi, A. Myeloid-Derived Suppressor Cells in COVID-19: The Paradox of Good. Front. Immunol. 2022, 13, 842949. [Google Scholar] [CrossRef]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef]
- Sacchi, A.; Grassi, G.; Notari, S.; Gili, S.; Bordoni, V.; Tartaglia, E.; Casetti, R.; Cimini, E.; Mariotti, D.; Garotto, G.; et al. Expansion of Myeloid Derived Suppressor Cells Contributes to Platelet Activation by L-Arginine Deprivation during SARS-CoV-2 Infection. Cells 2021, 10, 2111. [Google Scholar] [CrossRef]
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef] [Green Version]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Llitjos, J.F.; Leclerc, M.; Chochois, C.; Monsallier, J.M.; Ramakers, M.; Auvray, M.; Merouani, K. High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J. Thromb. Haemost. 2020, 18, 1743–1746. [Google Scholar] [CrossRef]
- Tan, B.K.; Mainbourg, S.; Friggeri, A.; Bertoletti, L.; Douplat, M.; Dargaud, Y.; Grange, C.; Lobbes, H.; Provencher, S.; Lega, J.C. Arterial and venous thromboembolism in COVID-19: A study-level meta-analysis. Thorax 2021, 76, 970–979. [Google Scholar] [CrossRef]
- Conway, E.M.; Mackman, N.; Warren, R.Q.; Wolberg, A.S.; Mosnier, L.O.; Campbell, R.A.; Gralinski, L.E.; Rondina, M.T.; van de Veerdonk, F.L.; Hoffmeister, K.M.; et al. Understanding COVID-19-associated coagulopathy. Nat. Rev. Immunol. 2022, 22, 639–649. [Google Scholar] [CrossRef]
- Lazzaroni, M.G.; Piantoni, S.; Masneri, S.; Garrafa, E.; Martini, G.; Tincani, A.; Andreoli, L.; Franceschini, F. Coagulation dysfunction in COVID-19: The interplay between inflammation, viral infection and the coagulation system. Blood Rev. 2021, 46, 100745. [Google Scholar] [CrossRef]
- Bunch, C.M.; Moore, E.E.; Moore, H.B.; Neal, M.D.; Thomas, A.V.; Zackariya, N.; Zhao, J.; Zackariya, S.; Brenner, T.J.; Berquist, M.; et al. Immuno-Thrombotic Complications of COVID-19: Implications for Timing of Surgery and Anticoagulation. Front. Surg. 2022, 9, 889999. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated with Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Gomez-Mesa, J.E.; Galindo-Coral, S.; Montes, M.C.; Munoz Martin, A.J. Thrombosis and Coagulopathy in COVID-19. Curr. Probl. Cardiol. 2021, 46, 100742. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Liu, F.; Han, K.; Blair, R.; Kenst, K.; Qin, Z.; Upcin, B.; Wörsdörfer, P.; Midkiff, C.C.; Mudd, J.; Belyaeva, E.; et al. SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front. Cell. Infect. Microbiol. 2021, 11, 701278. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, L.; Chew, K.Y.; Stocks, C.J.; Yordanov, T.E.; Essebier, P.; Kulasinghe, A.; Monkman, J.; dos Santos Miggiolaro, A.F.R.; Cooper, C.; de Noronha, L.; et al. Endothelial cells are not productively infected by SARS-CoV-2. Clin. Transl. Immunol. 2021, 10, e1350. [Google Scholar] [CrossRef] [PubMed]
- McCracken, I.R.; Saginc, G.; He, L.; Huseynov, A.; Daniels, A.; Fletcher, S.; Peghaire, C.; Kalna, V.; Andaloussi-Mäe, M.; Muhl, L.; et al. Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation 2021, 143, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Mendes, M.C.; Camargo Martins, A.P.; Borges, N.H.; Godoy, T.M.; Miggiolaro, A.; da Silva Dezidério, F.; Machado-Souza, C.; de Noronha, L. Endothelial Dysfunction and Thrombosis in Patients with COVID-19-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2404–2407. [Google Scholar] [CrossRef]
