1. Introduction
Enhanced lipid metabolism in liver cancer is an important feature in the oncogenic process [
1]. Interestingly, unlike most normal cells, even highly proliferative ones, their tumoral counterparts mostly display a preference to increased endogenous fatty acid (FA) biosynthesis in spite of dietary lipid abundancy [
2]. This de novo pathway has a significant impact on the qualitative composition of the membranes via the enrichment of phospholipids with saturated and/or monounsaturated fatty acid chains since mammalian cells have a limited ability to synthesize polyunsaturated FAs (PUFAs) [
3]. Because saturated FAs are less prone to lipid peroxidation than polyunsaturated ones, by shifting lipid acquisition from external sources toward de novo lipogenesis, cancer cells acquire specific protective features in their membranes to resist oxidative stress-induced cell insults.
This context highlights the importance of the proprotein convertase subtilisin/kexin type 9, or PCSK9. Targeting PCSK9 to treat cancer becomes attractive not only because of its critical role in lipid metabolism but also because of its growing place in the recent literature, which has reported its tight association with the incidence and progression of several cancers [
4]. Moreover, there is a diversity of therapeutic approaches targeting this enzyme that have been evaluated and, in some cases, approved for clinical use to lower cholesterol levels in patients suffering from hyperlipidemia.
Indeed, PCSK9 is a serine protease that belongs to the family of proprotein convertases, mainly involved in the degradation of the hepatic low-density lipoprotein receptor (LDLR) present on hepatocytes’ surface. This function interferes with the primary pathway of LDL-cholesterol (LDL-C) uptake from the circulation, thus leading to the increase in blood cholesterol levels. PCSK9 can target other members of LDLRs such as very low-density lipoprotein receptor (VLDLR), apolipoprotein E receptor 2 (ApoER2), cluster of differentiation 36 (CD36) and CD81 [
5].
Because of its essential role in lipid metabolism, many therapeutic approaches targeting PCSK9 have been implemented in combination with other hypolipidemic drugs such as statins to treat patients with hyperlipidemia and cardiovascular diseases.
Besides monoclonal antibodies (mAbs), one of the most recent approaches is the first-in-class chemically synthesized small interfering ribonucleic acid (siRNA) against PCSK9, inclisiran (Leqivo®; Novartis, Basel, Switzerland), which was approved for use in adults suffering from heterozygous familial hypercholesterolemia (FH) or non-familial and mixed dyslipidemia in Europe in late 2020 [
6,
7,
8].
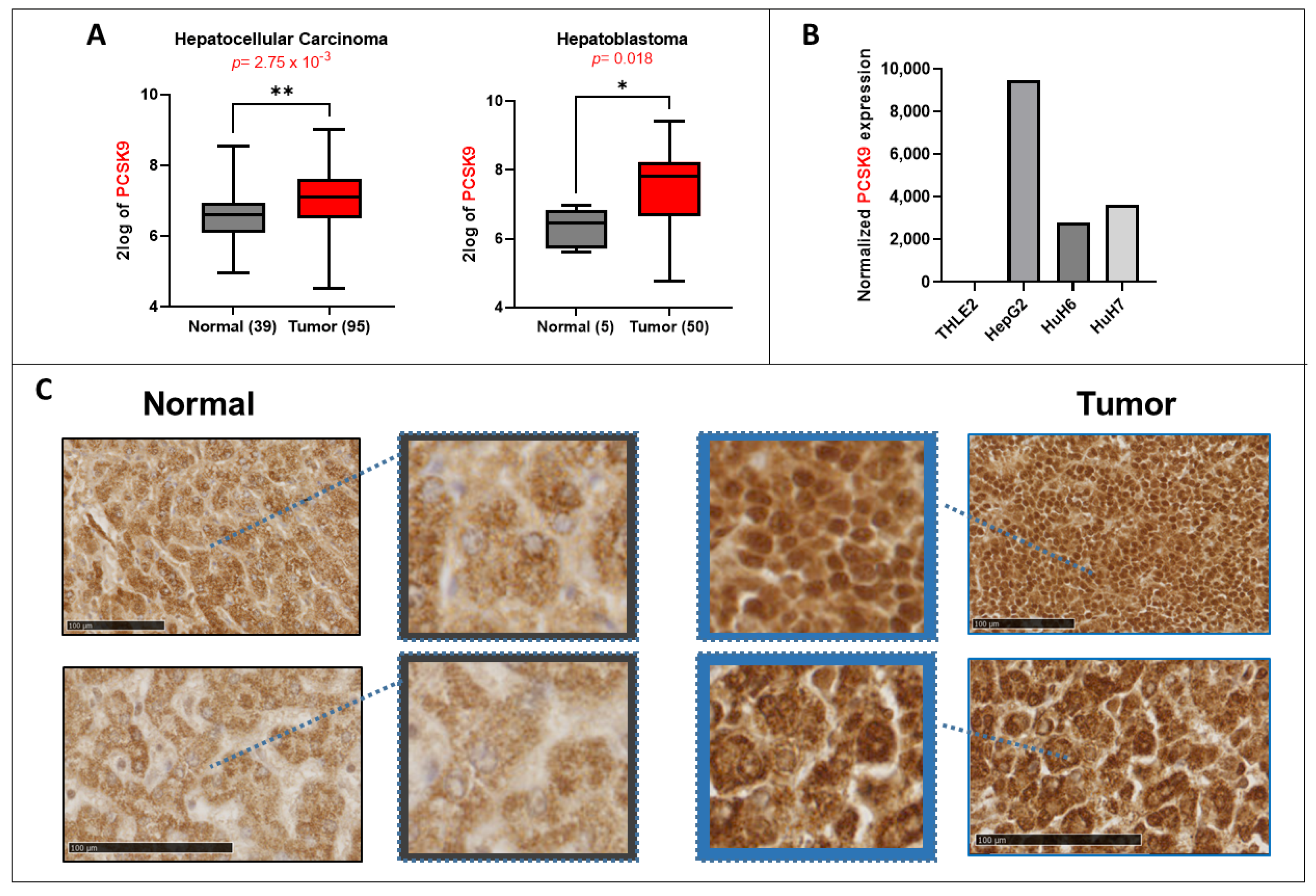
The expression of PCSK9 is deregulated in different types of cancers, including HCC neuroglioma, breast cancer, colorectal cancer and others [
4,
9], highlighting a major question about the therapeutic strategies that could be adopted to target PCSK9 in these cancers. More specifically, a very recent study reported the high expression of PCSK9 in tumor tissues in HCC patients. In this research, the expression of PCSK9 in tumors correlated with a poor prognosis after curative resection and was an independent risk factor for overall and disease-free survival. This illustrates the potential of PCSK9 as a prognostic marker for HCC [
10].
