
The E3 Ligase TRIM25 Impairs Apoptotic Cell Death in Colon Carcinoma Cells via Destabilization of Caspase-7 mRNA: A Possible Role of hnRNPH1

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Small Hairpin (sh)-Mediated Knockdown of TRIM25
2.4. Small Interference (si)RNA
2.5. Ectopic Expression of Plasmids in RKO Cells
2.6. Western Blot Analysis
2.7. Isolation of Nuclear and Cytoplasmic Fractions
2.8. qRT-PCR-Analysis
2.9. Sub-G1 Analysis by Flow Cytometry
2.10. Immunoprecipitation of Endogenous Proteins
2.11. Immunoprecipitation of Flag-Tagged Proteins
2.12. Ribonucleoprotein (RNP) IP RT-PCR Assay
2.13. Construction of Flag-Tagged TRIM25 Truncations
2.14. Generation of a Plasmid Bearing the 3′UTR of Caspase-7 for In Vitro Transcription
2.15. RNA Affinity Chromatography
2.16. Mass Spectrometry (LC-MS)
2.17. Indirect Immunofluorescence Microscopy
2.18. Statistical Analysis
3. Results
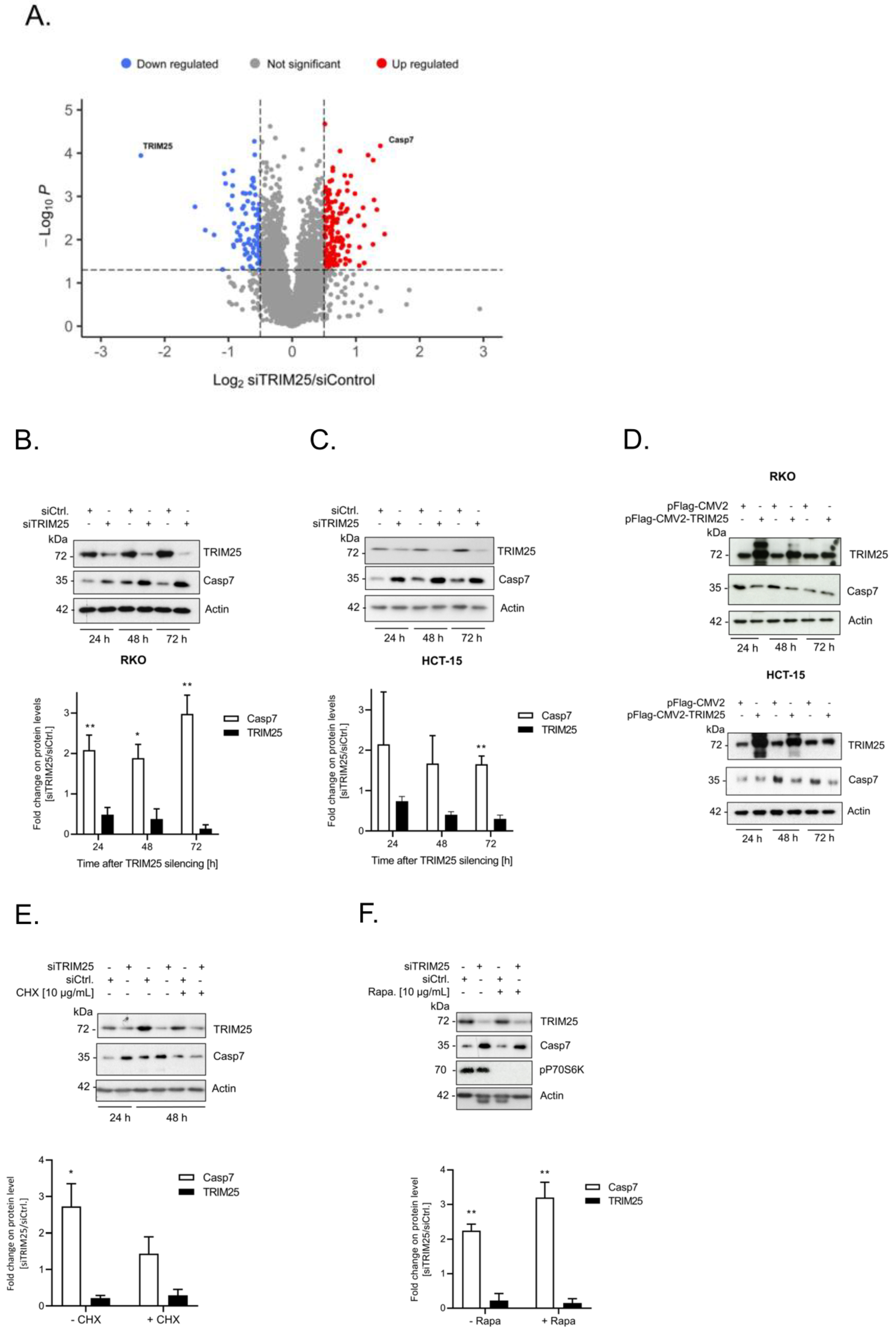
3.1. Identification of Caspase-7 as a Negative Target of TRIM25
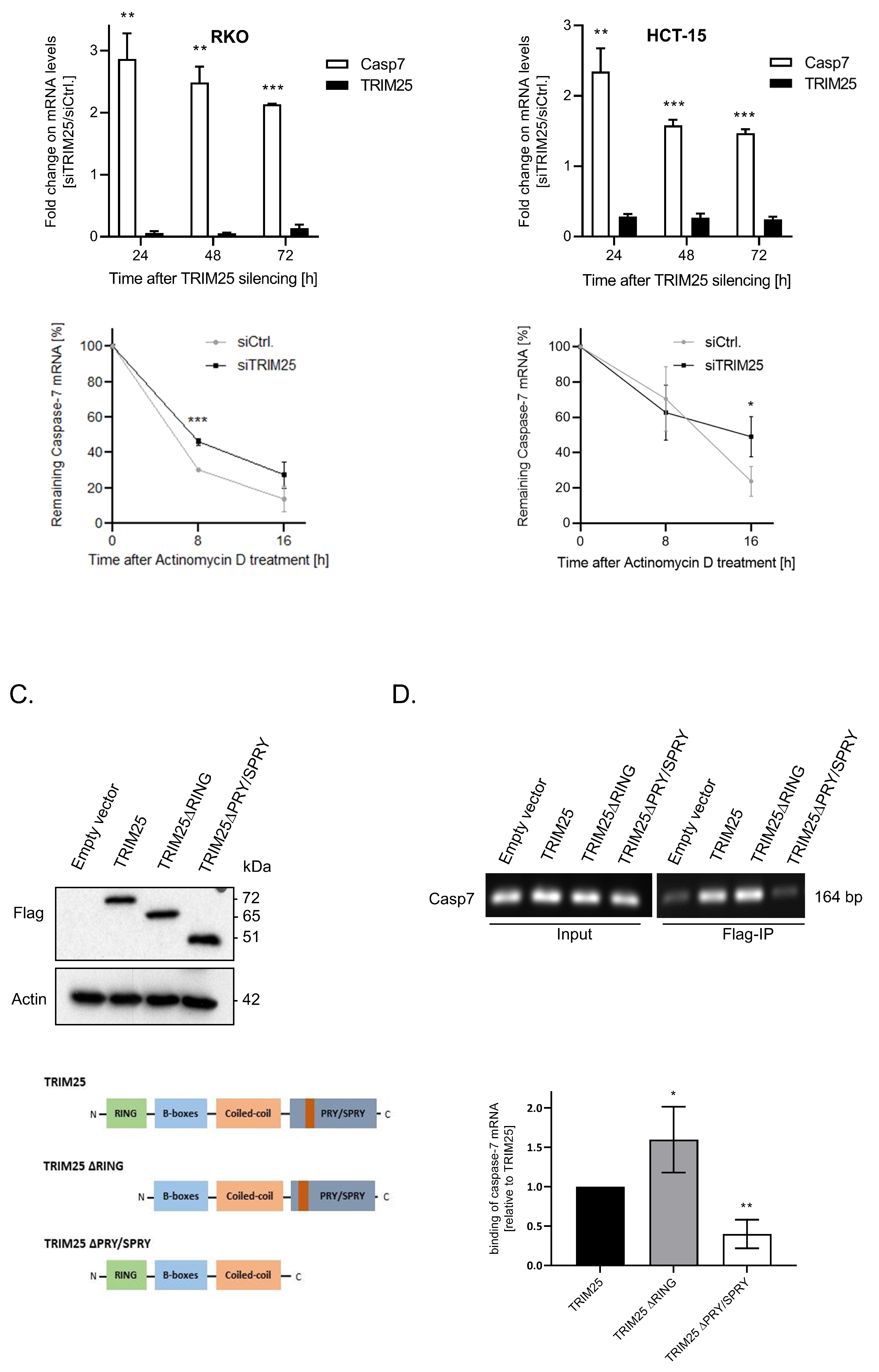
3.2. TRIM25 Knockdown-Dependent Elevation of Caspase-7 Is Due to an Increase in Caspase-7 mRNA Stability
3.3. TRIM25 Binding to Caspase-7 mRNA Is Mediated through the PRY/SPRY Domain
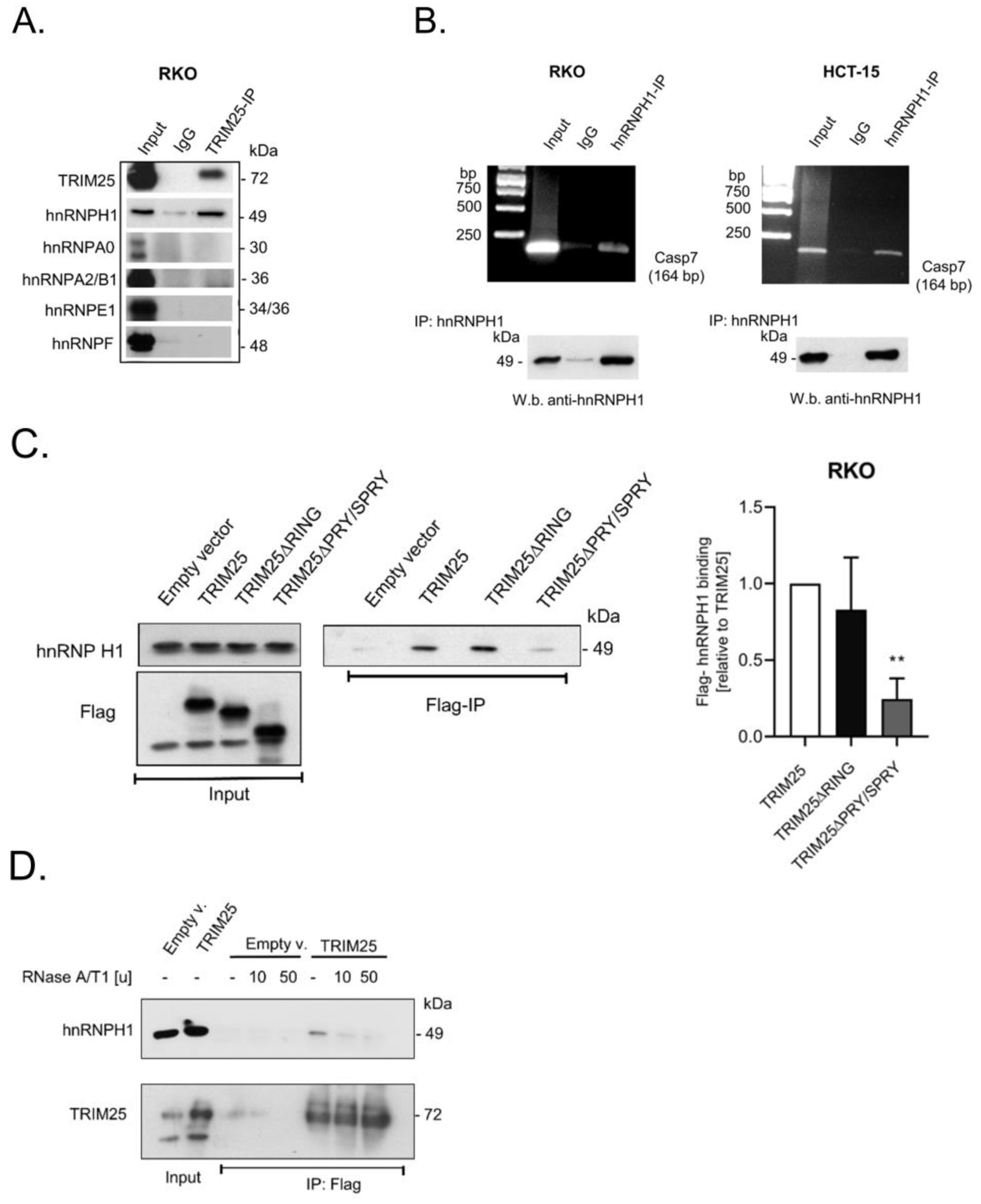
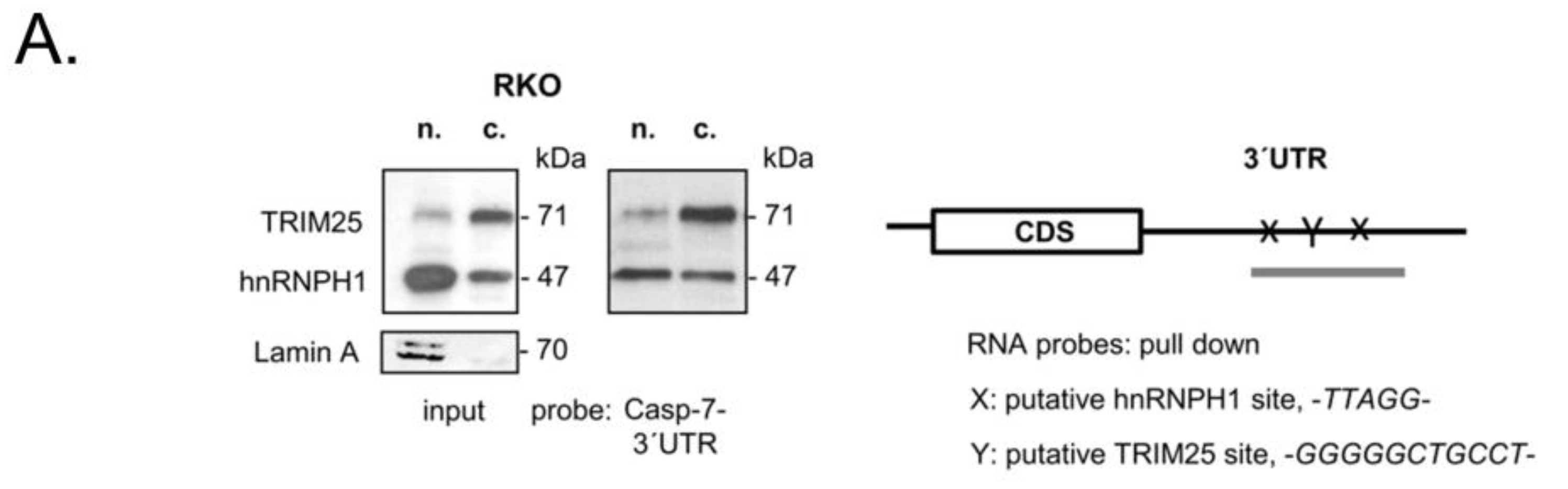
3.4. TRIM25 Interacts with hnRNPH1 through a Common Binding to Caspase-7 mRNA
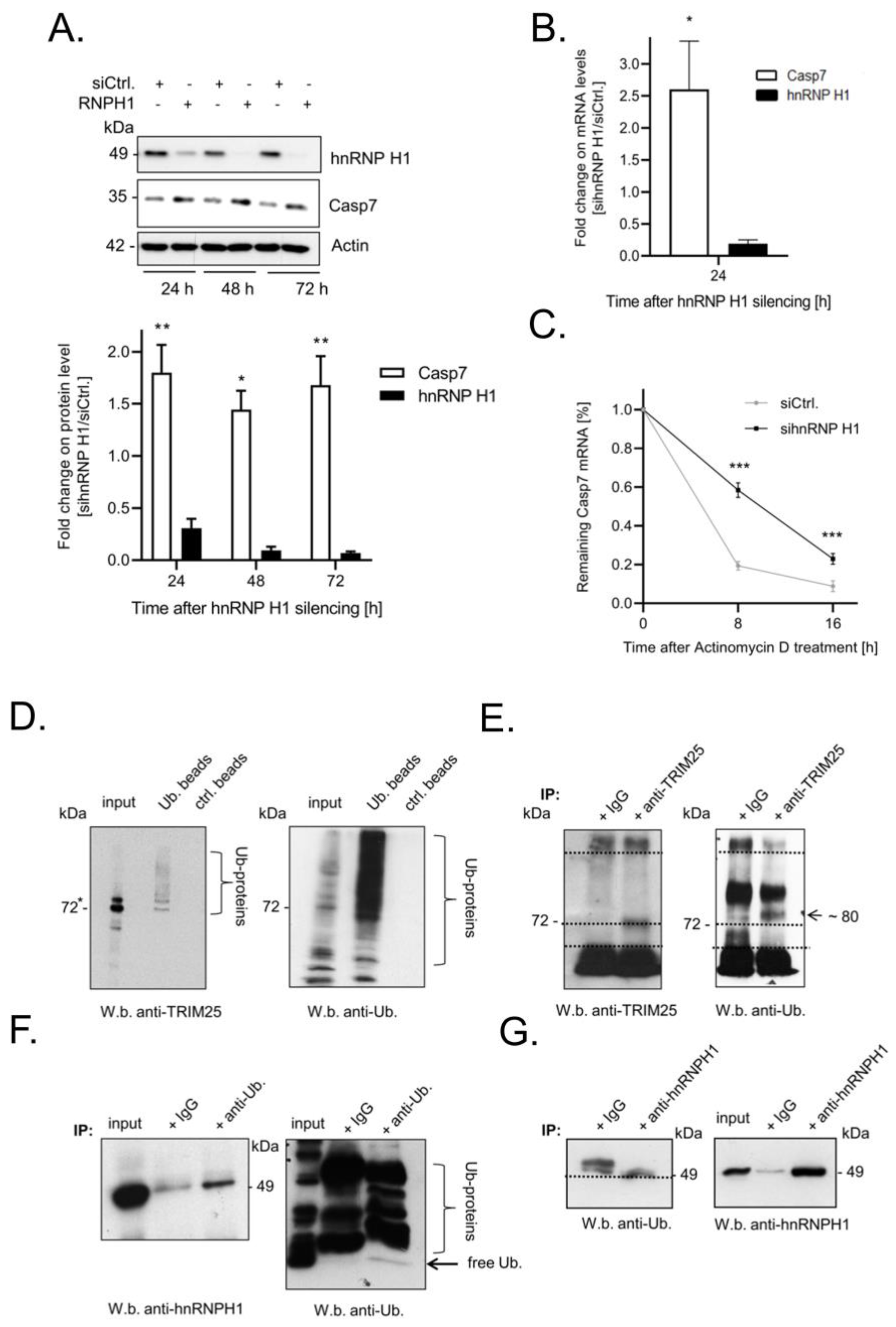
3.5. Elevated Expression of Caspase-7 upon Knockdown of hnRNPH1 Is Due to an Increase in mRNA Stability
3.6. TRIM25 and hnRNPH1 Are Both Targets of Permanent Ubiquitination
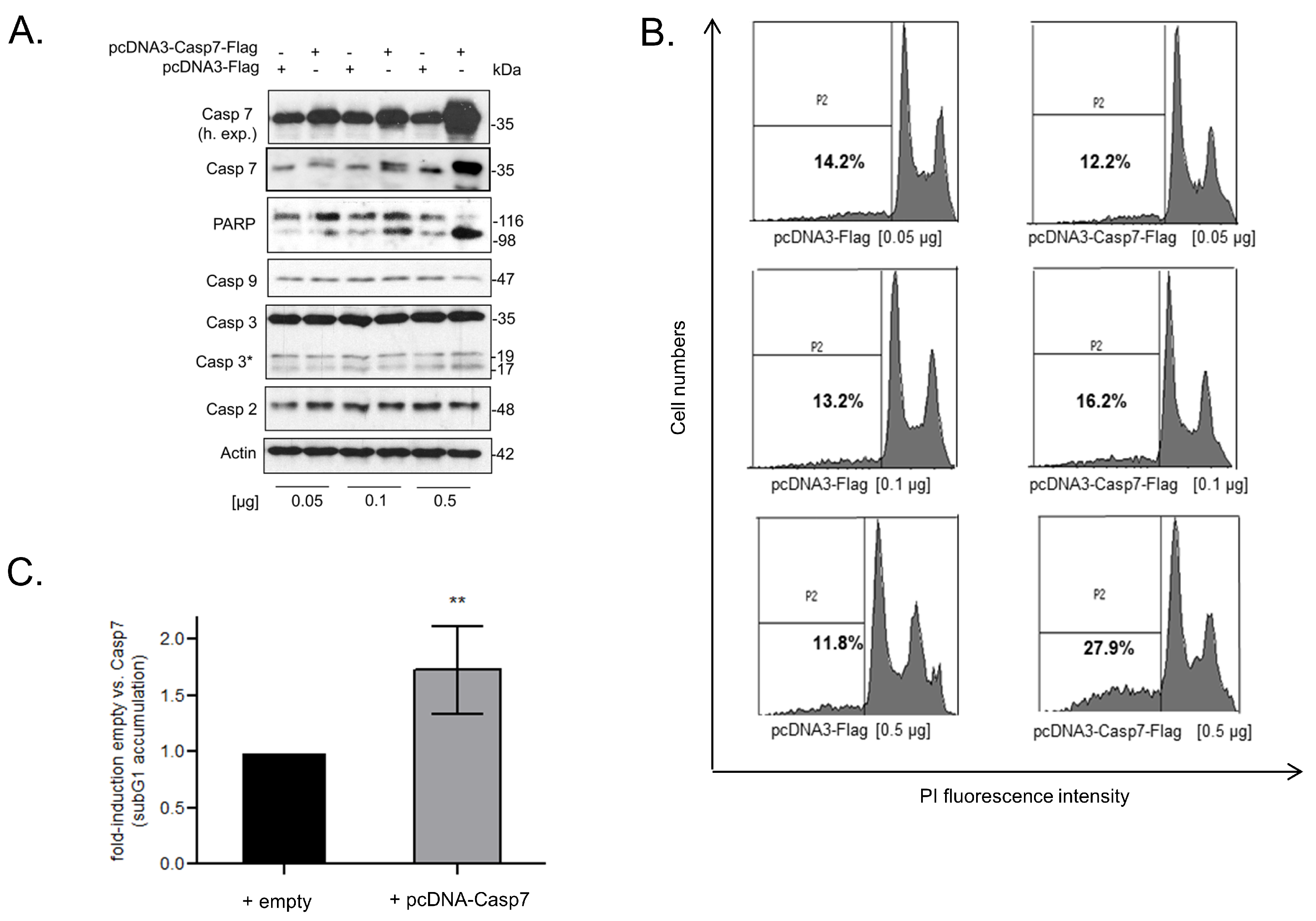
3.7. Functional Consequences of Caspase-7 for Apoptotic Cell Death
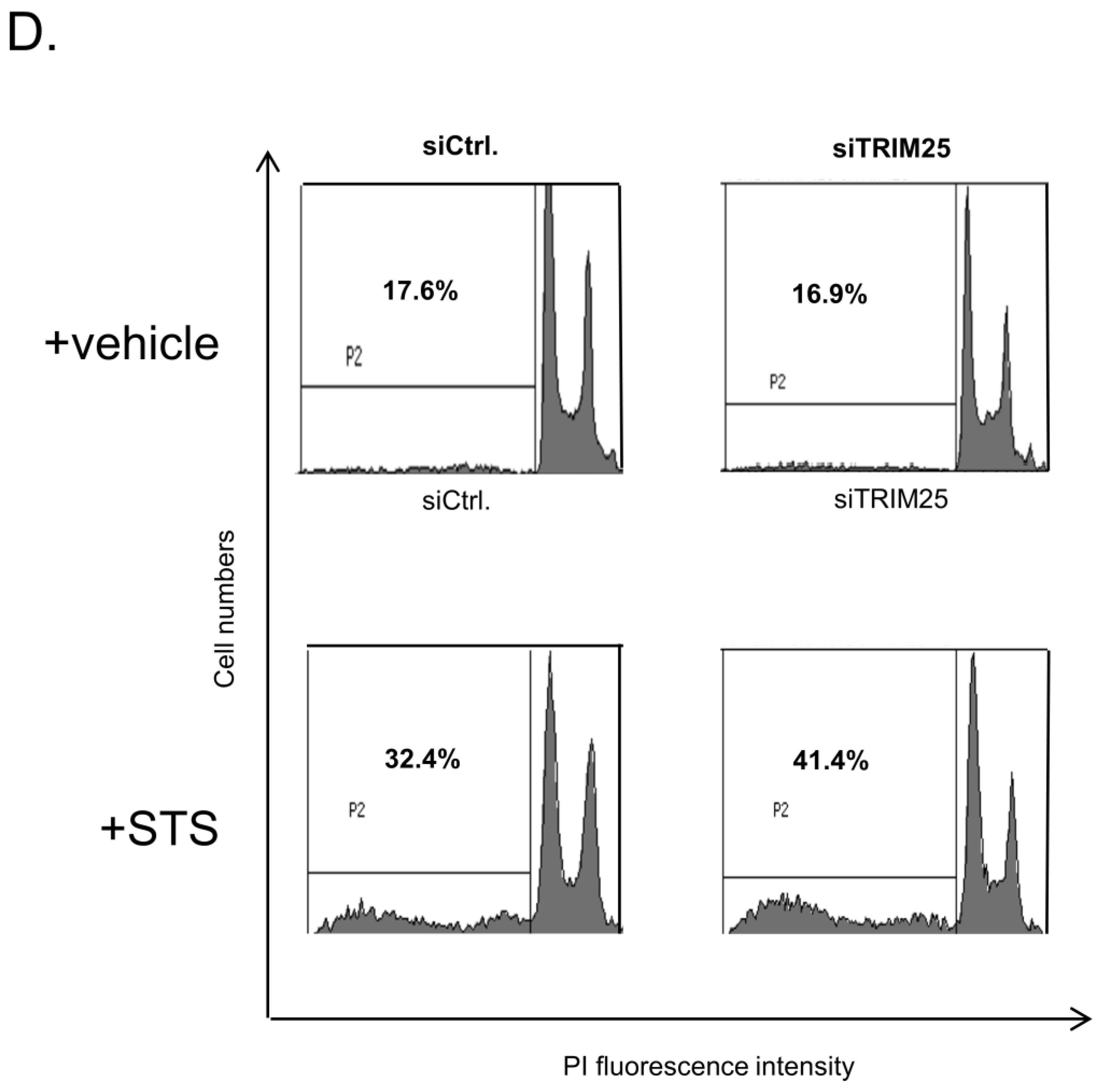
3.8. Knockdown of TRIM25 Sensitizes Colorectal Carcinoma Cells to Drug-Induced Apoptosis
