
CXCR3 Inhibition Blocks the NF-κB Signaling Pathway by Elevating Autophagy to Ameliorate Lipopolysaccharide-Induced Intestinal Dysfunction in Mice
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Handling and Sample Collection
2.2. Cell Culture
2.3. HE Staining
2.4. RNA Extraction and Real-Time Fluorescence Quantification (qPCR)
2.5. Western Blot
2.6. Indirect Immunofluorescence
2.7. Statistical Analysis
3. Results
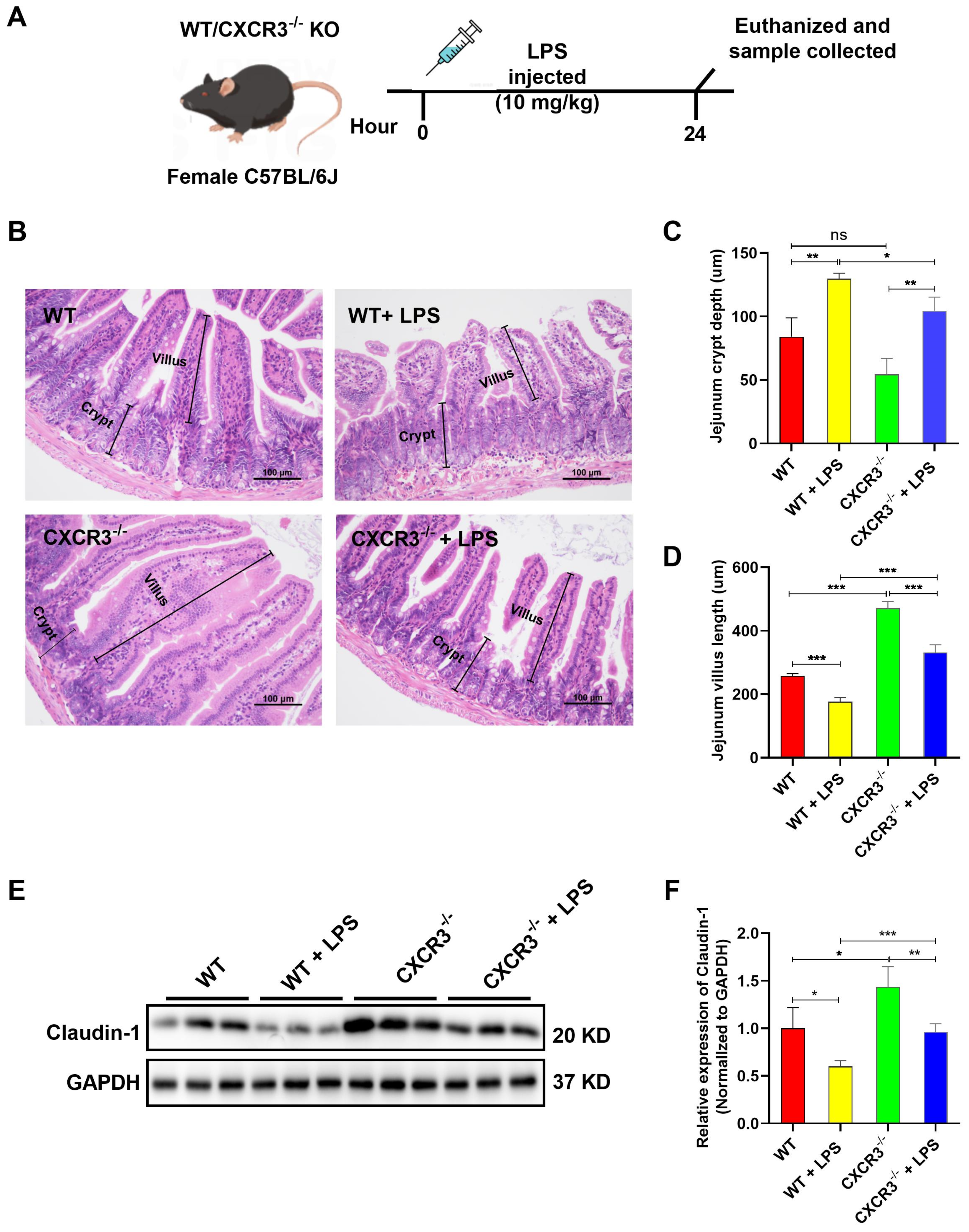
3.1. CXCR3 Knockout Attenuates Intestinal Mucosal Structural Damage and Increases Tight Junction Protein Expression in the Mouse Intestine
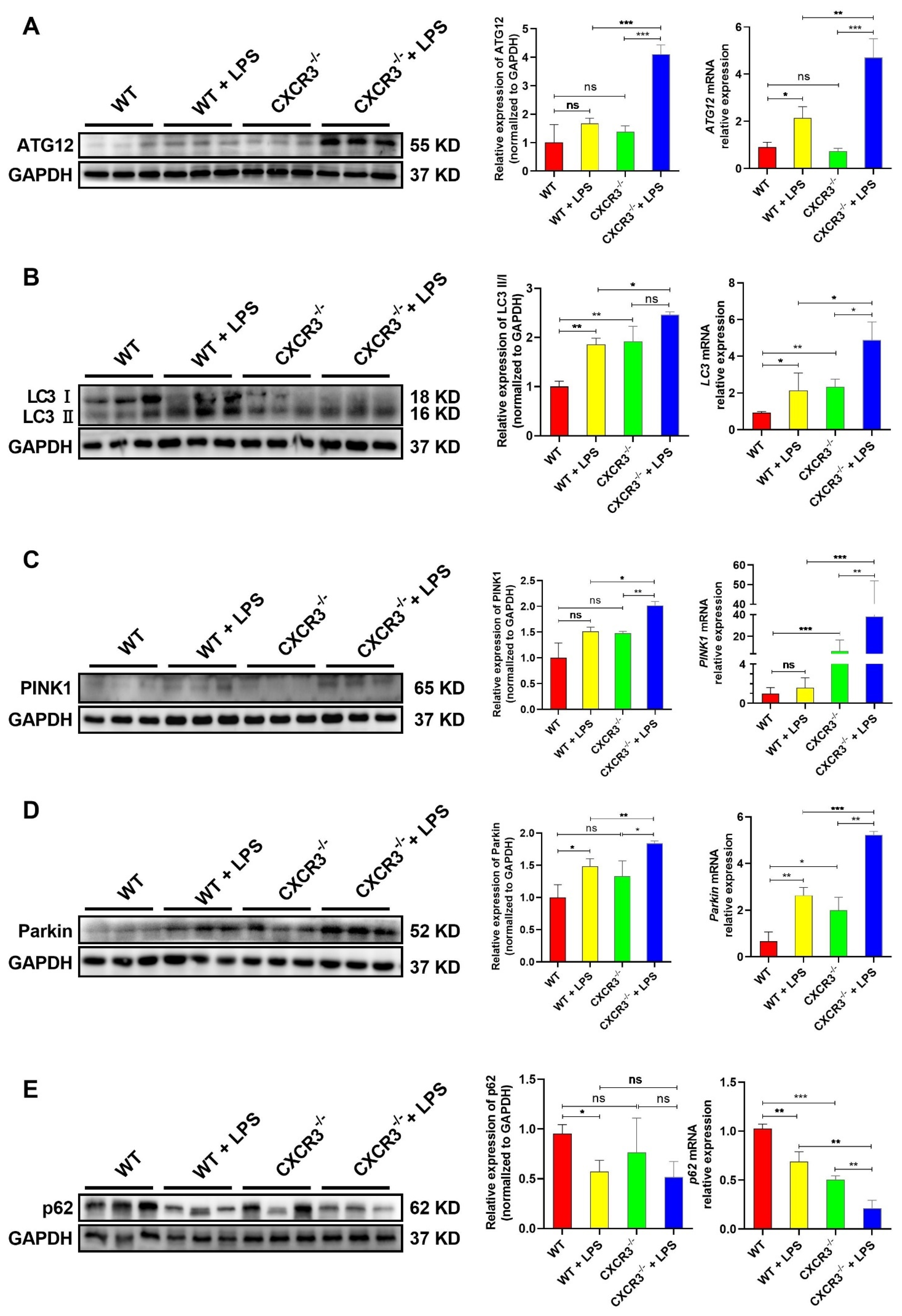
3.2. CXCR3 Knockout Promotes Autophagy in the Mouse Intestine
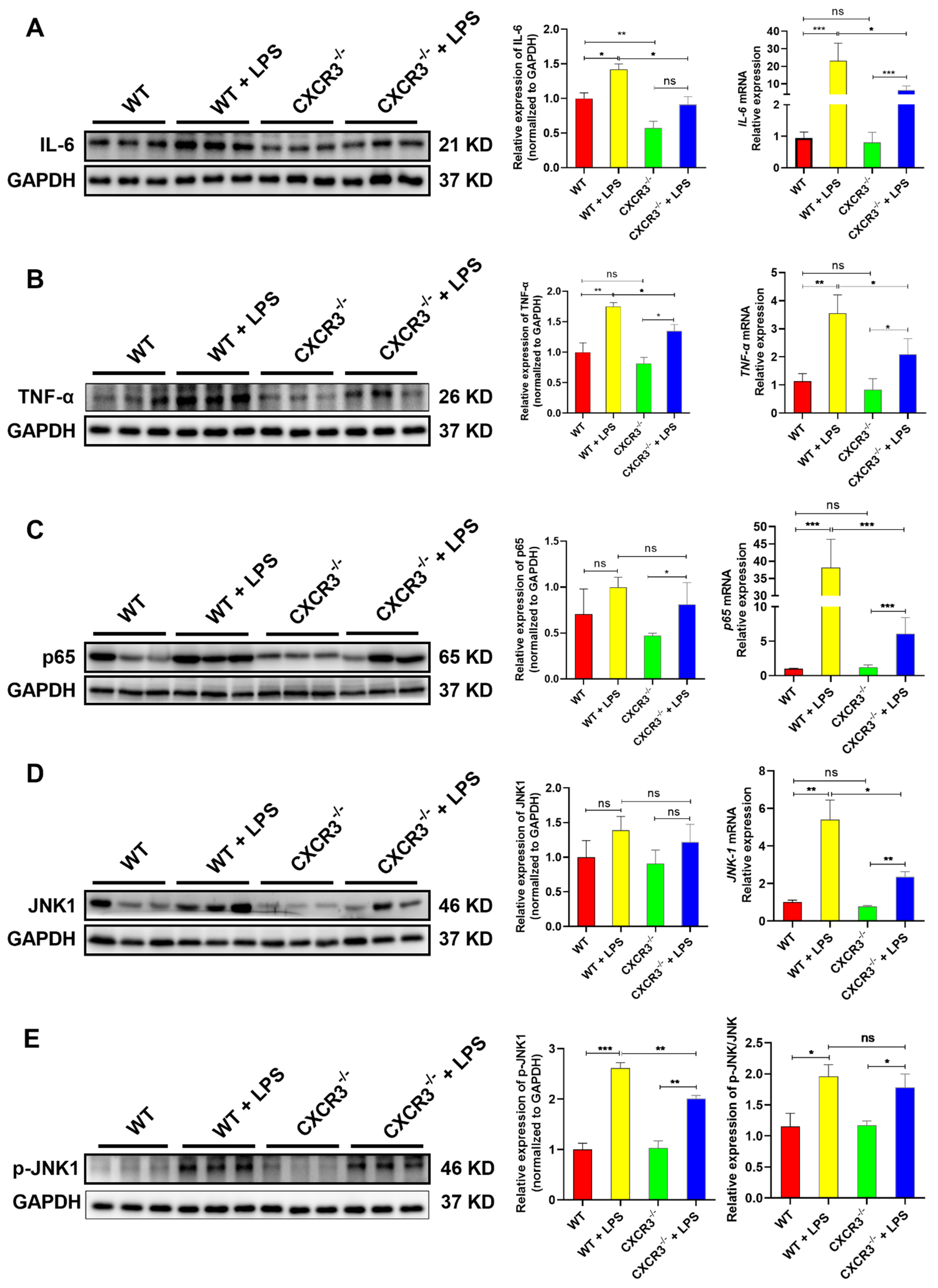
3.3. CXCR3 Knockout Blocks the Activation of the NF-κB Signaling Pathway in the Mouse Intestine
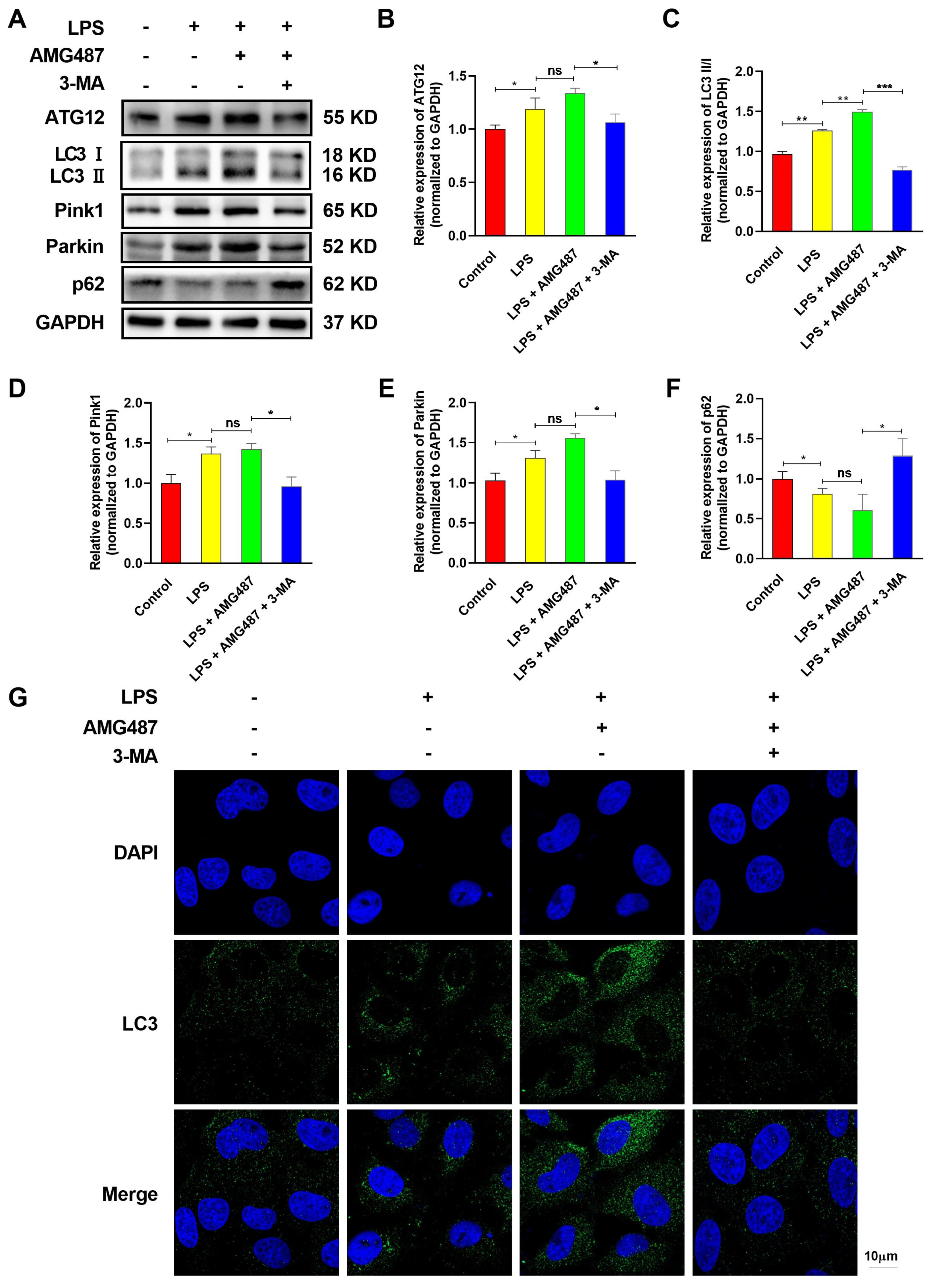
3.4. CXCR3 Inhibition Promotes Autophagy in IPEC-J2 Cells
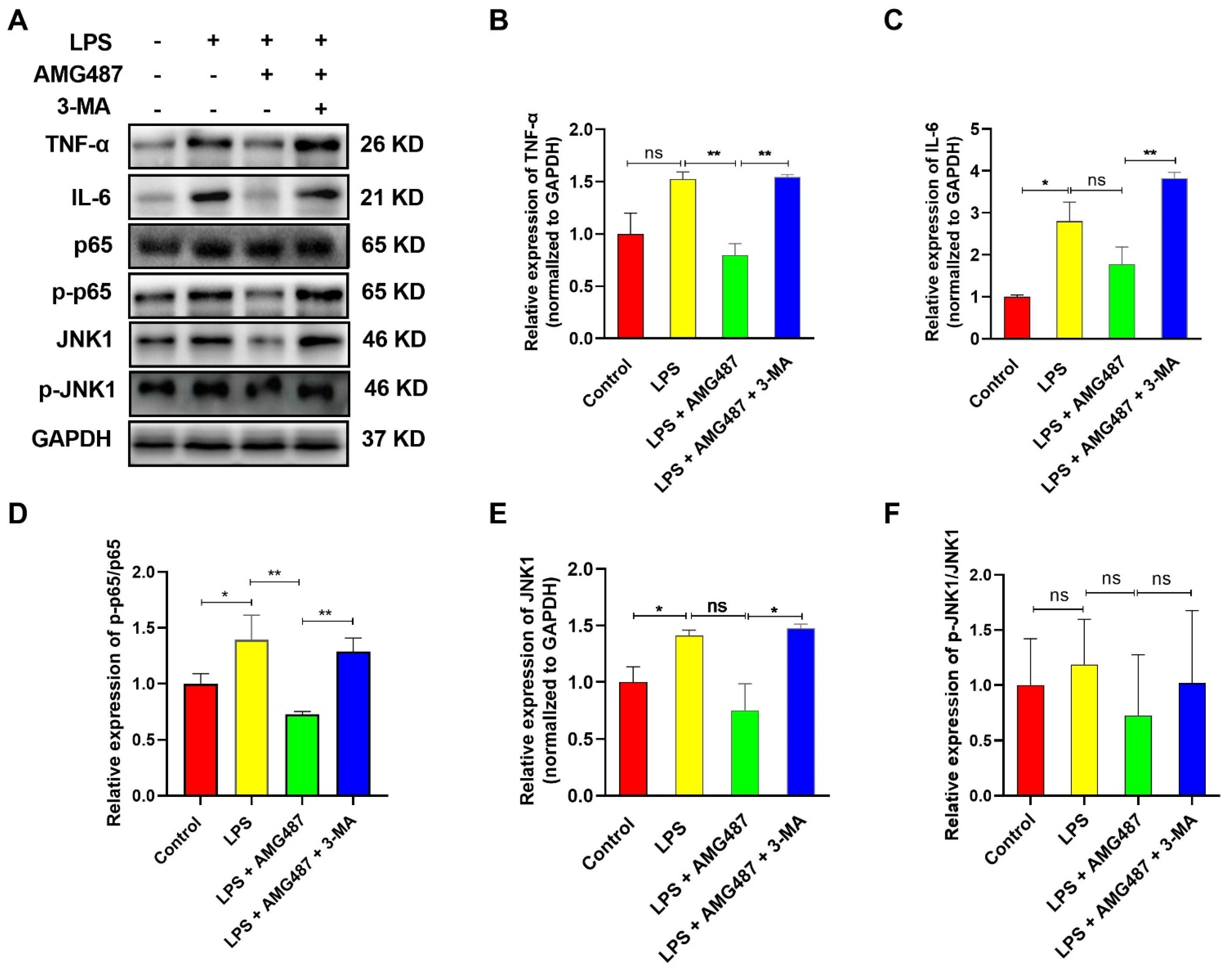
3.5. CXCR3 Inhibition Blocks the NF-κB Signaling Pathway by Promoting Autophagy in IPEC-J2 Cells
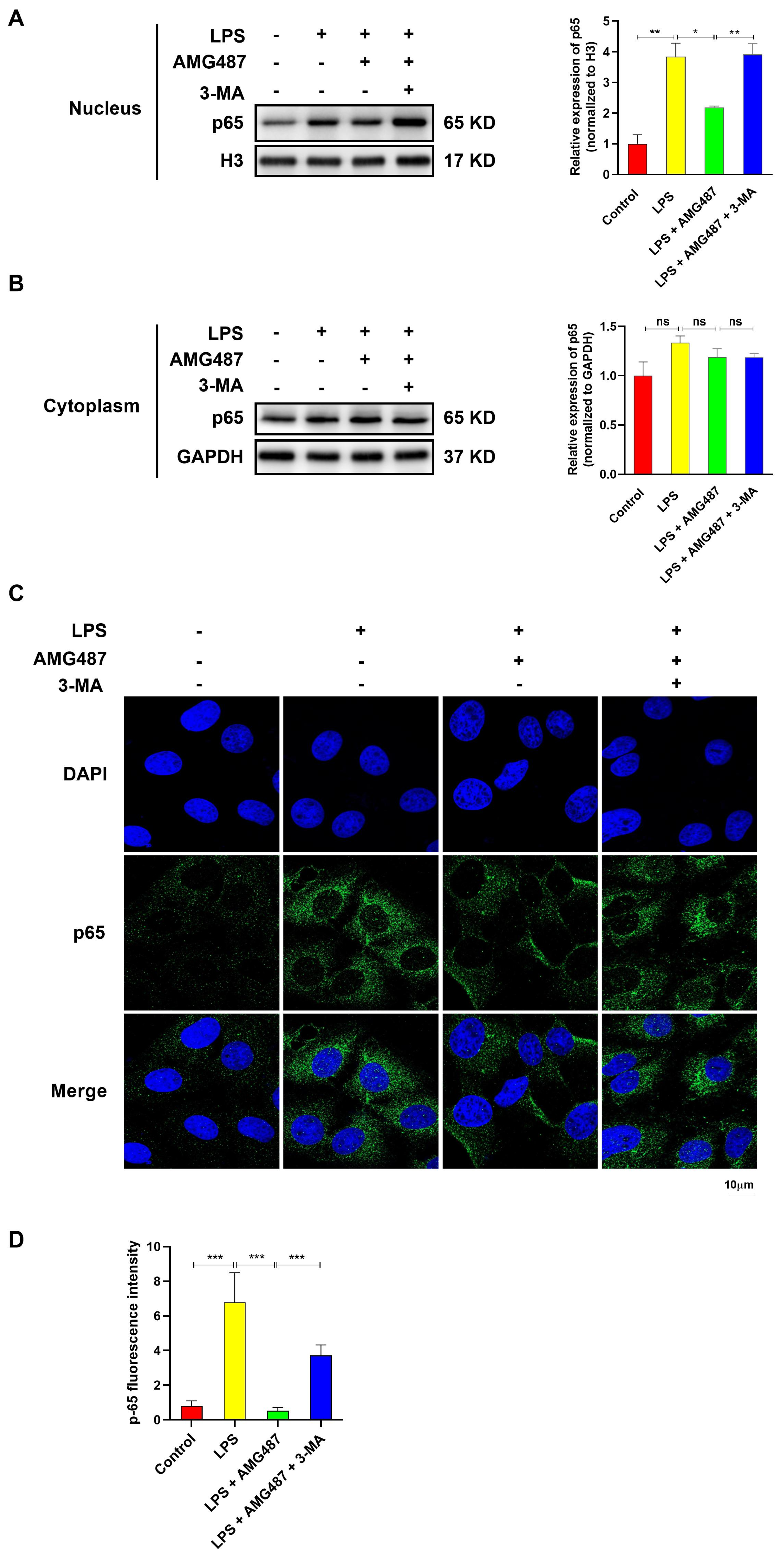
3.6. CXCR3 Inhibition Blocked p65 Nuclear Translocation by Promoting Autophagy in IPEC-J2 Cells
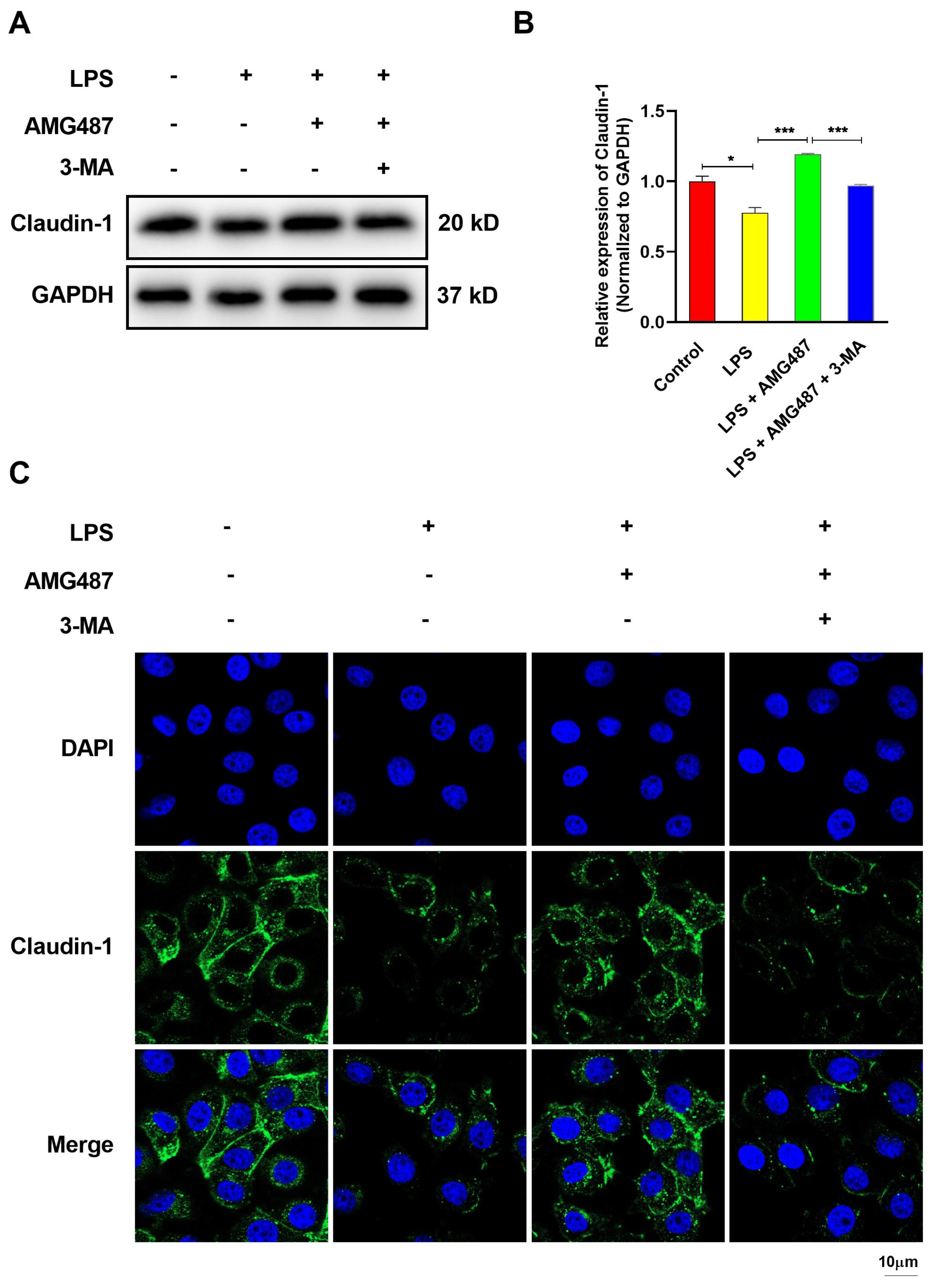
3.7. CXCR3 Inhibition Promotes Tight Junction Protein Expression in IPEC-J2 Cells
4. Discussion
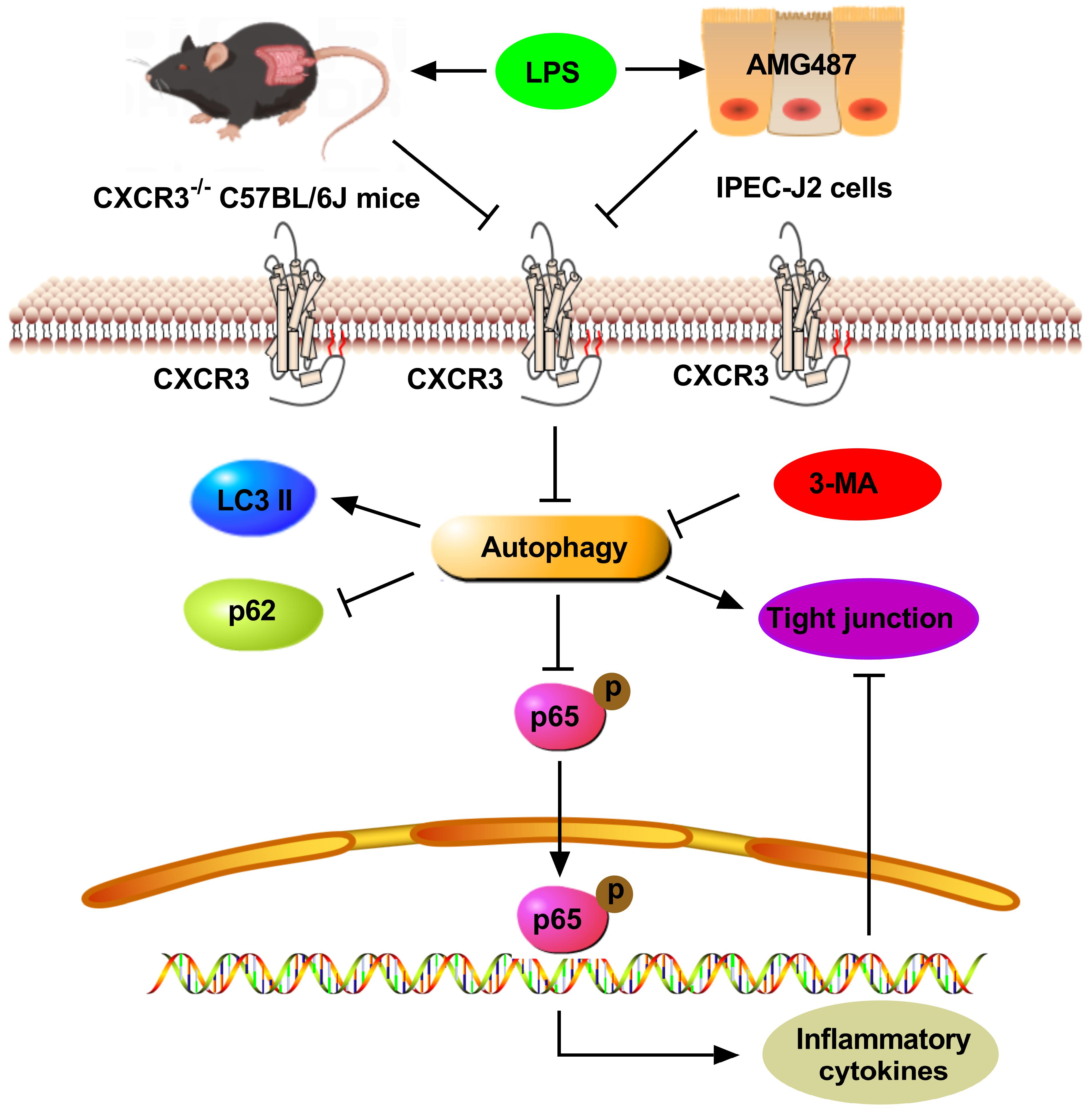
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alizadeh, A.; Akbari, P.; Garssen, J.; Fink-Gremmels, J.; Braber, S. Epithelial integrity, junctional complexes, and biomarkers associated with intestinal functions. Tissue Barriers 2021, 10, 1996830. [Google Scholar] [CrossRef]
- Aloyouny, A.Y.; Bepari, A.; Rahman, I. Evaluating the Role of CXCR3 in Pain Modulation: A Literature Review. J. Pain Res. 2020, 13, 1987–2001. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.D. The indigenous gastrointestinal microflora. Trends Microbiol. 1996, 4, 430–435. [Google Scholar] [CrossRef] [PubMed]