- Nicosia, R.F.; Ligresti, G.; Caporarello, N.; Akilesh, S.; Ribatti, D. COVID-19 Vasculopathy: Mounting Evidence for an Indirect Mechanism of Endothelial Injury. Am. J. Pathol. 2021, 191, 1374–1384. [Google Scholar] [CrossRef]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef]
- Cooper, S.L.; Boyle, E.; Jefferson, S.R.; Heslop, C.R.A. Role of the Renin-Angiotensin-Aldosterone and Kinin-Kallikrein Systems in the Cardiovascular Complications of COVID-19 and Long COVID. Int. J. Mol. Sci. 2021, 22, 8255. [Google Scholar] [CrossRef]
- Martens, C.P.; Van Mol, P.; Wauters, J.; Wauters, E.; Gangnus, T.; Noppen, B.; Callewaert, H.; Feyen, J.H.M.; Liesenborghs, L.; Heylen, E.; et al. Dysregulation of the kallikrein-kinin system in bronchoalveolar lavage fluid of patients with severe COVID-19. EBioMedicine 2022, 83, 104195. [Google Scholar] [CrossRef]
- Bonaffini, P.A.; Franco, P.N.; Bonanomi, A.; Giaccherini, C.; Valle, C.; Marra, P.; Norsa, L.; Marchetti, M.; Falanga, A.; Sironi, S. Ischemic and hemorrhagic abdominal complications in COVID-19 patients: Experience from the first Italian wave. Eur. J. Med. Res. 2022, 27, 165. [Google Scholar] [CrossRef]
- Paul, T.; Joy, A.R.; Alsoub, H.; Parambil, J.V. Case Report: Ischemic Colitis in Severe COVID-19 Pneumonia: An Unforeseen Gastrointestinal Complication. Am. J. Trop. Med. Hyg. 2021, 104, 63–65. [Google Scholar] [CrossRef]
- Wu, X.; Jing, H.; Wang, C.; Wang, Y.; Zuo, N.; Jiang, T.; Novakovic, V.A.; Shi, J. Intestinal Damage in COVID-19: SARS-CoV-2 Infection and Intestinal Thrombosis. Front. Microbiol. 2022, 13, 860931. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cao, J.; Wang, Q.; Shi, Q.; Liu, K.; Luo, Z.; Chen, X.; Chen, S.; Yu, K.; Huang, Z.; et al. D-dimer as a biomarker for disease severity and mortality in COVID-19 patients: A case control study. J. Intensive Care 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Poudel, A.; Poudel, Y.; Adhikari, A.; Aryal, B.B.; Dangol, D.; Bajracharya, T.; Maharjan, A.; Gautam, R. D-dimer as a biomarker for assessment of COVID-19 prognosis: D-dimer levels on admission and its role in predicting disease outcome in hospitalized patients with COVID-19. PLoS ONE 2021, 16, e0256744. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, L.; Wang, Y.; Dai, T.; Qin, Z.; Zhou, F.; Zhang, L. Alterations in microbiota of patients with COVID-19: Potential mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 143. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.; Zhang, F.; Liu, Q.; Li, A.Y.; Chung, A.C.; Cheung, C.P.; Tso, E.Y.; Fung, K.S.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Sun, Z.; Song, Z.-G.; Liu, C.; Tan, S.; Lin, S.; Zhu, J.; Dai, F.-H.; Gao, J.; She, J.-L.; Mei, Z.; et al. Gut microbiome alterations and gut barrier dysfunction are associated with host immune homeostasis in COVID-19 patients. BMC Med. 2022, 20, 24. [Google Scholar] [CrossRef]