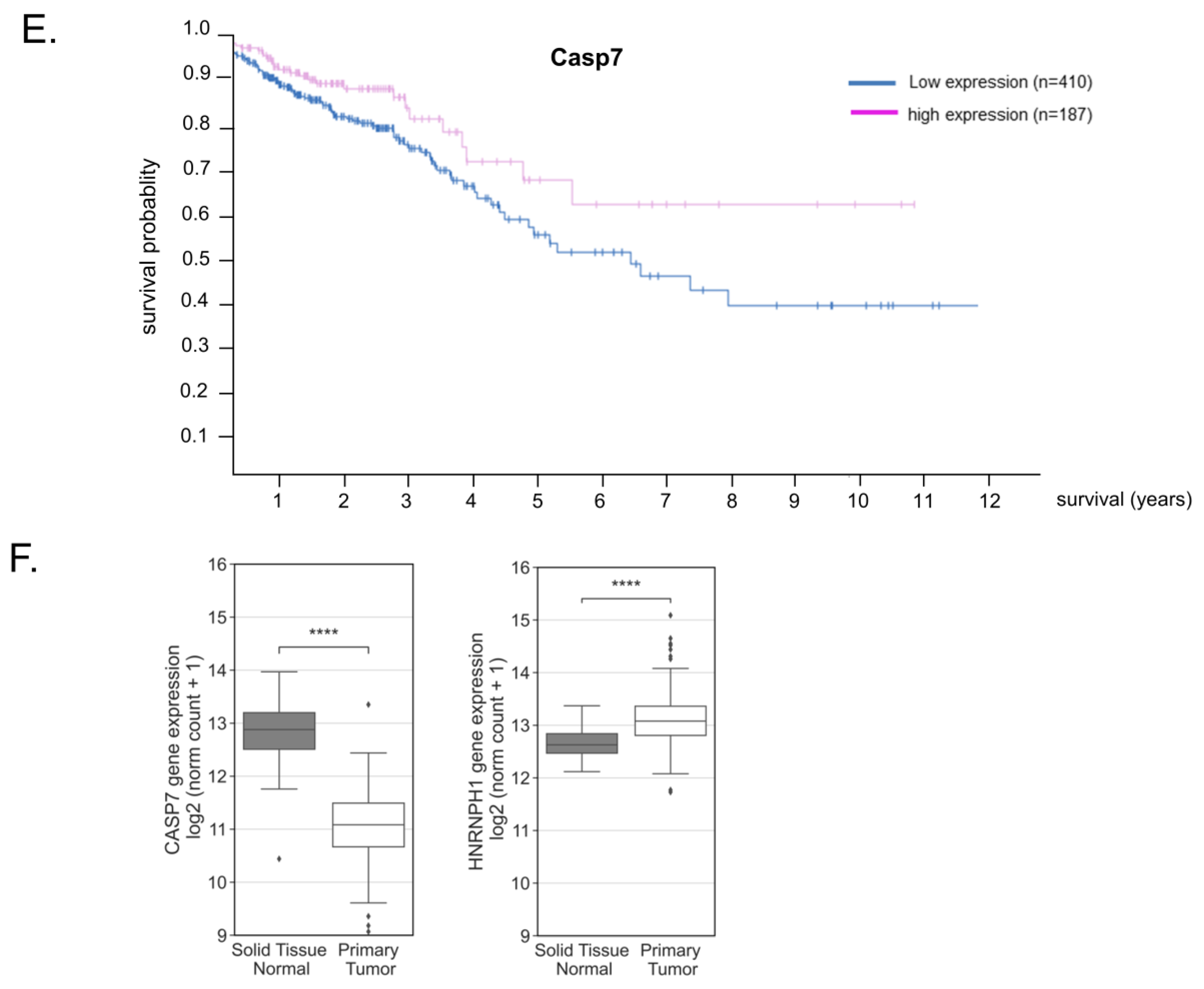
3.9. Reduced Caspase-7 Expression in Primary Colonic Tumor Samples Correlates with Poor Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lech, G.; Słotwiński, R.; Słodkowski, M.; Krasnodębski, I.W. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J. Gastroenterol. 2016, 22, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Marley, A.R.; Nan, H. Epidemiology of colorectal cancer. Int. J. Mol. Epidemiol. Genet. 2016, 7, 105–114. [Google Scholar] [PubMed]
- Papamichael, D.; Audisio, R.A.; Glimelius, B.; de Gramont, A.; Glynne-Jones, R.; Haller, D.; Köhne, C.H.; Rostoft, S.; Lemmens, V.; Mitry, E.; et al. Treatment of colorectal cancer in older patients: International Society of Geriatric Oncology (SIOG) consensus recommendations 2013. Ann. Oncol. 2015, 26, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, S.; Kyprianou, N. Apoptosis evasion: The role of survival pathways in prostate cancer progression and therapeutic resistance. J. Cell. Biochem. 2006, 97, 18–32. [Google Scholar] [CrossRef] [Green Version]
- Nasrullah, U.; Haeussler, K.; Biyanee, A.; Wittig, I.; Pfeilschifter, J.; Eberhardt, W. Identification of TRIM25 as a Negative Regulator of Caspase-2 Expression Reveals a Novel Target for Sensitizing Colon Carcinoma Cells to Intrinsic Apoptosis. Cells 2019, 8, 1622. [Google Scholar] [CrossRef] [Green Version]
- Cano, F.; Miranda-Saavedra, D.; Lehner, P.J. RNA-binding E3 ubiquitin ligases: Novel players in nucleic acid regulation. Biochem. Soc. Trans. 2010, 38, 1621–1626. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, N.R.; Heikel, G.; Trubitsyna, M.; Kubik, P.; Nowak, J.S.; Webb, S.; Granneman, S.; Spanos, C.; Rappsilber, J.; Castello, A.; et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 2017, 15, 105–125. [Google Scholar] [CrossRef] [Green Version]
