

eIF4A/PDCD4 Pathway, a Factor for Doxorubicin Chemoresistance in a Triple-Negative Breast Cancer Cell Model
 ,
,  , , , , and
, , , , and
Abstract
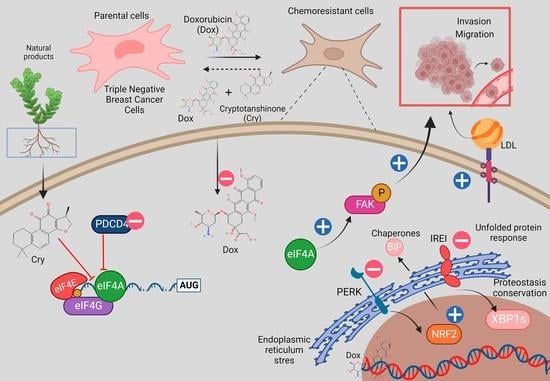
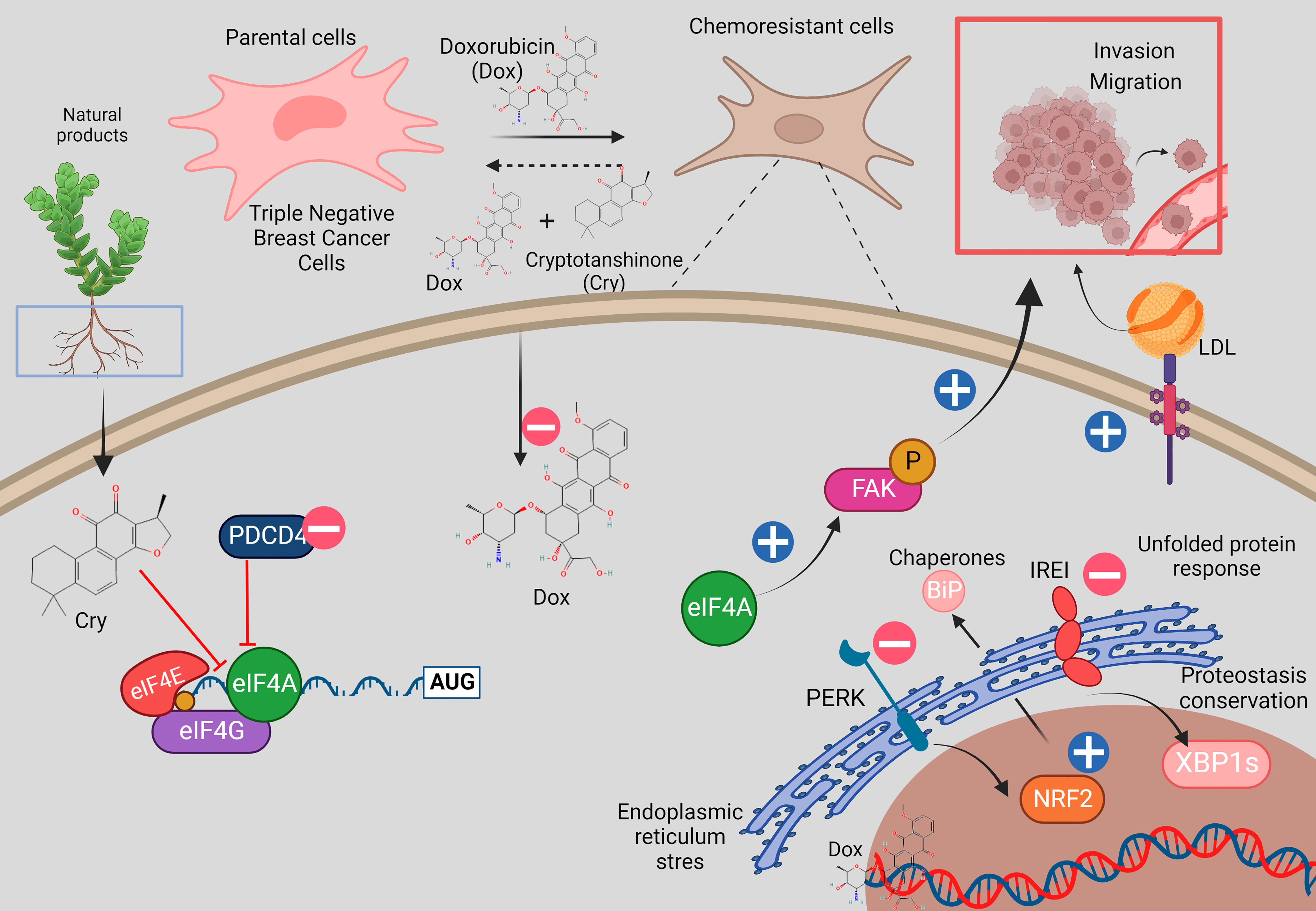
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Chemoresistance Protocols
2.4. Cell Viability
2.5. Western Blot (WB) Analysis
2.6. Characterization of Proteins in Extracellular Media
2.7. Immunoprecipitation Assays
2.8. Nucleus Isolation
2.9. Evaluation of Cellular Dox-Internalization
2.9.1. Cell Cytometer Assays
2.9.2. Dox Quantification in Supernatant Media
2.10. Scratch-Wound Assay
2.11. Cellular Invasion Assays
2.12. Zymography
2.13. Purification of Tanshinone Molecules
2.14. Isolation of LDL and Fluorescent Labeling
2.15. Molecular Docking
2.16. Small Interfering RNA (siRNA)
2.17. Overexpression of eIF4AI in MDA-MB-231 Variants
2.18. eIF4AI Overexpression and Purification
2.19. Statistical Analysis
3. Results