- Eroğlu, İ.; Eroğlu, B.; Güven, G.S. Altered tryptophan absorption and metabolism could underlie long-term symptoms in survivors of coronavirus disease 2019 (COVID-19). Nutrition 2021, 90, 111308. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, X.; Patel, A.; Potru, R.; Azizi-Ghannad, S.; Dolinger, M.; Cao, J.; Bartholomew, C.; Mazurkiewicz, J.; Conti, D.; et al. Rapamycin Inhibition of mTOR Reduces Levels of the Na+/H+ Exchanger 3 in Intestines of Mice and Humans, Leading to Diarrhea. Gastroenterology 2015, 149, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Cumhur Cure, M.; Cure, E. Effects of the Na(+)/H(+) Ion Exchanger on Susceptibility to COVID-19 and the Course of the Disease. J. Renin-Angiotensin-Aldosterone Syst. 2021, 2021, 4754440. [Google Scholar] [CrossRef]
- He, P.; Yun, C.C. Mechanisms of the regulation of the intestinal Na+/H+ exchanger NHE3. J. Biomed. Biotechnol. 2010, 2010, 238080. [Google Scholar] [CrossRef]
- Dhar, D.; Mohanty, A. Gut microbiota and COVID-19-possible link and implications. Virus Res. 2020, 285, 198018. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Zhang, G.; Wang, X.; Guo, M.; Zeng, W.; Xu, Z.; Cao, D.; Pan, A.; Wang, Y.; Zhang, K.; et al. Analysis of the intestinal microbiota in COVID-19 patients and its correlation with the inflammatory factor IL-18. Med. Microecol. 2020, 5, 100023. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients with COVID-19 during Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e948. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bin, P.; Tao, S.; Zhu, G.; Wu, Z.; Cheng, W.; Ren, W.; Wei, H. Evaluation of the Mechanisms Underlying Amino Acid and Microbiota Interactions in Intestinal Infections Using Germ-Free Animals. Infect. Microbes Dis. 2021, 3, 79–86. [Google Scholar] [CrossRef]
- Viana, S.D.; Nunes, S.; Reis, F. ACE2 imbalance as a key player for the poor outcomes in COVID-19 patients with age-related comorbidities—Role of gut microbiota dysbiosis. Ageing Res. Rev. 2020, 62, 101123. [Google Scholar] [CrossRef]
- Hundt, M.A.; Deng, Y.; Ciarleglio, M.M.; Nathanson, M.H.; Lim, J.K. Abnormal Liver Tests in COVID-19: A Retrospective Observational Cohort Study of 1,827 Patients in a Major U.S. Hospital Network. Hepatology 2020, 72, 1169–1176. [Google Scholar] [CrossRef]
- Lei, P.; Zhang, L.; Han, P.; Zheng, C.; Tong, Q.; Shang, H.; Yang, F.; Hu, Y.; Li, X.; Song, Y. Liver injury in patients with COVID-19: Clinical profiles, CT findings, the correlation of the severity with liver injury. Hepatol. Int. 2020, 14, 733–742. [Google Scholar] [CrossRef]
- Parohan, M.; Yaghoubi, S.; Seraji, A. Liver injury is associated with severe coronavirus disease 2019 (COVID-19) infection: A systematic review and meta-analysis of retrospective studies. Hepatol. Res. 2020, 50, 924–935. [Google Scholar] [CrossRef]
- Phipps, M.M.; Barraza, L.H.; LaSota, E.D.; Sobieszczyk, M.E.; Pereira, M.R.; Zheng, E.X.; Fox, A.N.; Zucker, J.; Verna, E.C. Acute Liver Injury in COVID-19: Prevalence and Association with Clinical Outcomes in a Large U.S. Cohort. Hepatology 2020, 72, 807–817. [Google Scholar] [CrossRef]
- Fan, Z.; Chen, L.; Li, J.; Tian, C.; Zhang, Y.; Huang, S.; Liu, Z.; Cheng, J. Clinical Features of COVID-19-Related Liver Damage. medRxiv 2020. [Google Scholar] [CrossRef]
- Kulkarni, A.V.; Kumar, P.; Tevethia, H.V.; Premkumar, M.; Arab, J.P.; Candia, R.; Talukdar, R.; Sharma, M.; Qi, X.; Rao, P.N.; et al. Systematic review with meta-analysis: Liver manifestations and outcomes in COVID-19. Aliment. Pharmacol. Ther. 2020, 52, 584–599. [Google Scholar] [CrossRef] [PubMed]