- Vaishali; Dimitrova-Paternoga, L.; Haubrich, K.; Sun, M.; Ephrussi, A.; Hennig, J. Validation and classification of RNA binding proteins identified by mRNA interactome capture. RNA 2021, 27, 1173–1185. [Google Scholar] [CrossRef]
- Eberhardt, W.; Haeussler, K.; Nasrullah, U.; Pfeilschifter, J. Multifaceted Roles of TRIM Proteins in Colorectal Carcinoma. Int. J. Mol. Sci. 2020, 21, 7532. [Google Scholar] [CrossRef]
- Walsh, L.A.; Alvarez, M.J.; Sabio, E.Y.; Reyngold, M.; Makarov, V.; Mukherjee, S.; Lee, K.W.; Desrichard, A.; Turcan, Ş.; Dalin, M.G.; et al. An Integrated Systems Biology Approach Identifies TRIM25 as a Key Determinant of Breast Cancer Metastasis. Cell Rep. 2017, 20, 1623–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Declercq, W.; Kalai, M.; Saelens, X.; Vandenabeele, P. Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ. 2002, 9, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Germain, M.; Affar, E.B.; D’Amours, D.; Dixit, V.M.; Salvesen, G.S.; Poirier, G.G. Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. J. Biol. Chem. 1999, 274, 28379–28384. [Google Scholar] [CrossRef] [Green Version]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, J.G.; Cullen, S.P.; Sheridan, C.; Lüthi, A.U.; Gerner, C.; Martin, S.J. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. USA 2008, 105, 12815–12819. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Kanneganti, T.D. Caspase-7: A protease involved in apoptosis and inflammation. Int. J. Biochem. Cell Biol. 2010, 42, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Palmerini, F.; Devilard, E.; Jarry, A.; Birg, F.; Xerri, L. Caspase 7 downregulation as an immunohistochemical marker of colonic carcinoma. Hum. Pathol. 2001, 32, 461–467. [Google Scholar] [CrossRef]