The present study aims at assessing the interest of inhibiting PCSK9 by siRNA/shRNA (short hairpin RNA) in liver cancer using different cell lines and a tumor xenograft approach. We show the critical role of this enzyme in controlling the homeostasis of lipid biosynthesis and traffic but also and for the first time, its place in maintaining redox homeostasis. Interference with this protein leads to aberrant lipid metabolism, high lipid peroxidation and the death of liver cancer cells by ferroptosis.
2. Materials and Methods
2.1. Patient Samples
All patients were recruited in accordance with European and French laws and institutional ethical guidelines. Non-tumoral, tumoral and adjacent non-tumoral livers were collected from patients treated at French university hospitals and centralized by the French HEPATOBIO network.
2.2. Transcriptomic Data Acquisition
The R2: Genomics Analysis and Visualization Platform (
http://r2.amc.nl, accessed on 23 September 2022 was used to generate the gene expression data from different available datasets. In this study, two different datasets were selected: Hepatoblastoma—López-Terrada—55- fRMA—u133p2 (GEO ID: gse75271) [
11] and Tumor HCC—Wu—134—MAS50 (GEO ID: gse45436) [
12]. Numeric data of gene expression were downloaded in Excel files and graphs were generated using GraphPad Prism 9 software (GraphPad Software, Inc., San Diego, CA, USA). In addition, the level of expression of lipid-related genes in the hepatic cell lines was generated by referring to the transcriptomic data obtained on these cell lines by Hooks et al. [
13].
2.3. RNA Sequencing Analysis
Total RNA from Huh7 and THLE-2 cell lines was extracted using the
mirVana kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the supplier’s protocol and the analysis was conducted by Hooks et al. [
13] in a manner similar to what they performed for the other cell lines and patient tissues.
2.4. Cell Culture
Human HCC (Huh7) and hepatoblastoma (HB) (HepG2, Huh6) derived cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM GlutaMAX™, Gibco, Thermo Fisher Scientific, Waltham, MA, USA, supplemented with high (4.5 g/L) for Huh7 and HepG2 or low (1 g/L) D-glucose for Huh6) supplemented with 10% fetal bovine serum (FBS), 100 µg/mL streptomycin and 100 U/mL penicillin. The cells were maintained at 37 °C in a humidified atmosphere of 5% CO2. Cell line authentication was performed in April 2021 using short tandem repeats (LGC, Molsheim, France) and the absence of mycoplasma contamination was tested on a monthly basis.
2.5. Lentivirus Production and Transduction
Lentivirus vector production was performed using the Vect’UB service platform (INSERM US 005-CNRS UMS 3427-TBM-Core, Université de Bordeaux, Bordeaux, France). Lentiviral particles were produced by transient transfection of human embryonic kidney cells (HEK293T) according to standard protocols. In brief, subconfluent HEK293T cells were co-transfected with lentiviral genome (psPAX2) (a gift from Didier Trono (Addgene plasmid # 12260)), with an envelope-coding plasmid (pMD2G-VSVG) and with vector constructs (305 pLKO-sh886 or 306 pLKO-shCTR) by calcium phosphate precipitation. LVs were harvested 48 h post-transfection and concentrated by ultrafiltration. Viral titers of VSV-g pseudotype pLV lentivectors were determined by transducing the HEK293T cells with serial dilutions of viral supernatant and lentiviral integration was evaluated by quantitative PCR using RRE primers. The following forward (F) and reverse (R) sequences of shPCSK9-886 were used:
F-5′ CCGGGGGTCATGGTCACCGACTTCGCTCGAGCGAAGTCGGTGACCATGACCCTTTTT-3′ and R-5′ AATTCAAAAAGGGTCATGGTCACCGACTTCGCTCGAGCGAAGTCGGTGACCATGACCC 3′. The hairpin sequence of negative control shRNA is:
HepG2 cells stably expressing a tomato transgene were generated by lentivirus transduction at a multiplicity of infection (MOI) of 10 with an MND-Tomato-265 virus (donated by the Vect’UB platform). Red fluorescent cells were sorted by Fluorescence-Activated Cell Sorting (FACS). Stable inhibition of PCSK9 expression was induced by cell transduction with the lentivirus 305 pLKO-sh886 (shPCSK9) or the control (306 pLKO-shCTR) at an MOI of 10. Transduced cells were selected using puromycin (P8833, Sigma-Aldrich, St. Louis, MO, USA) at 3 μg/mL.
2.6. siRNA Transfection
Small interfering siRNAs (si1 {sense: 5′ GUGCUCAACUGCCAAGGGA[dT][dT] 3′; anti-sense: 5′ UCCCUUGGCAGUUGAGCAC[dT][dT] 3′} and si2 {sense: 5′ GGGUCAUGGUCACCGACUU[dT][dT] 3′; anti-sense: 5′ AAGUCGGUGACCAUGACCC[dT][dT] 3′}) against PCSK9 (Sigma-Aldrich, St. Louis, MO, USA) were diluted in 1X siMAX dilution buffer (30 mM HEPES, 100 mM KCl, 1 mM MgCl2, pH 7.3, Eurofins, Luxembourg). Hepatic cancer cells were transfected independently with 20 nM si1 or 2 or control siCTR (AllStars Negative Control siRNA, Qiagen, Hilden, Germany) using a lipofectamine RNAi MAX transfection reagent (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions for reverse transfection. For transfection, lipofectamine RNAi MAX was diluted by 1/100th in a transfection medium (OptiMEM, GibcoTM, Thermo Fisher Scientific, Waltham, MA, USA).