- Chau, T.N.; Lee, K.C.; Yao, H.; Tsang, T.Y.; Chow, T.C.; Yeung, Y.C.; Choi, K.W.; Tso, Y.K.; Lau, T.; Lai, S.T.; et al. SARS-associated viral hepatitis caused by a novel coronavirus: Report of three cases. Hepatology 2004, 39, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zhou, X.; Qiu, Y.; Feng, F.; Feng, J.; Jia, Y.; Zhu, H.; Hu, K.; Liu, J.; Liu, Z.; et al. Clinical characteristics of 82 death cases with COVID-19. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Lagana, S.M.; Kudose, S.; Iuga, A.C.; Lee, M.J.; Fazlollahi, L.; Remotti, H.E.; Del Portillo, A.; De Michele, S.; de Gonzalez, A.K.; Saqi, A.; et al. Hepatic pathology in patients dying of COVID-19: A series of 40 cases including clinical, histologic, and virologic data. Mod. Pathol. 2020, 33, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Wanner, N.; Andrieux, G.; Badia-i-Mompel, P.; Edler, C.; Pfefferle, S.; Lindenmeyer, M.T.; Schmidt-Lauber, C.; Czogalla, J.; Wong, M.N.; Okabayashi, Y.; et al. Molecular consequences of SARS-CoV-2 liver tropism. Nat. Metab. 2022, 4, 310–319. [Google Scholar] [CrossRef]
- Cai, Q.; Huang, D.; Yu, H.; Zhu, Z.; Xia, Z.; Su, Y.; Li, Z.; Zhou, G.; Gou, J.; Qu, J.; et al. COVID-19: Abnormal liver function tests. J. Hepatol. 2020, 73, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Chu, H.K.; Bai, T. Liver damage at admission is an independent prognostic factor for COVID-19. J. Dig. Dis. 2020, 21, 512–518. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Liu, H.; Li, W.; Lin, F.; Jiang, L.; Li, X.; Xu, P.; Zhang, L.; Zhao, L.; et al. SARS-CoV-2 infection of the liver directly contributes to hepatic impairment in patients with COVID-19. J. Hepatol. 2020, 73, 807–816. [Google Scholar] [CrossRef]
- Paizis, G.; Tikellis, C.; Cooper, M.E.; Schembri, J.M.; Lew, R.A.; Smith, A.I.; Shaw, T.; Warner, F.J.; Zuilli, A.; Burrell, L.M.; et al. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut 2005, 54, 1790–1796. [Google Scholar] [CrossRef]
- McConnell, M.J.; Kondo, R.; Kawaguchi, N.; Iwakiri, Y. COVID-19 and Liver Injury: Role of Inflammatory Endotheliopathy, Platelet Dysfunction, and Thrombosis. Hepatol. Commun. 2022, 6, 255–269. [Google Scholar] [CrossRef]
- Dawood, D.R.M.; Salum, G.M.; El-Meguid, M.A. The Impact of COVID-19 on Liver Injury. Am. J. Med. Sci. 2022, 363, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Sookoian, S. SARS-CoV-2 virus and liver expression of host receptors: Putative mechanisms of liver involvement in COVID-19. Liver Int. 2020, 40, 2038–2040. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.H.L.; Zhang, Y.; Han, W.; Lu, Z.; Ke, A.; Zhou, J.; Shi, G.; Fang, N.; Fan, J.; Cai, J.; et al. Specific ACE2 Expression in Cholangiocytes May Cause Liver Damage After 2019-nCoV Infection. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Lax, S.F.; Skok, K.; Zechner, P.; Kessler, H.H.; Kaufmann, N.; Koelblinger, C.; Vander, K.; Bargfrieder, U.; Trauner, M. Pulmonary Arterial Thrombosis in COVID-19 with Fatal Outcome. Ann. Intern. Med. 2020, 173, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Shaveisi-Zadeh, F.; Nikkho, B.; Khadem Erfan, M.B.; Amiri, A.; Azizi, A.; Mansouri, N.; Tarlan, M.; Rostami-Far, Z. Changes in liver enzymes and association with prognosis in patients with COVID-19: A retrospective case-control study. J. Int. Med. Res. 2022, 50, 3000605221110067. [Google Scholar] [CrossRef]