- Cambien, B.; Karimdjee, B.F.; Richard-Fiardo, P.; Bziouech, H.; Barthel, R.; Millet, M.A.; Martini, V.; Birnbaum, D.; Scoazec, J.Y.; Abello, J.; et al. Organ-specific inhibition of metastatic colon carcinoma by CXCR3 antagonism. Br. J. Cancer 2009, 100, 1755–1764. [Google Scholar] [CrossRef] [Green Version]
- Camilleri, M.; Madsen, K.; Spiller, R.; Greenwood-Van Meerveld, B.; Verne, G.N. Intestinal barrier function in health and gastrointestinal disease. Neurogastroenterol. Motil. 2012, 24, 503–512. [Google Scholar] [CrossRef]
- Chami, B.; Yeung, A.W.; van Vreden, C.; King, N.J.; Bao, S. The role of CXCR3 in DSS-induced colitis. PLoS ONE 2014, 9, e101622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, K.L.; Kuballa, P.; Song, J.H.; Patel, K.K.; Castoreno, A.B.; Yilmaz, O.H.; Jijon, H.B.; Zhang, M.; Aldrich, L.N.; Villablanca, E.J.; et al. Atg16l1 is required for autophagy in intestinal epithelial cells and protection of mice from Salmonella infection. Gastroenterology 2013, 145, 1347–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, B.; Lin, H.; Yu, J.; Yu, J.; Hu, Z. Autophagy and the Immune Response. Adv. Exp. Med. Biol. 2019, 1206, 595–634. [Google Scholar] [CrossRef]
- Czaja, M.J. Functions of autophagy in hepatic and pancreatic physiology and disease. Gastroenterology 2011, 140, 1895–1908. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Zheng, L.; Xie, X.; Wei, H.; Peng, J. GPA peptide enhances Nur77 expression in intestinal epithelial cells to exert a protective effect against DSS-induced colitis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 15364–15378. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Zhang, X.; Han, J.; Man, K.; Zhang, Y.; Chu, E.S.; Nan, Y.; Yu, J. Pro-Inflammatory CXCR3 Impairs Mitochondrial Function in Experimental Non-Alcoholic Steatohepatitis. Theranostics 2017, 7, 4192–4203. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foerster, E.G.; Mukherjee, T.; Cabral-Fernandes, L.; Rocha, J.D.B.; Girardin, S.E.; Philpott, D.J. How autophagy controls the intestinal epithelial barrier. Autophagy 2022, 18, 86–103. [Google Scholar] [CrossRef]