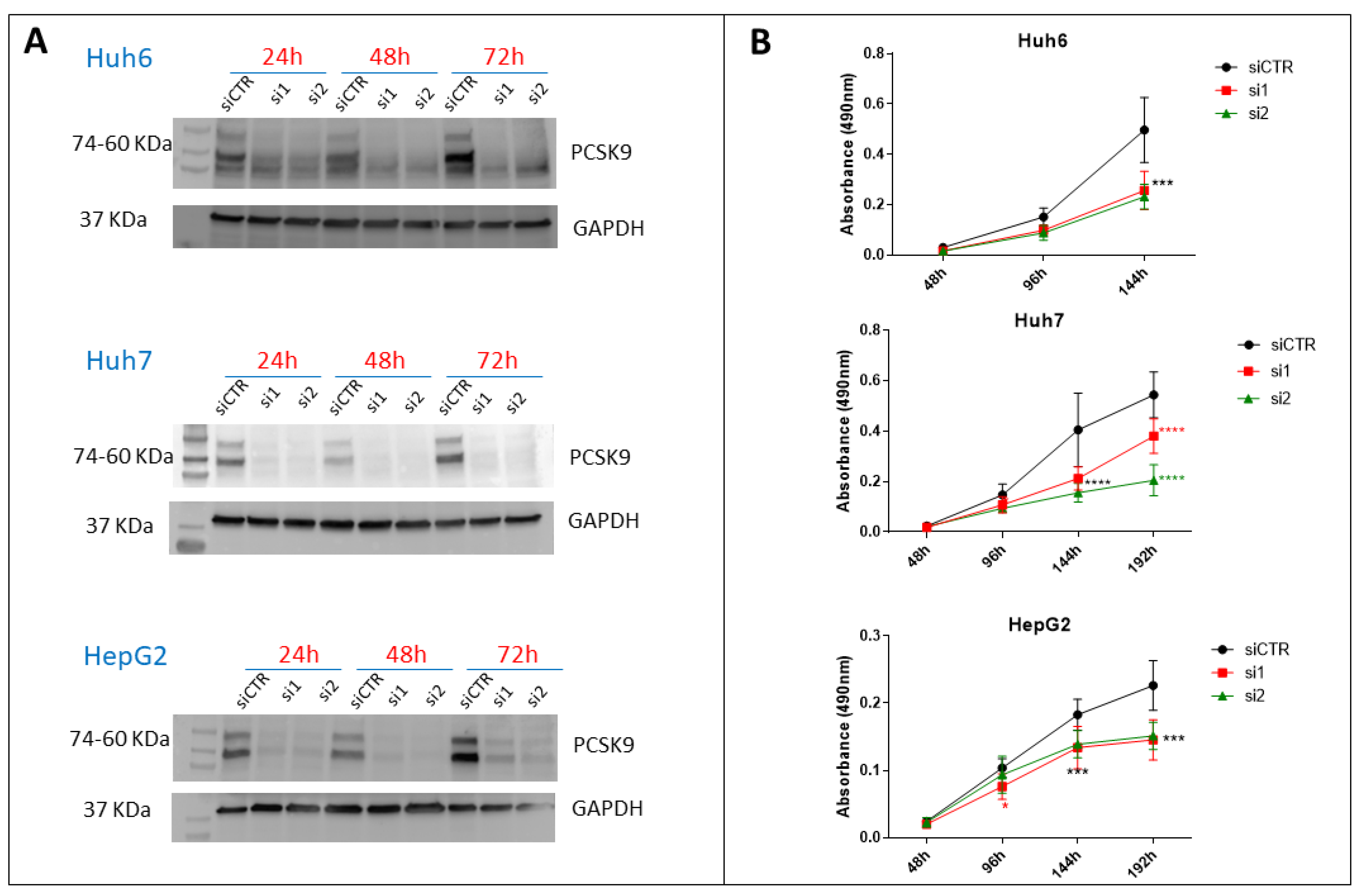
2.7. Proliferation Assay
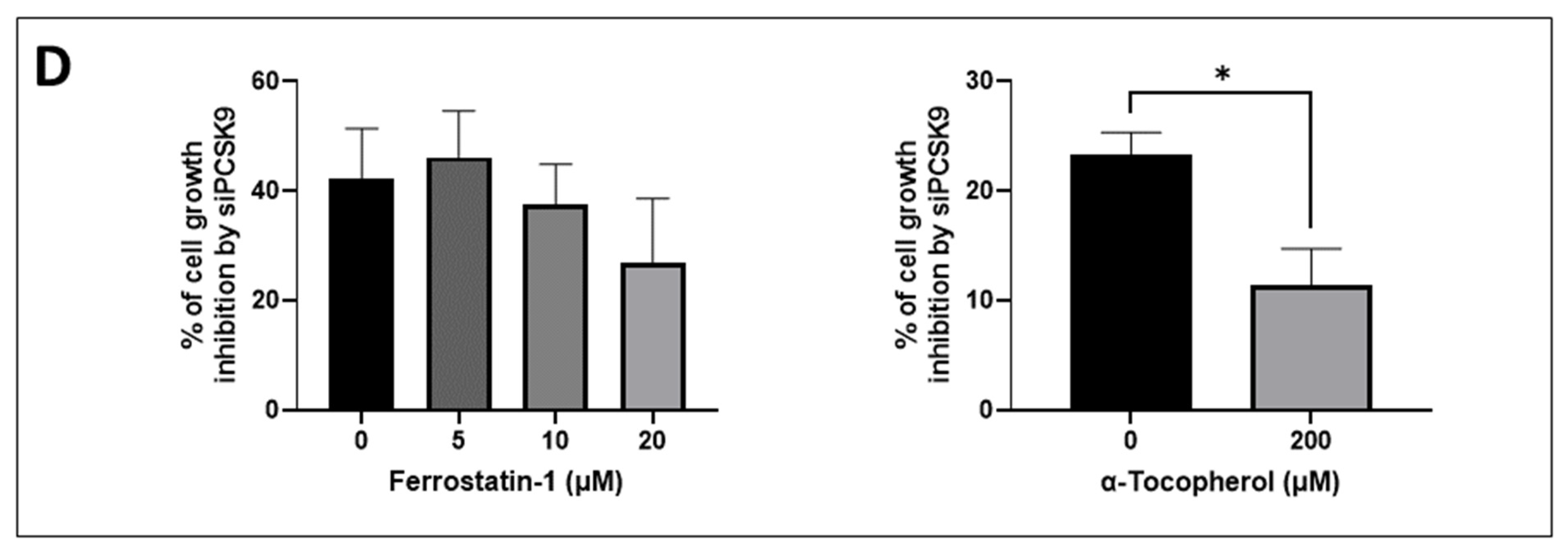
Cells were transfected with siRNAs for 24 h before seeding them into 96-well plates in triplicates at various densities (3000 cells/well for Huh7, 2000–2500 cells/well for HepG2, 700–2000 cells/well for Huh6) and then treated with various concentrations of ferrostatin-1 (0–20 µM) or α-tocopherol (0–200 µM). The experiment was followed for different time intervals, during which a fresh medium with the drug was changed every 2 days. The proliferation of cells was assessed using CellTiter 96® AQueous One Solution Reagent (Promega, Madison, WI, USA) and the absorbance was recorded at 490 nm using ClarioStar (BMG Labtech, Champigny-sur-Marne, France).