- Yan, S.; Li, Y.Z.; Zhu, X.W.; Liu, C.L.; Wang, P.; Liu, Y.L. HuGE systematic review and meta-analysis demonstrate association of CASP-3 and CASP-7 genetic polymorphisms with cancer risk. Genet. Mol. Res. 2013, 12, 1561–1573. [Google Scholar] [CrossRef]
- Eicher, M.; Distler, U.; Nasrullah, U.; Krishnan, A.; Kaulich, M.; Husnjak, K.; Eberhardt, W.; Rajalingam, K.; Tenzer, S.; Pfeilschifter, J.; et al. The caspase-2 substrate p54nrb exhibits a multifaceted role in tumor cell death susceptibility via gene regulatory functions. Cell Death Dis. 2022, 13, 386. [Google Scholar] [CrossRef]
- Zou, W.; Zhang, D.E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef]
- Stennicke, H.R.; Salvesen, G.S. Biochemical characteristics of caspases-3, -6, -7, and -8. J. Biol. Chem. 1997, 272, 25719–25723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, C.; Doller, A.; Imre, G.; Badawi, A.; Schmid, T.; Schulz, S.; Steinmeyer, N.; Pfeilschifter, J.; Rajalingam, K.; Eberhardt, W. Attenuation of the ELAV1-like protein HuR sensitizes adenocarcinoma cells to the intrinsic apoptotic pathway by increasing the translation of caspase-2L. Cell Death Dis. 2014, 5, e1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, E.; Matthias, P.; Müller, M.M.; Schaffner, W. Rapid detection of octamer binding proteins with “mini-extracts” prepared from a small number of cells. Nucleic Acids Res. 1989, 17, 6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawi, A.; Hehlgans, S.; Pfeilschifter, J.; Rödel, F.; Eberhardt, W. Silencing of the mRNA-binding protein HuR increases the sensitivity of colorectal cancer cells to ionizing radiation through upregulation of caspase-2. Cancer Lett. 2017, 393, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Garcia, C.; Hartmann, O.; Reissland, M.; Braun, F.; Bozkurt, S.; Pahor, N.; Fuss, C.; Schirbel, A.; Schülein-Völk, C.; Buchberger, A.; et al. USP28 enables oncogenic transformation of respiratory cells, and its inhibition potentiates molecular therapy targeting mutant EGFR, BRAF and PI3K. Mol. Oncol. 2022, 16, 3082–3106. [Google Scholar] [CrossRef]