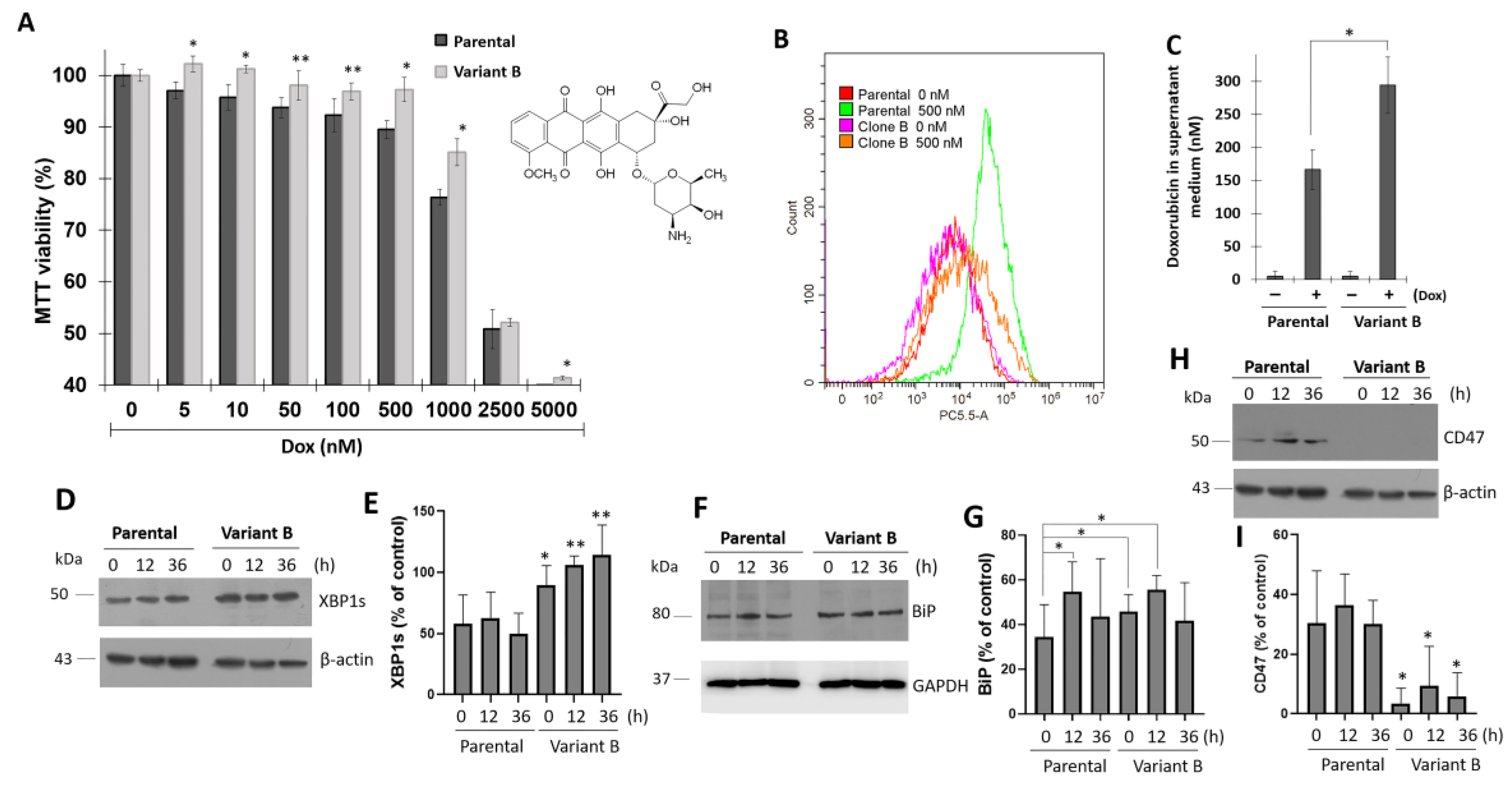
3.1. Association between UPR and Dox-Induced Chemoresistance
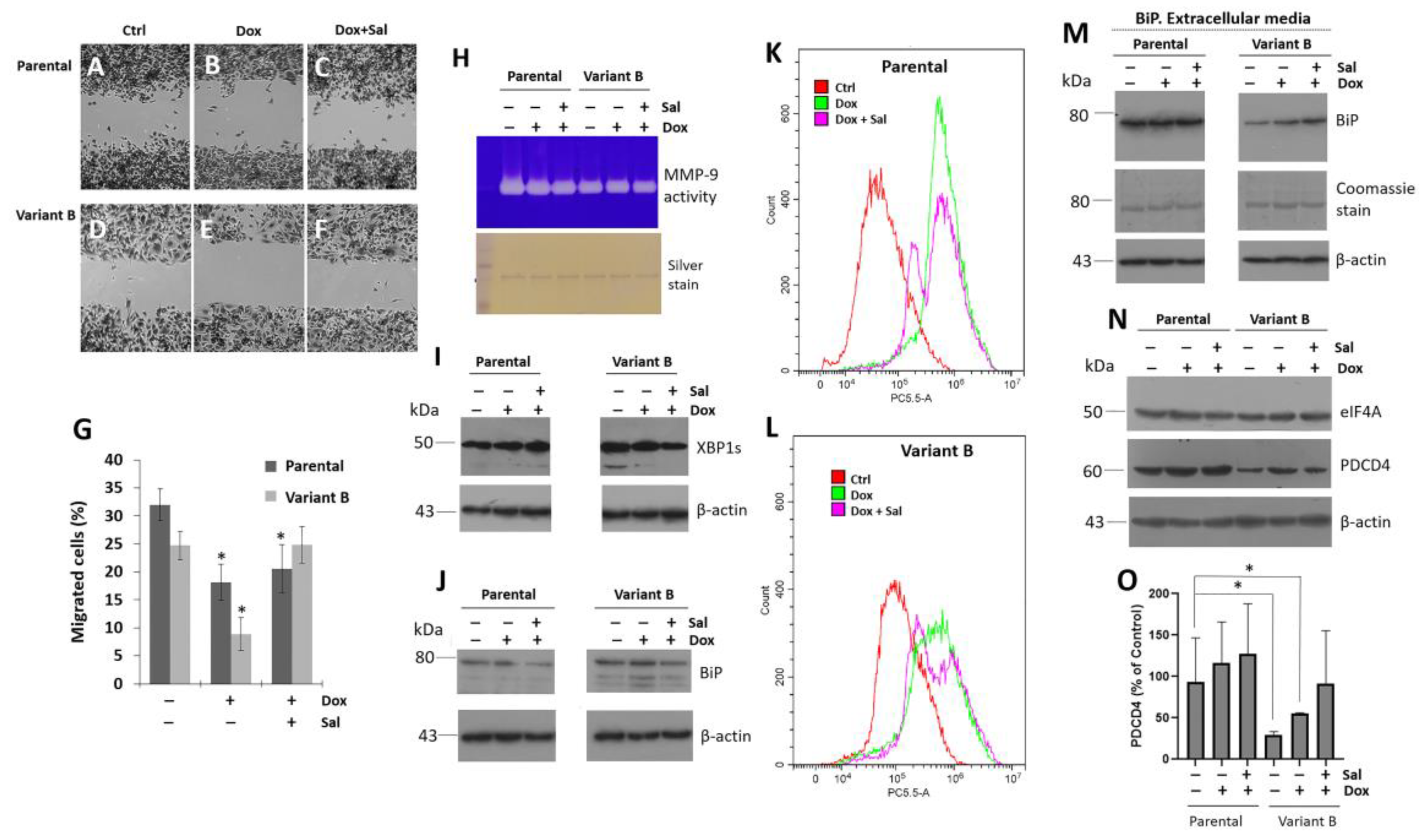
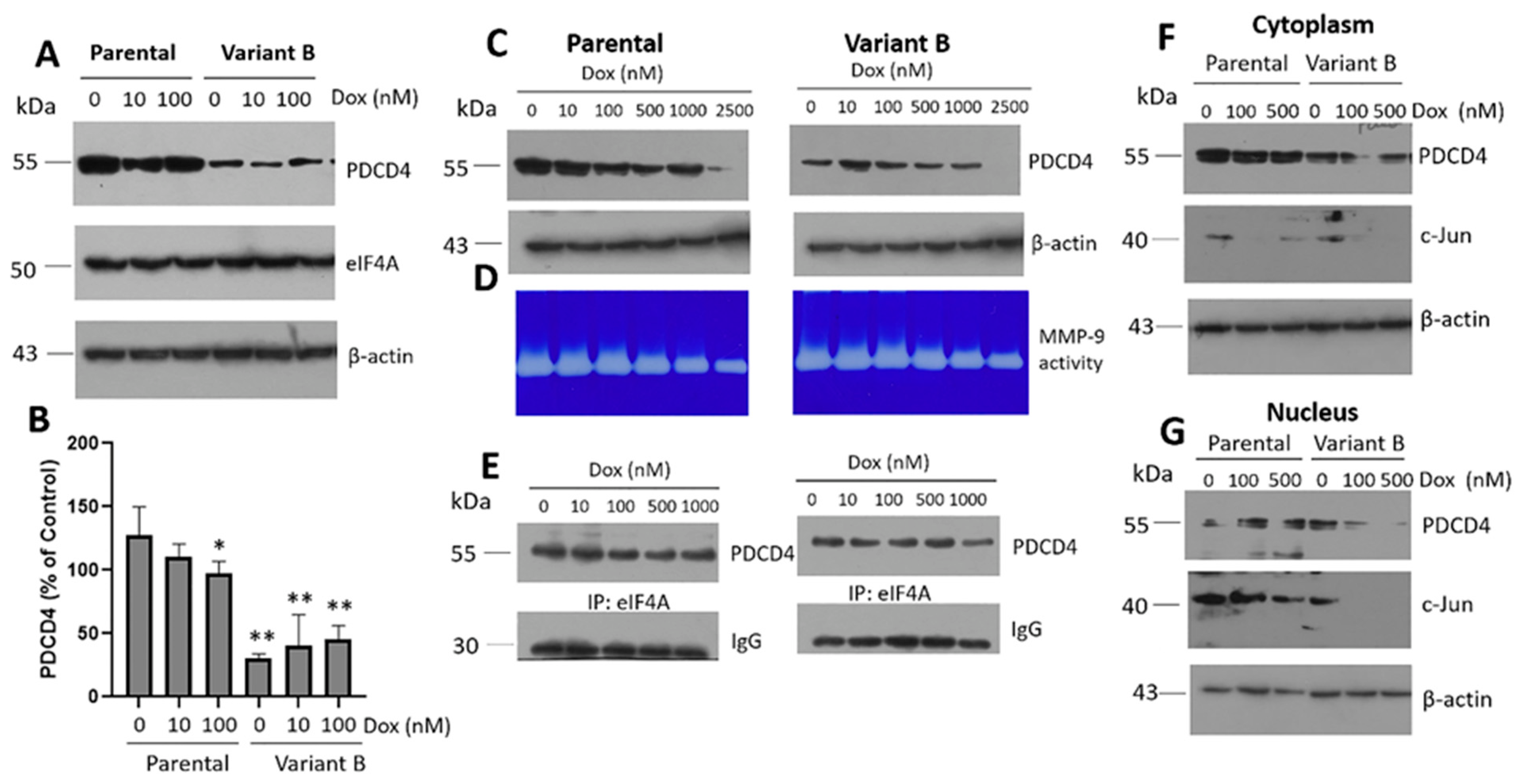
3.2. Cell Migratory Capacity Is Associated with the Disruption of the PDCD4 Tumor Suppressor Gene
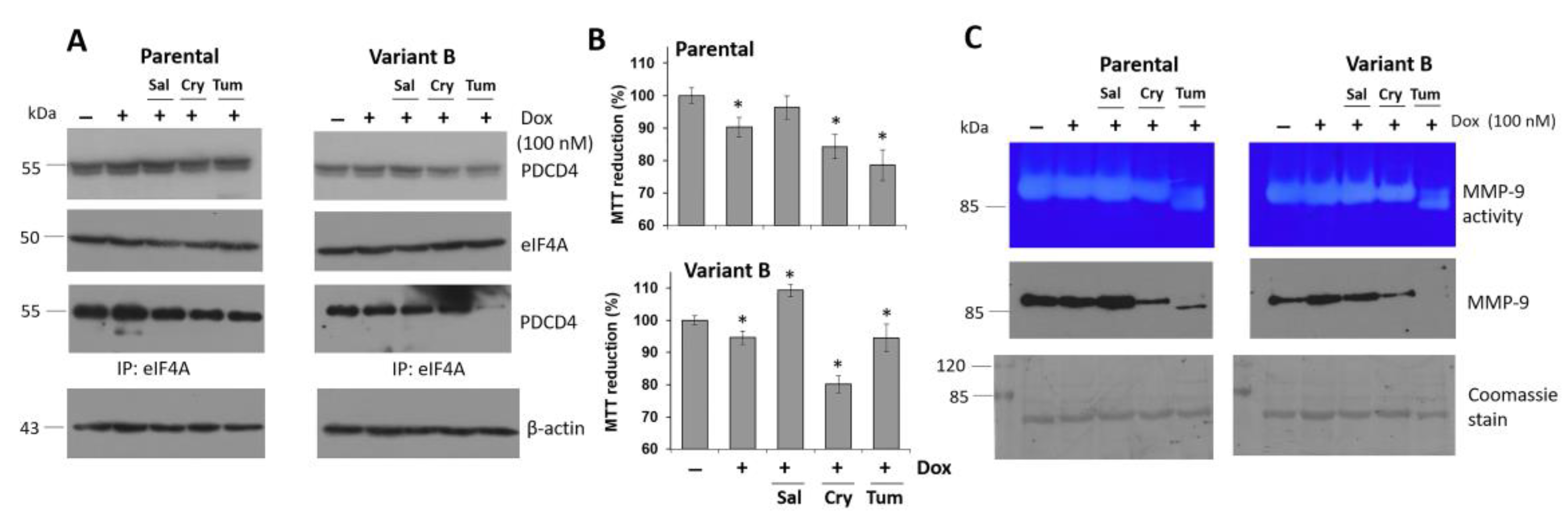
3.3. Characterization of the Role of the Tumor Suppressor Gene PDCD4
3.4. Small Molecule Treatment Can Desensitize the Chemoresistance of BC Cells
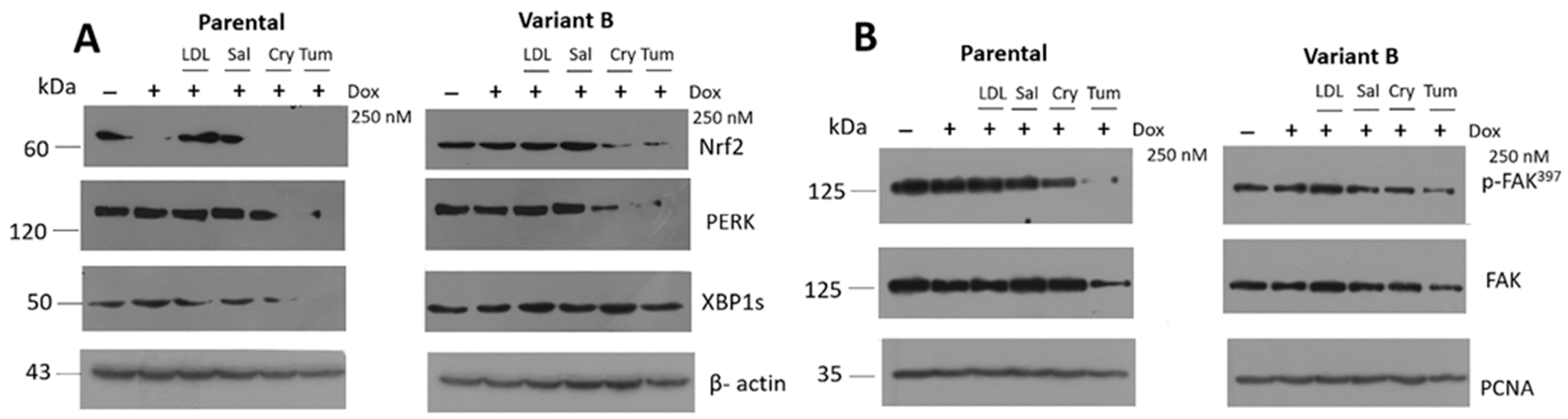
3.5. Relevance of PERK/Nrf2 and Regulation by Terpene-Derived Molecules
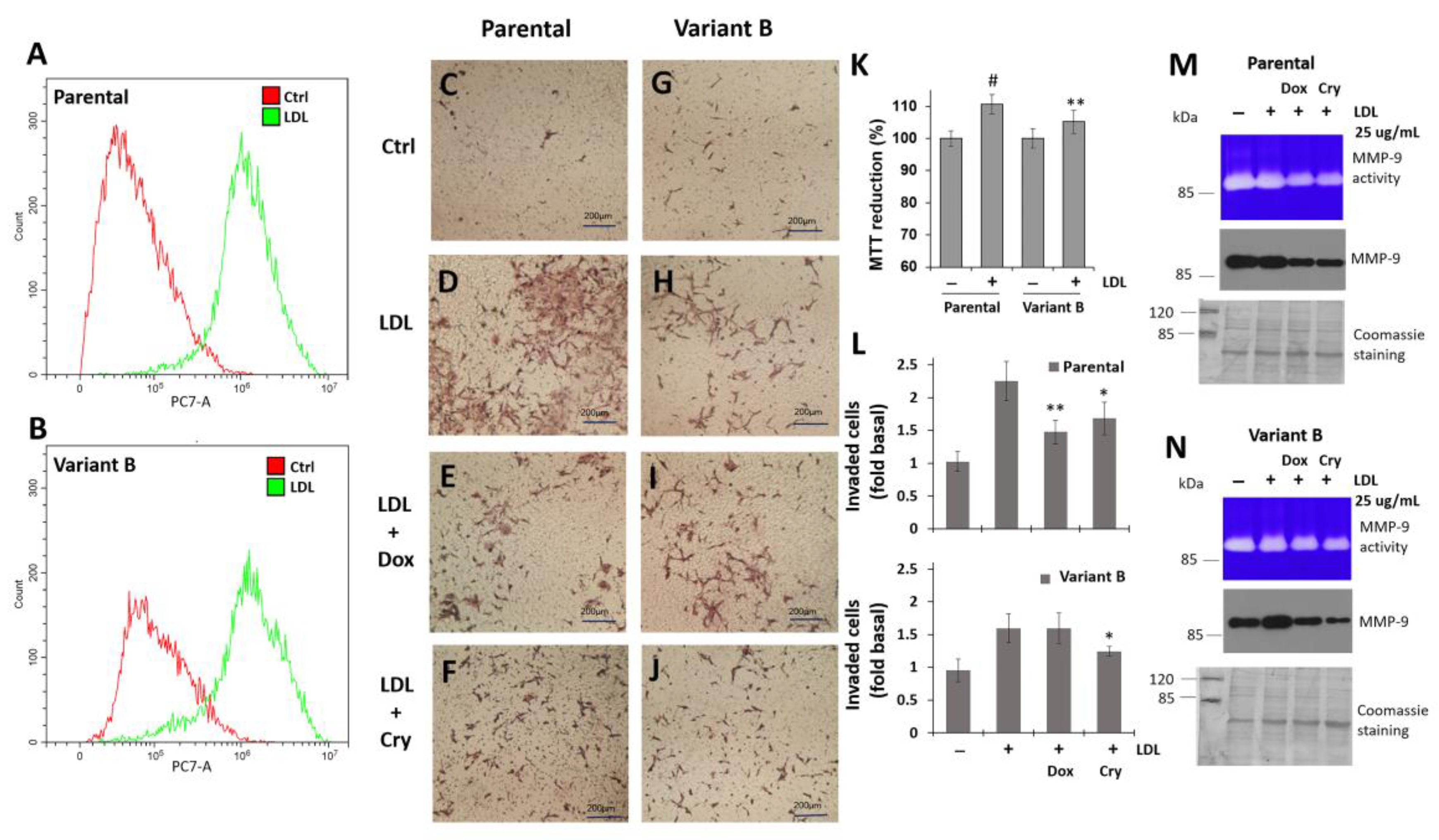
3.6. Regulation of Cell Invasion by Cry Treatment
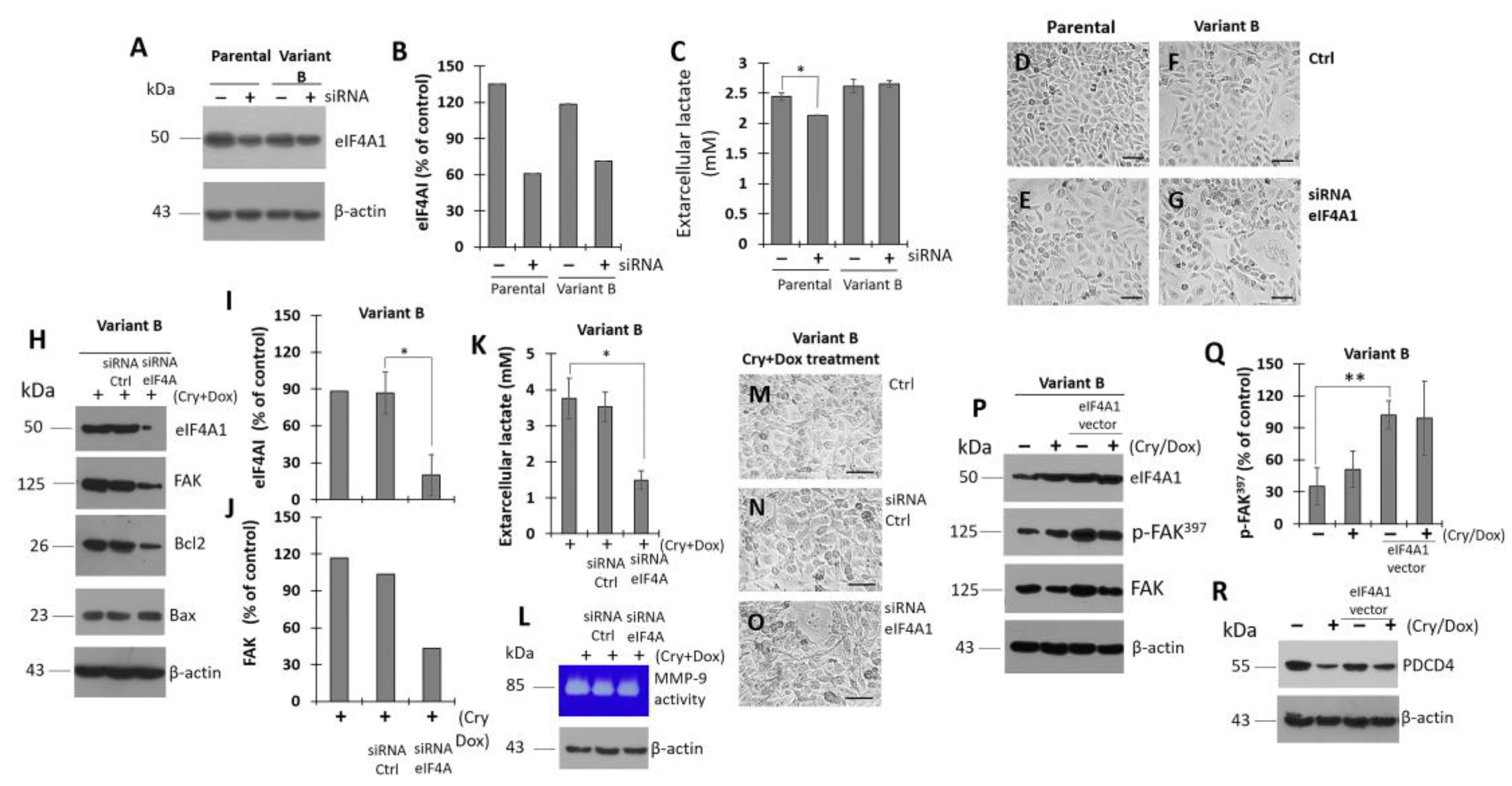
3.7. Modulation of eIF4A on Oncogenic Factor FAK
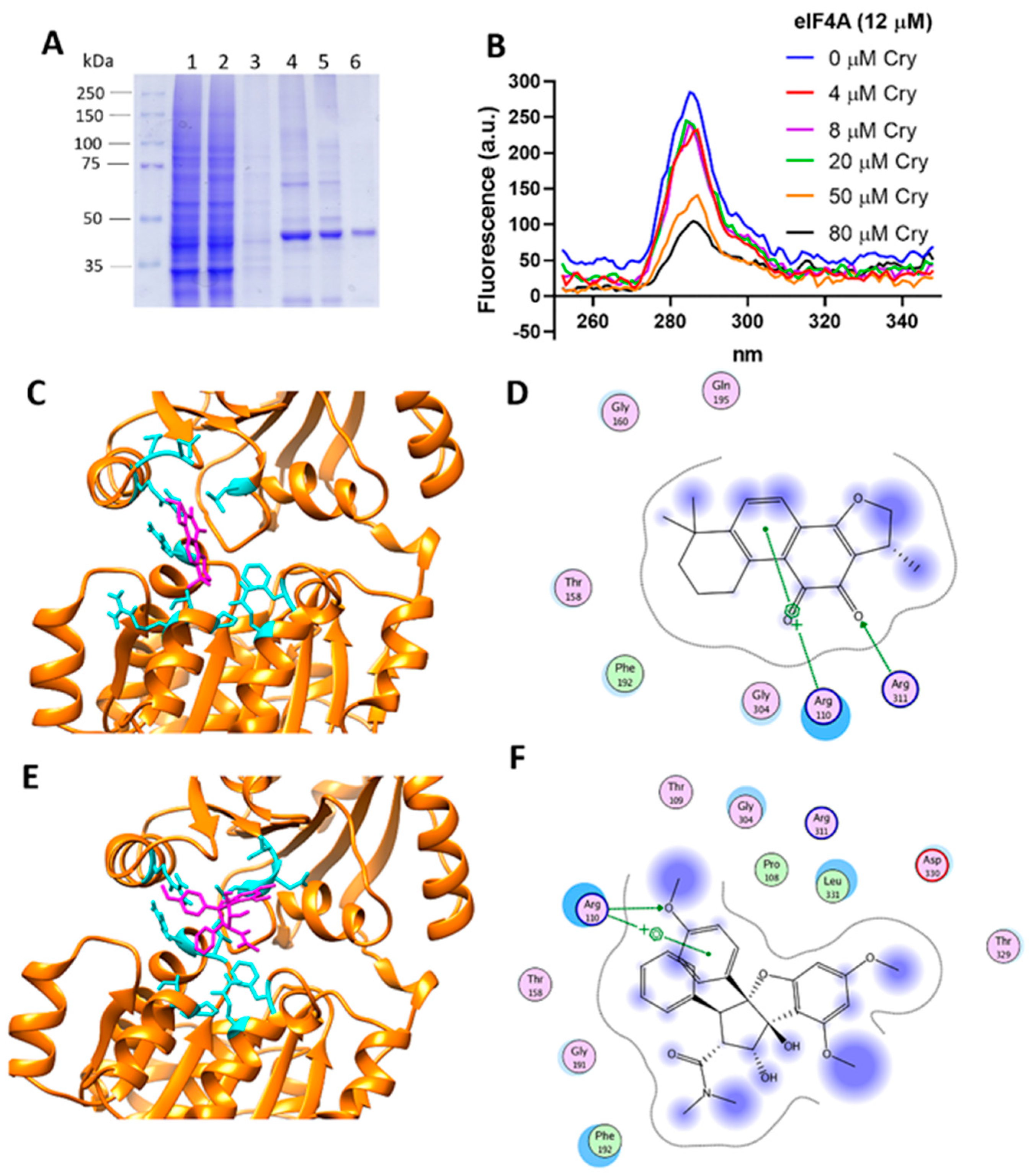
3.8. Cryptotanshinone Mechanism Is Mediated by eIF4A Interaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rigiracciolo, D.C.; Nohata, N.; Lappano, R.; Cirillo, F.; Talia, M.; Scordamaglia, D.; Gutkind, J.S.; Maggiolini, M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020, 9, 1010. [Google Scholar] [CrossRef] [Green Version]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [Green Version]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The Landscape of Targeted Therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Pan, H.; Chen, Y.; Xu, Y.H.; Yang, W.; Wu, Z. A review of current progress in triple-negative breast cancer therapy. Open Med. 2020, 15, 1143–1149. [Google Scholar] [CrossRef]
- Hua, Z.; White, J.; Zhou, J. Cancer stem cells in TNBC. Semin. Cancer Biol. 2022, 82, 26–34. [Google Scholar] [CrossRef]
- Diaz-Villanueva, J.F.; Diaz-Molina, R.; Garcia-Gonzalez, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef] [Green Version]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Plygawko, A.T.; Kan, S.; Campbell, K. Epithelial-mesenchymal plasticity: Emerging parallels between tissue morphogenesis and cancer metastasis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20200087. [Google Scholar] [CrossRef]
- Liang, D.; Khoonkari, M.; Avril, T.; Chevet, E.; Kruyt, F.A.E. The unfolded protein response as regulator of cancer stemness and differentiation: Mechanisms and implications for cancer therapy. Biochem. Pharmacol. 2021, 192, 114737. [Google Scholar] [CrossRef]
- Lehman, S.L.; Wilson, E.D.; Camphausen, K.; Tofilon, P.J. Translation Initiation Machinery as a Tumor Selective Target for Radiosensitization. Int. J. Mol. Sci. 2021, 22, 10664. [Google Scholar] [CrossRef]
- Zindy, P.; Berge, Y.; Allal, B.; Filleron, T.; Pierredon, S.; Cammas, A.; Beck, S.; Mhamdi, L.; Fan, L.; Favre, G.; et al. Formation of the eIF4F translation-initiation complex determines sensitivity to anticancer drugs targeting the EGFR and HER2 receptors. Cancer Res. 2011, 71, 4068–4073. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.S.; Jansen, A.P.; Komar, A.A.; Zheng, X.; Merrick, W.C.; Costes, S.; Lockett, S.J.; Sonenberg, N.; Colburn, N.H. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell Biol. 2003, 23, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Waters, L.C.; Strong, S.L.; Ferlemann, E.; Oka, O.; Muskett, F.W.; Veverka, V.; Banerjee, S.; Schmedt, T.; Henry, A.J.; Klempnauer, K.H.; et al. Structure of the tandem MA-3 region of Pdcd4 protein and characterization of its interactions with eIF4A and eIF4G: Molecular mechanisms of a tumor suppressor. J. Biol. Chem. 2011, 286, 17270–17280. [Google Scholar] [CrossRef] [Green Version]
- Moustafa-Kamal, M.; Kucharski, T.J.; El-Assaad, W.; Abbas, Y.M.; Gandin, V.; Nagar, B.; Pelletier, J.; Topisirovic, I.; Teodoro, J.G. The mTORC1/S6K/PDCD4/eIF4A Axis Determines Outcome of Mitotic Arrest. Cell Rep. 2020, 33, 108230. [Google Scholar] [CrossRef]
- Wen, Y.H.; Shi, X.; Chiriboga, L.; Matsahashi, S.; Yee, H.; Afonja, O. Alterations in the expression of PDCD4 in ductal carcinoma of the breast. Oncol. Rep. 2007, 18, 1387–1393. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Wan, L.; Tang, Z.; Yao, C.; Zhang, D.; Jiang, M.; Wang, C.; Liu, Y.; Xue, C.; Wang, X.; et al. TRIM27 regulates the expression of PDCD4 by the ubiquitin-proteasome pathway in ovarian and endometrial cancer cells. Oncol. Rep. 2022, 48, 120. [Google Scholar] [CrossRef]
- Gerson-Gurwitz, A.; Young, N.P.; Goel, V.K.; Eam, B.; Stumpf, C.R.; Chen, J.; Fish, S.; Barrera, M.; Sung, E.; Staunton, J.; et al. Zotatifin, an eIF4A-Selective Inhibitor, Blocks Tumor Growth in Receptor Tyrosine Kinase Driven Tumors. Front. Oncol. 2021, 11, 766298. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006, 38, 317–332. [Google Scholar] [CrossRef]
- Hwang, S.K.; Jeong, Y.J.; Chang, Y.C. PDCD4 inhibits lung tumorigenesis by the suppressing p62-Nrf2 signaling pathway and upregulating Keap1 expression. Am. J. Cancer Res. 