2.8. Western Blot
Cells were lysed in RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA) supplemented with protease and phosphatase inhibitor cocktails (Roche, Basel, Switzerland) and centrifuged at 13,000 rpm for 15 min at 4 °C. Protein concentration was determined using the Pierce™ BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Approximately 40 μg of proteins were loaded per lane for Western blot analyses in 4–15% precast polyacrylamide gel (Bio-Rad, Hercules, CA, USA) and blotted onto 0.2 μm nitrocellulose membrane (Bio-Rad, Hercules, CA, USA). The membranes were blocked in 5% BSA in TBST (20 mM Tris, 150 mM NaCl, 0.1% Tween 20), then incubated with each of the following specific primary antibodies: a sheep anti-PCSK9 (1 μg/mL, AF3888, R&D Systems, Minneapolis, MN, USA), ferroptosis antibody sampler kit (Cell signaling, 29650), rabbit anti-HO-1/HMOX1 (1:3000, 10701-1-AP, ProteinTech Group, Inc., Rosemont, IL, USA), mouse anti-NQO1 (1:7000, 67240-1-Ig, ProteinTech Group, Inc., Rosemont, IL, USA), mouse anti-GAPDH HRP conjugated (1:10,000, BLE649203, BioLegend, San Diego, CA, USA) and goat anti-vinculin (1:1000, sc-7649, Santa Cruz Biotechnology, Dallas, TX, USA) overnight at 4 °C. After incubation with the appropriate secondary antibody coupled with horseradish peroxidase (goat anti-mouse IgG (H+L) HRP, 1:3000, 170-6516, Bio-Rad; rabbit anti-goat IgG HRP, 1:1000, HAF017, R&D Systems; goat anti-rabbit IgG HRP, 1:5000, A0545, Sigma-Aldrich; rabbit anti-sheep IgG HRP, 1:3000, 402100, Calbiochem, San Diego, CA, USA), all blots were revealed with Fusion FX (Vilber Lourmat, Marne-la-Vallée, France) following incubation with the ECL reagents from Bio-Rad. Quantification was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
2.9. Lipidomics
The extraction of lipids from cell pellets was performed using chloroform:methanol (2:1, v/v), following the original Folch method, with a CHCl3:MeOH:H2O ratio of 8:4:3 (v/v). For this purpose, 0.5 mL methanol and 1 mL chloroform were added directly to the cell pellet. The suspension was incubated for 30 min on ice with repeated vortexing. After the addition of water to separate the aqueous and organic phases, the mixture was incubated on ice for an additional 10 min. The samples were then centrifuged at 1000× g for 5 min. The organic phase was transferred to a new tube. The aqueous layer was re-extracted with 2 mL chloroform:methanol (2:1, v/v). The chloroform layers were combined, evaporated to dryness and resuspended in 100 µL chloroform:methanol (1:1, v/v).
Phospholipids were analyzed by loading 25 µL of total lipids onto HPTLC plates (60F254, Merck, Darmstadt, Germany) and developed with methyl acetate/n-propanol/chloroform/methanol/0.25% aqueous KCl (5:5:5:2:1.8, v/v) as solvent. Neutral lipids were analyzed by loading 25 µL of total lipids onto HPTLC plates and developed with hexane/ethyl ether/formic acid (10:5:0.5, v/v).
For lipid quantification, the plates were then immersed in a copper acetate solution (3% copper acid + 8% phosphoric acid in distilled water) and heated at 115 °C for 30 min. Lipids were identified by co-migration with known standards and quantified by densitometric analysis using a TLC scanner (CAMAG, Muttenz, Switzerland).
2.10. Radiolabeling Experiment
For radiolabeling experiments, the counted cells of each sample were transferred to a glass tube in 6 mL of DMEM medium. To start the reaction, 200 nmol (10 µCi) of [1-14C] acetate (PerkinElmer Life Sciences, Waltham, MA, USA) were added to each tube and the tubes were incubated at 37 °C in 5% CO2. The uptake of acetate was studied for each sample at 3 different time points (1 h, 2 h and 4 h). To stop the reaction, the samples were centrifuged at 1000× g for 5 min and the supernatants were removed. After the addition of 2 mL chloroform/methanol (2:1, v/v), the cells were incubated overnight at −20 °C. To separate the aqueous and organic phases, 1 mL of 0.9% NaCl was added and the mixtures were centrifuged at 1000× g for 5 min. The organic phases were transferred to a new tube. The aqueous layer was re-extracted with 2 mL chloroform/methanol (2:1, v/v). The chloroform layers were combined and washed one time with 1 mL 0.9% NaCl. The organic phases were evaporated to dryness, resuspended in 100 µL chloroform/methanol (2:1, v/v) and stored at −20 °C. Radiolabeled products were analyzed by thin-layer chromatography using HPTLC Silica Gel 60 plates (Merck, Darmstadt, Germany).