- McAlister, G.C.; Nusinow, D.P.; Jedrychowski, M.P.; Wühr, M.; Huttlin, E.L.; Erickson, B.K.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisen-acher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Schulz, S.; Doller, A.; Pendini, N.R.; Wilce, J.A.; Pfeilschifter, J.; Eberhardt, W. Domain-specific phosphomimetic mutation allows dissection of different protein kinase C (PKC) isotype-triggered activities of the RNA binding protein HuR. Cell. Signal. 2013, 25, 2485–2495. [Google Scholar] [CrossRef]
- Kwon, S.C.; Yi, H.; Eichelbaum, K.; Föhr, S.; Fischer, B.; You, K.T.; Castello, A.; Krijgsveld, J.; Hentze, M.W.; Kim, V.N. The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1122–1130. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Nowak, J.S.; Zuo, J.; Rappsilber, J.; Spoel, S.H.; Michlewski, G. Trim25 Is an RNA-Specific Activator of Lin28a/TuT4-Mediated Uridylation. Cell Rep. 2014, 9, 1265–1272. [Google Scholar] [CrossRef]
- Han, N.; Li, W.; Zhang, M. The function of the RNA-binding protein hnRNP in cancer metastasis. J. Cancer Res. Ther. 2013, 9, S129–S134. [Google Scholar] [CrossRef] [PubMed]
- Uren, P.J.; Bahrami-Samani, E.; de Araujo, P.R.; Vogel, C.; Qiao, M.; Burns, S.C.; Smith, A.D.; Penalva, L.O. High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol. 2016, 13, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Fujiya, M.; Konishi, H.; Murakami, Y.; Iwama, T.; Sasaki, T.; Kunogi, T.; Sakatani, A.; Ando, K.; Ueno, N.; et al. Heterogenous Nuclear Ribonucleoprotein H1 Promotes Colorectal Cancer Progression through the Stabilization of mRNA of Sphingosine-1-Phosphate Lyase 1. Int. J. Mol. Sci. 2020, 21, 4514. [Google Scholar] [CrossRef] [PubMed]