- Diaz-Louzao, C.; Barrera-Lopez, L.; Lopez-Rodriguez, M.; Casar, C.; Vazquez-Agra, N.; Pernas-Pardavila, H.; Marques-Afonso, A.; Vidal-Vazquez, M.; Montoya, J.G.; Andrade, A.H.; et al. Longitudinal relationship of liver injury with inflammation biomarkers in COVID-19 hospitalized patients using a joint modeling approach. Sci. Rep. 2022, 12, 5547. [Google Scholar] [CrossRef]
- Sonzogni, A.; Previtali, G. Liver histopathology in severe COVID 19 respiratory failure is suggestive of vascular alterations. Liver Int. 2020, 40, 2110–2116. [Google Scholar] [CrossRef] [PubMed]
- Alkattan, W.; Yaqinuddin, A.; Shafqat, A.; Kashir, J. NET-Mediated Pathogenesis of COVID-19: The Role of NETs in Hepatic Manifestations. J. Health Allied Sci. NU 2022, 12, 235–242. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, X.; Liu, X. NETosis and Neutrophil Extracellular Traps in COVID-19: Immunothrombosis and Beyond. Front. Immunol. 2022, 13, 838011. [Google Scholar] [CrossRef]
- Liu, F.; Long, X.; Zhang, B.; Zhang, W.; Chen, X.; Zhang, Z. ACE2 Expression in Pancreas May Cause Pancreatic Damage After SARS-CoV-2 Infection. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, 18, 2128–2130.e2122. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, W.J.; Dabrowska, A.; Milewska, A.; Ziojla, N.; Blaszczyk, K.; Barreto-Duran, E.; Sanak, M.; Surmiak, M.; Owczarek, K.; Grzanka, D.; et al. SARS-CoV-2 infects an in vitro model of the human developing pancreas through endocytosis. iScience 2022, 25, 104594. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Lidsky, P.V.; Xiao, Y.; Lee, I.T.; Cheng, R.; Nakayama, T.; Jiang, S.; Demeter, J.; Bevacqua, R.J.; Chang, C.A.; et al. SARS-CoV-2 infects human pancreatic β cells and elicits β cell impairment. Cell Metab. 2021, 33, 1565–1576.e1565. [Google Scholar] [CrossRef]
- Correia de Sá, T.; Soares, C.; Rocha, M. Acute pancreatitis and COVID-19: A literature review. World J. Gastrointest. Surg. 2021, 13, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, P.; Szakacs, Z.; Sahin-Toth, M. Lipotoxicity and Cytokine Storm in Severe Acute Pancreatitis and COVID-19. Gastroenterology 2020, 159, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Kusmartseva, I.; Wu, W.; Syed, F.; Van Der Heide, V.; Jorgensen, M.; Joseph, P.; Tang, X.; Candelario-Jalil, E.; Yang, C.; Nick, H.; et al. Expression of SARS-CoV-2 Entry Factors in the Pancreas of Normal Organ Donors and Individuals with COVID-19. Cell Metab. 2020, 32, 1041–1051.e1046. [Google Scholar] [CrossRef]
- Müller, J.A.; Groß, R.; Conzelmann, C.; Krüger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat. Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef]
- Shaharuddin, S.H.; Wang, V.; Santos, R.S.; Gross, A.; Wang, Y.; Jawanda, H.; Zhang, Y.; Hasan, W.; Garcia, G.; Arumugaswami, V.; et al. Deleterious Effects of SARS-CoV-2 Infection on Human Pancreatic Cells. Front. Cell. Infect. Microbiol. 2021, 11, 678482. [Google Scholar] [CrossRef]