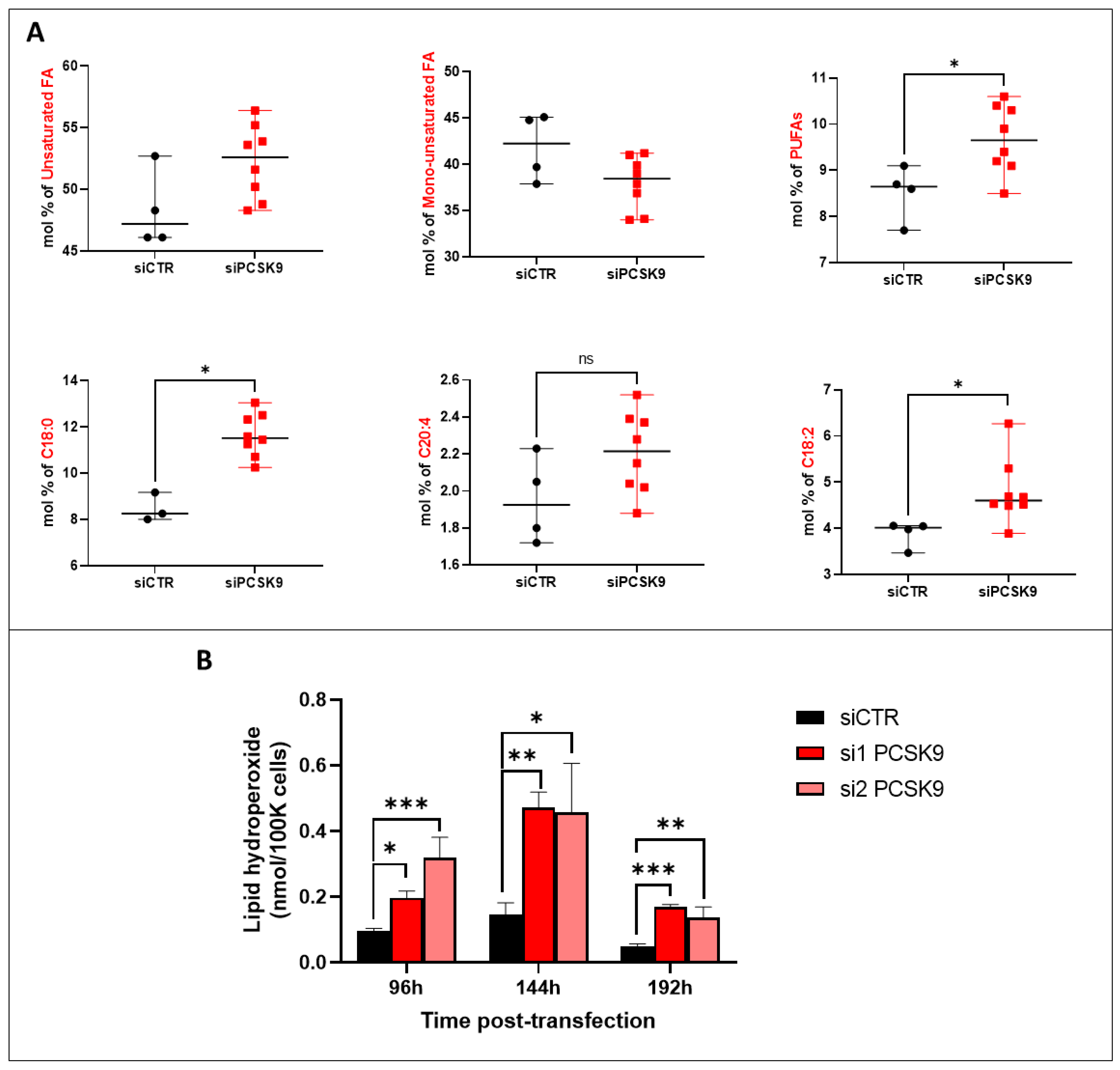
2.11. FA Saturation and Lipid Peroxide Analysis
Cell pellets of counted cells were directly used for fatty acid analysis. Fatty acid methyl esters were obtained by transmethylation at 90 °C for 1 h with 0.5 M sulfuric acid in methanol containing 2% (
v/
v) dimethoxypropane and 50 μg of heptadecanoic acid (C17:0) as internal standards. After cooling, 1 mL of NaCl (2.5%,
w/v) was added and fatty acyl chains were extracted with 1 mL hexane. Samples were subsequently analyzed by GC-MS as described by Domergue et al. [
14]. The measurement of lipid hydroperoxide was performed using the Lipid Hydroperoxide (LPO) Assay Kit (Cayman Chemical, Ann Arbor, MI, USA) as instructed by the manufacturer.
2.12. Immunohistochemistry (IHC)
The 3.5-µm thick sections of hepatoblastoma tumors were deparaffinized, rehydrated and antigen retrieval was performed in 0.01 M citrate buffer pH 6 solution. All staining procedures were performed by an autostainer (Dako-Agilent, Santa Clara, CA, USA) using standard reagents provided by the manufacturer. The sections were blocked using EnVision™ Flex peroxidase-blocking reagent (SM801, Dako-Agilent) to block endogenous peroxidase, then washed and incubated with rabbit anti-PCSK9 (1:100, 55206-1-AP, ProteinTech Group, Inc., Rosemont, IL, USA). Incubation in horseradish peroxidase (EnVision Flex/HRP, SM802, Dako-Agilent, Santa Clara, CA, USA) was used for signal amplification. A 3,3′-diaminobenzidine (DAB, Dako-Agilent, Santa Clara, CA, USA) development was used for detecting primary antibodies by producing a crisp brown end product at the site of the target antigen. The slides were counterstained with hematoxylin, dehydrated and mounted. Each immunohistochemical run contained a negative control (buffer, no primary antibody). Sections were visualized with a Hamamatsu NANOZOOMER 2.0 HT at 20× magnification in the Photonic Unit of the Bordeaux Imaging Center (BIC).
2.13. Transmission Electron Microscopy (TEM)
HepG2 cells transduced with shCTR and shPCSK9 were seeded in a Nunc™ Lab-Tek™ 8-chamber slide system (Thermo Fisher Scientific, Waltham, MA, USA) to a confluence of 80%. The cells were fixed with 2.5% (v/v) glutaraldehyde and 4% (v/v) paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) during 2 h at room temperature (RT), washed in 0.1 M phosphate buffer (pH 7.4) and then postfixed in 1% osmium tetroxide in water during 1 h. Then, samples were washed in water, dehydrated through a series of graded ethanol and embedded in a mixture of pure ethanol and epoxy resin (Epon 812; Delta Microscopy, Toulouse, France) 50/50 (v/v) during 2 h and then in 100% resin overnight at RT. The polymerization of the resin was carried out over a period of 48 h at 60 °C. Samples were then sectioned using a diamond knife (Diatome, Biel-Bienne, Switzerland) on an ultramicrotome (EM UC7, Leica Microsystems, Vienna, Austria). Ultrathin sections (70 nm) were picked up on copper grids. Grids were examined with a transmission electron microscope (H7650, Hitachi, Tokyo, Japan) at 80 kV.