- Ross, J. Control of messenger RNA stability in higher eukaryotes. Trends Genet. 1996, 12, 171–175. [Google Scholar] [CrossRef]
- Eberhardt, W.; Doller, A.; Akool, E.-S.; Pfeilschifter, J. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 2007, 114, 56–73. [Google Scholar] [CrossRef]
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Stalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [Green Version]
- Inn, K.S.; Gack, M.U.; Tokunaga, F.; Shi, M.; Wong, L.Y.; Iwai, K.; Jung, J.U. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol. Cell 2011, 41, 354–365. [Google Scholar] [CrossRef] [Green Version]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikel, G.; Choudhury, N.R.; Michlewski, G. The role of Trim25 in development, disease and RNA metabolism. Biochem. Soc. Trans. 2016, 44, 1045–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Xu, T.; Chen, D.; Tan, X. Targeting SREBP1 chemosensitizes colorectal cancer cells to gemcitabine by caspase-7 upregulation. Bioengineered 2019, 10, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, M.Y.; Pozharissky, K.M.; Hanson, K.P.; Imyanitov, E.N.; Zhivotovsky, B. Expression of caspase-3 and -7 does not correlate with the extent of apoptosis in primary breast carcinomas. Cell Cycle 2002, 1, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Erener, S.; Pétrilli, V.; Kassner, I.; Minotti, R.; Castillo, R.; Santoro, R.; Hassa, P.O.; Tschopp, J.; Hottiger, M.O. Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-κB target genes. Mol. Cell 2012, 46, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.I.; Suzuki, T.; Tanaka, T.; Fujimura, T.; Takahashi, S.; Urano, T.; Ikeda, K.; Inoue, S. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 2018, 37, 2165–2180. [Google Scholar] [CrossRef]