- Haghbin, M.; Rostami-Nejad, M.; Forouzesh, F.; Sadeghi, A.; Rostami, K.; Aghamohammadi, E.; Asadzadeh-Aghdaei, H.; Masotti, A.; Zali, M.R. The role of CXCR3 and its ligands CXCL10 and CXCL11 in the pathogenesis of celiac disease. Medicine 2019, 98, e15949. [Google Scholar] [CrossRef]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haq, S.; Grondin, J.; Banskota, S.; Khan, W.I. Autophagy: Roles in intestinal mucosal homeostasis and inflammation. J. Biomed. Sci. 2019, 26, 19. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-κB in immunobiology. Cell Res. 2011, 21, 223–244. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Deng, J.; Hu, X.; Zhou, S.; Wu, J.; Xiao, D.; Darko, K.O.; Huang, Y.; Tao, T.; Peng, M.; et al. Vitamin A inhibits the action of LPS on the intestinal epithelial barrier function and tight junction proteins. Food Funct. 2019, 10, 1235–1242. [Google Scholar] [CrossRef]
- He, F.; Wu, C.; Li, P.; Li, N.; Zhang, D.; Zhu, Q.; Ren, W.; Peng, Y. Functions and Signaling Pathways of Amino Acids in Intestinal Inflammation. Biomed. Res. Int. 2018, 2018, 9171905. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Song, J.; Xu, Y.; Liu, C.; Qian, W.; Bai, T.; Hou, X. Piezo1 regulates intestinal epithelial function by affecting the tight junction protein claudin-1 via the ROCK pathway. Life Sci. 2021, 275, 119254. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Leal-Ekman, J.S.; Lu, Q.; Stappenbeck, T.S. Atg14 protects the intestinal epithelium from TNF-triggered villus atrophy. Autophagy 2019, 15, 1990–2001. [Google Scholar] [CrossRef] [PubMed]
- Kabat, A.M.; Pott, J.; Maloy, K.J. The Mucosal Immune System and Its Regulation by Autophagy. Front. Immunol. 2016, 7, 240. [Google Scholar] [CrossRef] [Green Version]