2.14. In Vivo Zebrafish Model
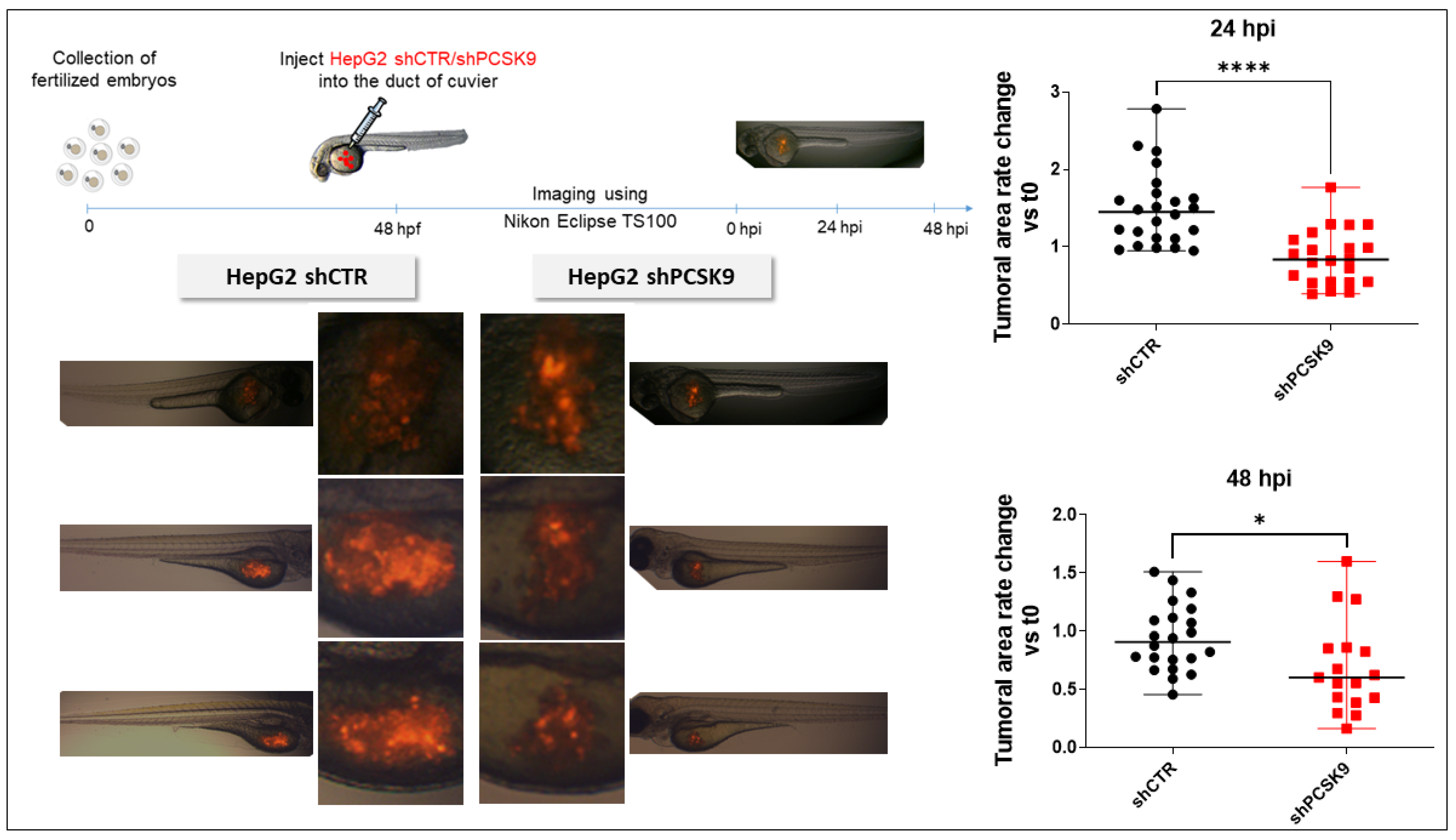
Zebrafish were maintained at 28 °C and in light cycle conditions (12 h). The Casper mutant fish line was purchased from the Zebrafish International Resource Center (ZIRC). For zebrafish xenotransplantation, 48 hours post-fertilization (hpf) zebrafish embryos were dechorionated and anesthetized in egg water solution containing 0.04 mg/mL tricaine (Sigma-Aldrich, St. Louis, MO, USA) before human cell injection. Approximately 200 to 500 fluorescent cells were injected (Eppendorf® Femtojet® microinjector) into the ducts of Cuvier of each embryo, and the zebrafish were maintained in 0.3X Danieau’s solution for 1 h at 28 °C. After confirmation of a visible cell mass at the injection site, the zebrafish were transferred to a 24-well plate in 500 μL of a 0.3X Danieau’s solution incubator and maintained at 34 °C. The zebrafish with already formed metastasis at 1 hour post-injection (hpi) were discarded.
After 24 hpi and 48 hpi, living zebrafish embryos were anesthetized using 0.04 mg/mL tricaine and observed under an inverted fluorescence microscope (Nikon Eclipse TS100). Low magnification (X4 objective) was used to provide an overview of the tumor cell metastasis pattern throughout the fish. Pictures were taken by using Archimed (Microvision Instruments) software. Fiji software was used for automated tumor area evaluation. Briefly, a 30–225-intensity threshold was set to select cells and the ‘Analyze’ particle tool was used with default selection of the cell size and cell shape during counting. A Fiji macro was generated using the ‘Record’ function to streamline the analyses.
2.15. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 9 software (GraphPad Software, Inc., San Diego, CA, USA). For two-group comparison, we used the t-test when values were ≥15; otherwise, the Mann–Whitney rank-sum test was used. For quantitative comparisons of more than two samples, the one-way ANOVA test was used followed by the Bonferroni post hoc test. Two-way ANOVA followed by the Bonferroni post hoc test was used for experiments containing three or more groups at different time points. For correlation graphs, a two-tailed Pearson correlation test was used. The experiments were carried out independently at least 3 times unless otherwise stated and each time, we included 3 technical replicates. In this case, n = number of independent experiments. A p-value of < 0.05 was considered to be statistically significant. For all data in figures, *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 or exact p-values were indicated. All tests were two-sided.