- Zhou, S.; Peng, J.; Xiao, L.; Zhou, C.; Fang, Y.; Ou, Q.; Qin, J.; Liu, M.; Pan, Z.; Hou, Z. TRIM25 regulates oxaliplatin resistance in colorectal cancer by promoting EZH2 stability. Cell Death Dis. 2021, 12, 463. [Google Scholar] [CrossRef]
- Li, M.M.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M.; et al. TRIM25 Enhances the Antiviral Action of Zinc-Finger Antiviral Protein (ZAP). PLoS Pathog. 2017, 13, e1006145. [Google Scholar] [CrossRef]
- Choudhury, N.R.; Trus, I.; Heikel, G.; Wolczyk, M.; Szymanski, J.; Bolembach, A.; Dos Santos Pinto, R.M.; Smith, N.; Trubitsyna, M.; Gaunt, E.; et al. TRIM25 inhibits influenza A virus infection, destabilizes viral mRNA, but is redundant for activating the RIG-I pathway. Nucleic Acids Res. 2022, 50, 7097–7114. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.; O’Neill, E.; Mack, B.; Matthias, C.; Munz, M.; Kolch, W.; Gires, O. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res. 2010, 70, 1679–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefave, C.V.; Squatrito, M.; Vorlova, S.; Rocco, G.L.; Brennan, C.W.; Holland, E.C.; Pan, Y.X.; Cartegni, L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011, 30, 4084–4097. [Google Scholar] [CrossRef] [PubMed]
- Han, S.P.; Tang, Y.H.; Smith, R. Functional diversity of the hnRNPs: Past, present and perspectives. Biochem. J. 2010, 430, 379–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelga, S.C.; Vu, A.Q.; Arnold, J.D.; Liang, T.Y.; Liu, P.P.; Yan, B.Y.; Donohue, J.P.; Shiue, L.; Hoon, S.; Brenner, S.; et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012, 1, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Turunen, J.J.; Verma, B.; Nyman, T.A.; Frilander, M.J. HnRNPH1/H2, U1 snRNP, and U11 snRNP cooperate to regulate the stability of the U11-48K pre-mRNA. RNA 2013, 19, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Gao, F.H. The Molecular Mechanisms and the Role of hnRNP K Protein Post-Translational Modification in DNA Damage Repair. Curr. Med. Chem. 2017, 24, 614–621. [Google Scholar] [CrossRef]