- Kuenzig, M.E.; Yim, J.; Coward, S.; Eksteen, B.; Seow, C.H.; Barnabe, C.; Barkema, H.W.; Silverberg, M.S.; Lakatos, P.L.; Beck, P.L.; et al. The NOD2-Smoking Interaction in Crohn’s Disease is likely Specific to the 1007fs Mutation and may be Explained by Age at Diagnosis: A Meta-Analysis and Case-Only Study. EBioMedicine 2017, 21, 188–196. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Moon, K.M.; Kim, C.Y. Tight Junction in the Intestinal Epithelium: Its Association with Diseases and Regulation by Phytochemicals. J. Immunol. Res. 2018, 2018, 2645465. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Liang, D.; Zhuo, Y.; Guo, Z.; He, L.; Wang, X.; He, Y.; Li, L.; Dai, H. SIRT1/PGC-1 pathway activation triggers autophagy/mitophagy and attenuates oxidative damage in intestinal epithelial cells. Biochimie 2020, 170, 10–20. [Google Scholar] [CrossRef]
- Lucas, C.; Salesse, L.; Hoang, M.H.T.; Bonnet, M.; Sauvanet, P.; Larabi, A.; Godfraind, C.; Gagnière, J.; Pezet, D.; Rosenstiel, P.; et al. Autophagy of Intestinal Epithelial Cells Inhibits Colorectal Carcinogenesis Induced by Colibactin-Producing Escherichia coli in Apc(Min/+) Mice. Gastroenterology 2020, 158, 1373–1388. [Google Scholar] [CrossRef] [Green Version]
- Metzemaekers, M.; Vanheule, V.; Janssens, R.; Struyf, S.; Proost, P. Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front. Immunol. 2017, 8, 1970. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Sándor, G.O.; Juhász, G. Autophagy maintains stem cells and intestinal homeostasis in Drosophila. Sci. Rep. 2018, 8, 4644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nighot, P.K.; Hu, C.A.; Ma, T.Y. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J. Biol. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef] [Green Version]
- Nishida, A.; Ikeda, R.; Furuoka, H.; Nishikawa, Y. CXCR3-Dependent Immune Pathology in Mice following Infection with Toxoplasma gondii during Early Pregnancy. Infect. Immun. 2021, 89, e00253-20. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Oghumu, S.; Varikuti, S.; Terrazas, C.; Kotov, D.; Nasser, M.W.; Powell, C.A.; Ganju, R.K.; Satoskar, A.R. CXCR3 deficiency enhances tumor progression by promoting macrophage M2 polarization in a murine breast cancer model. Immunology 2014, 143, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Fleming-Martinez, A.; Bastea, L.; Doeppler, H.R.; Eisenhauer, J.; Le, T.; Edenfield, B.; Storz, P. CXCL10/CXCR3 signaling contributes to an inflammatory microenvironment and its blockade enhances progression of murine pancreatic precancerous lesions. eLife 2021, 10, e60646. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wang, Y.; Li, H.; Fan, J.; Shen, J.; Yu, X.; Zhou, Y.; Mao, H. ATG5-mediated autophagy suppresses NF-κB signaling to limit epithelial inflammatory response to kidney injury. Cell Death Dis. 2019, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Petrisko, T.J.; Konat, G.W. Peripheral viral challenge increases c-fos level in cerebral neurons. Metab. Brain Dis. 2021, 36, 1995–2002. [Google Scholar] [CrossRef]
- Qin, C.; Liu, H.; Tang, B.; Cao, M.; Yu, Z.; Liu, B.; Liu, W.; Dong, Y.; Ren, H. In Vitro Immunological Effects of CXCR3 Inhibitor AMG487 on Dendritic Cells. Arch. Immunol. Ther. Exp. 2020, 68, 11. [Google Scholar] [CrossRef]
- Ran, X.; Li, Y.; Chen, G.; Fu, S.; He, D.; Huang, B.; Wei, L.; Lin, Y.; Guo, Y.; Hu, G. Farrerol Ameliorates TNBS-Induced Colonic Inflammation by Inhibiting ERK1/2, JNK1/2, and NF-κB Signaling Pathway. Int. J. Mol. Sci. 2018, 19, 2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoultz, I.; Keita, Å.V. The Intestinal Barrier and Current Techniques for the Assessment of Gut Permeability. Cells 2020, 9, 1909. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M. Multifunctions of dietary polyphenols in the regulation of intestinal inflammation. J. Food Drug Anal. 2017, 25, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderholm, A.T.; Pedicord, V.A. Intestinal epithelial cells: At the interface of the microbiota and mucosal immunity. Immunology 2019, 158, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Li, L.; Liang, D.; Zhuo, Y.; Wang, X.; Dai, H. Knockdown of long noncoding RNA urothelial carcinoma associated 1 inhibits colorectal cancer cell proliferation and promotes apoptosis via modulating autophagy. J. Cell Physiol. 