4. Discussion
Lipid metabolism plays a central role in liver oncogenesis and the important role of PCSK9 in lipid homeostasis places it at the center of the stage as an attractive target in liver cancers [
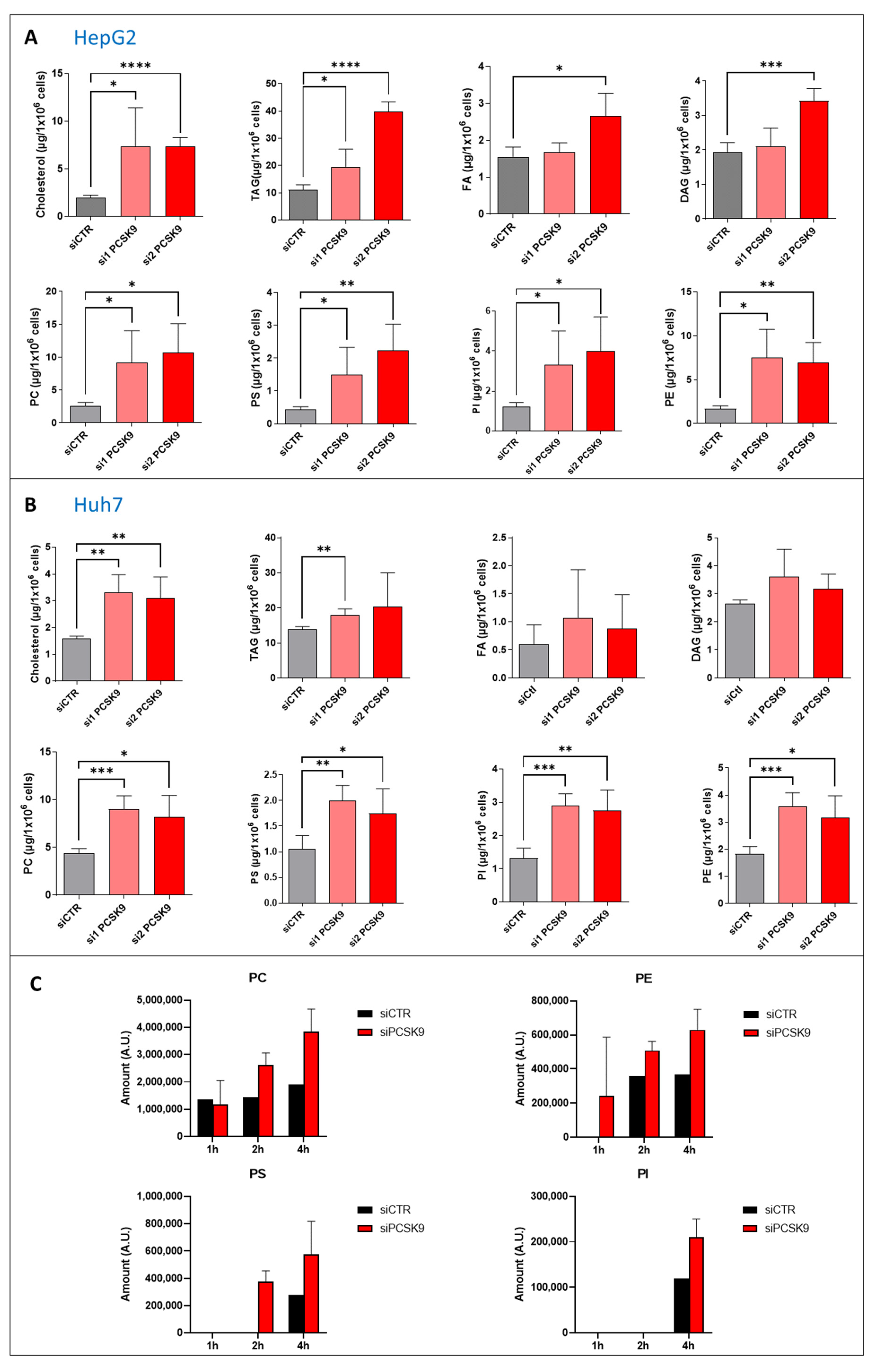
1]. PCSK9 modulation of lipid metabolism arises not only from LDLR binding and targeting for lysosomal destruction, but also from the degradation of other lipoprotein receptors such as VLDLR, ApoER2, CD36 and LRP1. Therefore, it was not inconceivable to expect the alteration of lipid homeostasis we have observed in liver cancer cells in the absence of PCSK9; lipid receptors madly play when PCSK9 is away.
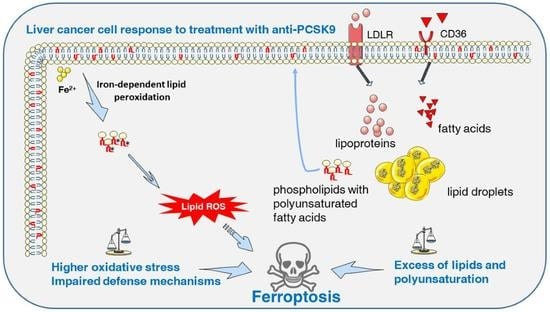
We report in this paper many anti-oncogenic features of anti-PCSK9 approaches, including the reduction in cell proliferation and the triggering of lipid cytotoxicity in liver tumor cells. Lipid metabolism is strongly affected by this approach, by which disruption probably leads to a massive accumulation in intracellular lipid and higher peroxidation of fatty acids, probably reflecting some metabolic exhaustion leading to overwhelming oxidative stress. The increased oxidative burden and failure of protective measures trigger irreversible cell damage and death by ferroptosis.
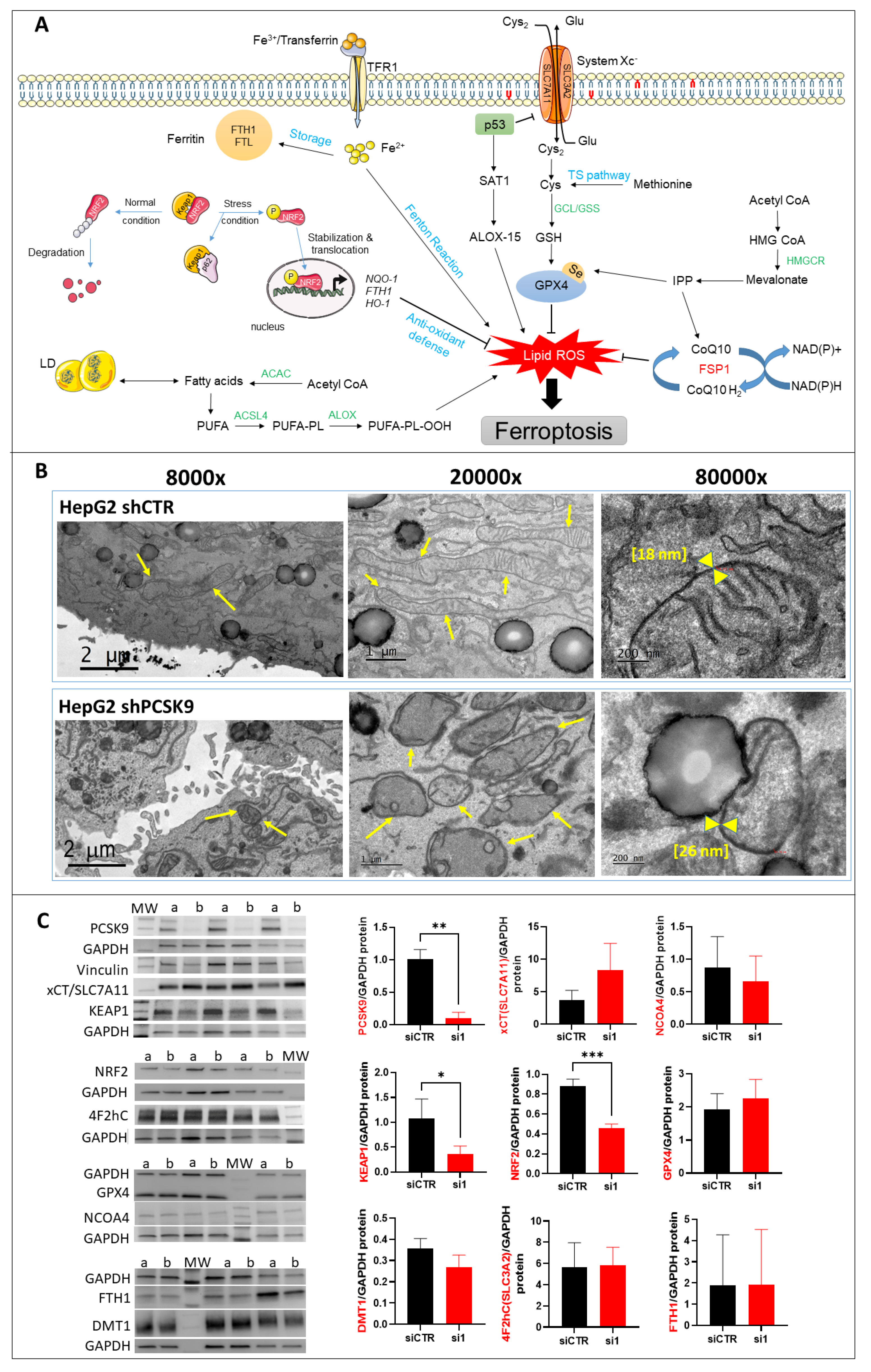
The ferroptotic mode of cell death was discovered in recent years (
Figure 5A). It is linked to iron-dependent lipid peroxidation triggering the death process [
15]. Unlike other modes of cell death, the distinctive morphological features of ferroptosis are obvious at the mitochondrial level with shrinkage of the organite, increased membrane density and the reduction or disappearance of mitochondrial cristae. The integrity of the cell membrane is not affected and the nucleus is normal in size with no chromatin condensation [
15,
16,
17].