- Yoon, J.H.; Abdelmohsen, K.; Kim, J.; Yang, X.; Martindale, J.L.; Tominaga-Yamanaka, K.; White, E.J.; Orjalo, A.V.; Rinn, J.L.; Kreft, S.G.; et al. Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat. Commun. 2013, 4, 2939. [Google Scholar] [CrossRef] [Green Version]
- Gerstmeier, J.; Possmayer, A.L.; Bozkurt, S.; Hoffmann, M.E.; Dikic, I.; Herold-Mende, C.; Burger, M.C.; Münch, C.; Kögel, D.; Linder, B. Calcitriol Promotes Differentiation of Glioma Stem-Like Cells and Increases Their Susceptibility to Temozolomide. Cancers 2021, 13, 3577. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasrullah, U.; Stanke, K.; Recknagel, V.; Bozkurt, S.; Wurzel, P.; Gauer, S.; Imre, G.; Münch, C.; Pfeilschifter, J.; Eberhardt, W. The E3 Ligase TRIM25 Impairs Apoptotic Cell Death in Colon Carcinoma Cells via Destabilization of Caspase-7 mRNA: A Possible Role of hnRNPH1. Cells 2023, 12, 201. https://doi.org/10.3390/cells12010201
Nasrullah U, Stanke K, Recknagel V, Bozkurt S, Wurzel P, Gauer S, Imre G, Münch C, Pfeilschifter J, Eberhardt W. The E3 Ligase TRIM25 Impairs Apoptotic Cell Death in Colon Carcinoma Cells via Destabilization of Caspase-7 mRNA: A Possible Role of hnRNPH1. Cells. 2023; 12(1):201. https://doi.org/10.3390/cells12010201
Chicago/Turabian StyleNasrullah, Usman, Kristina Stanke, Victoria Recknagel, Süleyman Bozkurt, Patrick Wurzel, Stefan Gauer, Gergely Imre, Christian Münch, Josef Pfeilschifter, and Wolfgang Eberhardt. 2023. "The E3 Ligase TRIM25 Impairs Apoptotic Cell Death in Colon Carcinoma Cells via Destabilization of Caspase-7 mRNA: A Possible Role of hnRNPH1" Cells 12, no. 1: 201. https://doi.org/10.3390/cells12010201