2019, 234, 7420–7434. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Foerster, E.G.; Tsalikis, J.; Abdel-Nour, M.; Mangiapane, J.; Sirluck-Schroeder, I.; Tattoli, I.; van Dalen, R.; Isenman, D.E.; Rohde, J.R.; et al. Complement C3 Drives Autophagy-Dependent Restriction of Cyto-invasive Bacteria. Cell Host Microbe 2018, 23, 644–652.e645. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. = Nihon Chikusan Gakkaiho 2020, 91, e13357. [Google Scholar] [CrossRef] [Green Version]
- Tan, P.; He, L.; Xing, C.; Mao, J.; Yu, X.; Zhu, M.; Diao, L.; Han, L.; Zhou, Y.; You, M.J.; et al. Myeloid loss of Beclin 1 promotes PD-L1hi precursor B cell lymphoma development. J. Clin. Invest. 2019, 129, 5261–5277. [Google Scholar] [CrossRef]
- Tang, Y.; Clayburgh, D.R.; Mittal, N.; Goretsky, T.; Dirisina, R.; Zhang, Z.; Kron, M.; Ivancic, D.; Katzman, R.B.; Grimm, G.; et al. Epithelial NF-kappaB enhances transmucosal fluid movement by altering tight junction protein composition after T cell activation. Am. J. Pathol. 2010, 176, 158–167. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Walser, T.C.; Rifat, S.; Ma, X.; Kundu, N.; Ward, C.; Goloubeva, O.; Johnson, M.G.; Medina, J.C.; Collins, T.L.; Fulton, A.M. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res. 2006, 66, 7701–7707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Guo, S.; Zhang, Y.; Zhang, Y.; Zhang, H. Remifentanil attenuates sepsis-induced intestinal injury by inducing autophagy. Bioengineered 2021, 12, 9575–9584. [Google Scholar] [CrossRef]
- Wu, W.; Wang, S.; Liu, Q.; Shan, T.; Wang, Y. Metformin Protects against LPS-Induced Intestinal Barrier Dysfunction by Activating AMPK Pathway. Mol. Pharm. 2018, 15, 3272–3284. [Google Scholar] [CrossRef]