Herein, we recognize all these ferroptotic morphological features in cells silenced for PCSK9. Moreover, our search for a potential ferroptotic signaling pathway singled out the Keap1/Nrf2 axis. Indeed, PCSK9 depletion led to a significant reduction in Nrf2, which is the chief coordinator of the antioxidant response machinery [
18]. Under normal conditions, Keap1 controls the basal cytoplasmic level of Nrf2 by promoting its degradation by the ubiquitin–proteasome system. Under oxidative stress conditions, Keap1, as a redox sensor, gets oxidized and dissociates from Nrf2, which becomes activated and translocates into the nucleus, where it can induce the transcription of different branches of the antioxidant defense system (
Figure 5A). In our experimental context, Nrf2 was downregulated in the absence of PCSK9 and failed to launch effective antioxidative responses, as seen by the lack of activation of FTH1, one of the Nrf2 downstream targets.
It is unclear whether PCSK9 nuclear localization, which we reported here, has any direct connection with Nrf2 regulation and activity. The hypothesis of a possible causative correlation may be attractive, but the question deserves further exploration.
Of note, Nrf2 was reported to be hyperactivated in HCC, promoting the survival of cancer cells and also conferring cellular resistance to chemotherapeutic drugs [
19,
20,
21]. Interestingly, among the main mechanisms accounting for the biological effects of sorafenib, the only approved first-line drug for advanced HCC is the induction of ferroptosis [
22], probably by inhibiting the SLC7A11 transporter [
15]. Although sorafenib treatment improves survival to some extent, unfortunate severe adverse effects and emerging resistance make it an unsatisfactory therapeutic approach [
23]. Interestingly, many strategies to improve sorafenib resistance are aimed at modulating ferroptosis, which is thus emerging as a potential new weapon in the fight against cancer. Some of these strategies are targeting Nrf2 itself. Indeed, the genetic or pharmacological inhibition of Nrf2 in HCC cells makes them more sensitive to the action of sorafenib and even overcomes chemoresistance through the induction of ferroptosis [
24].
Above all, since the disruption of PCSK9 inhibits the anti-ferroptosis p62-Keap1-Nrf2 pathway, one could speculate that a combination therapy of anti-PCSK9 with sorafenib would alleviate drug resistance and improve prognosis.
In parallel to its involvement in ferroptosis through the p62-Keap1-Nrf2 pathway, PCSK9 may have an impact on this death process through a mechanism that depends on its cholesterol-regulating functions. Indeed, by enhancing lipid/lipoprotein uptake, PCSK9 deficiency may enrich the membrane using PUFAs, which are sensitive to lipid peroxidation and are one of the essential elements for ferroptosis [
25]. Free PUFAs can be esterified into membrane phospholipids before being oxidized and thereby inducing ferroptotic signals. Moreover, among some of the highly produced/imported phospholipids following PCSK9 depletion is phosphatidylethanolamine (PE), which was increased 2 to 4 times (
Figure 4). Oxidized PEs are the key phospholipids acting as death signals to induce ferroptosis in cells [
26]. In this process, 15-lipoxygenase is an important contributor to pro-ferroptosis PE peroxidation, capable of generating doubly- and triply-oxygenated diacylated PE species. Tocopherols and vitamin E suppress this oxygenation and protect against ferroptosis [
26]. Therefore, PCSK9 inhibition may trigger ferroptosis in lipid-dependent and -independent fashions.
Apart from its role in lipid homeostasis, PCSK9 is involved in various signaling pathways including antiviral activity, apoptosis and more recently, anti-tumor immune responses. Indeed, Liu et al. [
27] demonstrated that the blockade of PCSK9 can boost the immunotherapeutic efficacy of the anti-programmed death 1 (PD-1) approach. The research involved PCSK9 in the degradation of the major histocompatibility complex class 1 (MHC1), hindering its recycling at the cell surface. Hence, the depletion of this enzyme results in high cellular expression of MHC1, leading to massive infiltration of cytotoxic T cells.
As per our results, PCSK9 may now modulate cancer survival and resistance by an additional mechanism connected to anti-oxidative housekeeping activities. Besides its critical role in modulating lipid metabolism and fluxes, our research brings out a previously unknown function of this intriguing enzyme, that is, maintaining the redox homeostasis via the p62/Keap1/Nrf2 axis.
The inhibition of PCSK9 led to excessive lipid accumulation and created a void in the defense against oxidative stress, thereby enhancing the vulnerability of cancer cells to ferroptosis. Specific targeting of PCSK9 in hepatic cancer cells showed strong beneficial outcomes and novel mechanistic insights highlighting a new player in cancer cell death by ferroptosis. Taking into account all these anti-tumoral effects of anti-PCSK9 approaches and the existence of a wide variety of therapeutic strategies of the PCSK9 blockade (monoclonal antibodies, small molecule and peptide inhibitors, antisense oligonucleotides, siRNA, etc.), we believe this enzyme is a very valuable and attractive target for the potential treatment of liver cancers.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