- Steenblock, C.; Richter, S.; Berger, I.; Barovic, M.; Schmid, J.; Schubert, U.; Jarzebska, N.; von Mässenhausen, A.; Linkermann, A.; Schürmann, A.; et al. Viral infiltration of pancreatic islets in patients with COVID-19. Nat. Commun. 2021, 12, 3534. [Google Scholar] [CrossRef]
- Qadir, M.M.F.; Bhondeley, M.; Beatty, W.; Gaupp, D.D.; Doyle-Meyers, L.A.; Fischer, T.; Bandyopadhyay, I.; Blair, R.V.; Bohm, R.; Rappaport, J.; et al. SARS-CoV-2 infection of the pancreas promotes thrombofibrosis and is associated with new-onset diabetes. JCI Insight 2021, 6, e151551. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef] [Green Version]
- Memon, B.; Abdelalim, E.M. ACE2 function in the pancreatic islet: Implications for relationship between SARS-CoV-2 and diabetes. Acta Physiol. 2021, 233, e13733. [Google Scholar] [CrossRef] [PubMed]
- Aloysius, M.M.; Thatti, A.; Gupta, A.; Sharma, N.; Bansal, P.; Goyal, H. COVID-19 presenting as acute pancreatitis. Pancreatology 2020, 20, 1026–1027. [Google Scholar] [CrossRef] [PubMed]
- Anand, E.R.; Major, C.; Pickering, O.; Nelson, M. Acute pancreatitis in a COVID-19 patient. Br. J. Surg. 2020, 107, e182. [Google Scholar] [CrossRef] [PubMed]
- Hadi, A.; Werge, M.; Kristiansen, K.T.; Pedersen, U.G.; Karstensen, J.G.; Novovic, S.; Gluud, L.L. Coronavirus Disease-19 (COVID-19) associated with severe acute pancreatitis: Case report on three family members. Pancreatology 2020, 20, 665–667. [Google Scholar] [CrossRef]
- Kataria, S.; Sharif, A.; Ur Rehman, A.; Ahmed, Z.; Hanan, A. COVID-19 Induced Acute Pancreatitis: A Case Report and Literature Review. Cureus 2020, 12, e9169. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef]
- Rao, S.A.; Kunte, A.R. Interleukin-6: An Early Predictive Marker for Severity of Acute Pancreatitis. Indian J. Crit. Care Med. Peer-Rev. Off. Publ. Indian Soc. Crit. Care Med. 2017, 21, 424–428. [Google Scholar] [CrossRef]
- Wang, F.; Wang, H.; Fan, J.; Zhang, Y.; Wang, H.; Zhao, Q. Pancreatic Injury Patterns in Patients With Coronavirus Disease 19 Pneumonia. Gastroenterology 2020, 159, 367–370. [Google Scholar] [CrossRef]
- De Oliveira, C.; Khatua, B.; Noel, P.; Kostenko, S.; Bag, A.; Balakrishnan, B.; Patel, K.S.; Guerra, A.A.; Martinez, M.N.; Trivedi, S.; et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J. Clin. Investig. 2020, 130, 1931–1947. [Google Scholar] [CrossRef]
- El-Kurdi, B.; Khatua, B.; Rood, C.; Snozek, C.; Cartin-Ceba, R.; Singh, V.P. Mortality From Coronavirus Disease 2019 Increases with Unsaturated Fat and May Be Reduced by Early Calcium and Albumin Supplementation. Gastroenterology 2020, 159, 1015–1018.e4. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khreefa, Z.; Barbier, M.T.; Koksal, A.R.; Love, G.; Del Valle, L. Pathogenesis and Mechanisms of SARS-CoV-2 Infection in the Intestine, Liver, and Pancreas. Cells 2023, 12, 262. https://doi.org/10.3390/cells12020262
Khreefa Z, Barbier MT, Koksal AR, Love G, Del Valle L. Pathogenesis and Mechanisms of SARS-CoV-2 Infection in the Intestine, Liver, and Pancreas. Cells. 2023; 12(2):262. https://doi.org/10.3390/cells12020262
Chicago/Turabian StyleKhreefa, Zaid, Mallory T. Barbier, Ali Riza Koksal, Gordon Love, and Luis Del Valle. 2023. "Pathogenesis and Mechanisms of SARS-CoV-2 Infection in the Intestine, Liver, and Pancreas" Cells 12, no. 2: 262. https://doi.org/10.3390/cells12020262