- Wu, X.X.; Huang, X.L.; Chen, R.R.; Li, T.; Ye, H.J.; Xie, W.; Huang, Z.M.; Cao, G.Z. Paeoniflorin Prevents Intestinal Barrier Disruption and Inhibits Lipopolysaccharide (LPS)-Induced Inflammation in Caco-2 Cell Monolayers. Inflammation 2019, 42, 2215–2225. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, L.; Wang, B.; Sun, Q.; Zhao, P.; Li, W. The role of autophagy in maintaining intestinal mucosal barrier. J. Cell Physiol. 2019, 234, 19406–19419. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Han, J.; Man, K.; Li, X.; Du, J.; Chu, E.S.; Go, M.Y.; Sung, J.J.; Yu, J. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J. Hepatol. 2016, 64, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Li, M.; Sun, K.; Su, S.; Geng, T.; Sun, H. Hippophae rhamnoides polysaccharides protect IPEC-J2 cells from LPS-induced inflammation, apoptosis and barrier dysfunction in vitro via inhibiting TLR4/NF-kappaB signaling pathway. Int. J. Biol. Macromol. 2020, 155, 1202–1215. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.Y.; He, X.Y.; Li, W.; Song, T.Z.; Han, J.B.; Yang, X.; Liu, F.L.; Luo, R.H.; Tian, R.R.; Feng, X.L.; et al. Pro-inflammatory microenvironment and systemic accumulation of CXCR3+ cell exacerbate lung pathology of old rhesus macaques infected with SARS-CoV-2. Signal Transduct. Target. Ther. 2021, 6, 328. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine 2018, 35, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Cao, Q.; Peng, Y.; Zhang, Q.J.; Castrillon, D.H.; DePinho, R.A.; Liu, Z.P. FoxO4 inhibits NF-kappaB and protects mice against colonic injury and inflammation. Gastroenterology 2009, 137, 1403–1414. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Y.; Li, C. Autophagy plays a positive role in zinc-induced apoptosis in intestinal porcine epithelial cells. Toxicol. Vitr. 2017, 44, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Vergote, D.; Pardo, C.; Noorbakhsh, F.; McArthur, J.C.; Hollenberg, M.D.; Overall, C.M.; Power, C. CXCR3 activation by lentivirus infection suppresses neuronal autophagy: Neuroprotective effects of antiretroviral therapy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 2928–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, L.; Kuo, W.T.; Turner, J.R. Tight Junctions as Targets and Effectors of Mucosal Immune Homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 327–340. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sense (5′-3′) | Antisense (5′-3′) |
|---|---|---|
| Pink1 | TGTCAGGAGATCCAGGCAATTTTTA | CTTCAGGACGACATCCGGGC |
| Parkin | ACAGAGACCACGGAGGAGAA | TAACTGGACCTCTGGCTGCT |
| LC3 | TCCTGGATAAGACCAAGTTTCTG | ATAGATGTCAGCGATGGGTGTG |
| p62 | ACGGAGTACCTGAACCCTCT | CTCGAGTCACAGTGGACCCT |
| IL-6 | AACGATGATGCACTTGCAGA | TGGTACTCCAGAAGAAGACCAGAGG |
| TNF-α | GAGGCACTCCCCCAAAAGAT | CACTTGGTGGTTTGCTACGAC |
| p65 | TGAACTTGTGGGGAAGGACTG | AGGTCTGTTTGGAAACTGGAGA |
| JNK1 | TCTCCTTTAGCACAGGTGCAG | CTGCTGTCTGTATCCGAGGC |
| ATG12 | CTTACGGATGTCTCCCCCAGA | ATGAGTCCTTGGATGGTTGG |
| GAPDH | AAATGGTGAAGGTCGGTGTGAAC | CAACAATCTCCACTTTGCCACTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Deng, Y.; Zhang, Y.; Ba, T.; Niu, S.; Chen, Y.; Gao, Y.; Dai, H. CXCR3 Inhibition Blocks the NF-κB Signaling Pathway by Elevating Autophagy to Ameliorate Lipopolysaccharide-Induced Intestinal Dysfunction in Mice. Cells 2023, 12, 182. https://doi.org/10.3390/cells12010182
Zhang C, Deng Y, Zhang Y, Ba T, Niu S, Chen Y, Gao Y, Dai H. CXCR3 Inhibition Blocks the NF-κB Signaling Pathway by Elevating Autophagy to Ameliorate Lipopolysaccharide-Induced Intestinal Dysfunction in Mice. Cells. 2023; 12(1):182. https://doi.org/10.3390/cells12010182
Chicago/Turabian StyleZhang, Cheng, Yian Deng, Yingsi Zhang, Tongtong Ba, Sai Niu, Yiqin Chen, Yuan Gao, and Hanchuan Dai. 2023. "CXCR3 Inhibition Blocks the NF-κB Signaling Pathway by Elevating Autophagy to Ameliorate Lipopolysaccharide-Induced Intestinal Dysfunction in Mice" Cells 12, no. 1: 182. https://doi.org/10.3390/cells12010182