2020, 10, 424–439. [Google Scholar]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Ajzashokouhi, A.H.; Bostan, H.B.; Jomezadeh, V.; Hayes, A.W.; Karimi, G. A review on the cardioprotective mechanisms of metformin against doxorubicin. Hum. Exp. Toxicol. 2020, 39, 237–248. [Google Scholar] [CrossRef]
- Pilco-Ferreto, N.; Calaf, G.M. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int. J. Oncol. 2016, 49, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and doxorubicin resistance in cancer. Anticancer Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef]
- Al-Malky, H.S.; Al Harthi, S.E.; Osman, A.M. Major obstacles to doxorubicin therapy: Cardiotoxicity and drug resistance. J. Oncol. Pharm. Pract. 2020, 26, 434–444. [Google Scholar] [CrossRef]
- Martin, M.; Villar, A.; Sole-Calvo, A.; Gonzalez, R.; Massuti, B.; Lizon, J.; Camps, C.; Carrato, A.; Casado, A.; Candel, M.T.; et al. Doxorubicin in combination with fluorouracil and cyclophosphamide (i.v. FAC regimen, day 1, 21) versus methotrexate in combination with fluorouracil and cyclophosphamide (i.v. CMF regimen, day 1, 21) as adjuvant chemotherapy for operable breast cancer: A study by the GEICAM group. Ann. Oncol. 2003, 14, 833–842. [Google Scholar] [CrossRef]
- Isakoff, S.J. Triple-negative breast cancer: Role of specific chemotherapy agents. Cancer J. 2010, 16, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Akman, M.; Belisario, D.C.; Salaroglio, I.C.; Kopecka, J.; Donadelli, M.; De Smaele, E.; Riganti, C. Hypoxia, endoplasmic reticulum stress and chemoresistance: Dangerous liaisons. J. Exp. Clin. Cancer Res. 2021, 40, 28. [Google Scholar] [CrossRef]
- Xu, Y.; Lou, Z.; Lee, S.H. Arctigenin represses TGF-beta-induced epithelial mesenchymal transition in human lung cancer cells. Biochem. Biophys. Res. Commun. 2017, 493, 934–939. [Google Scholar] [CrossRef]
- Giacomelli, C.; Daniele, S.; Natali, L.; Iofrida, C.; Flamini, G.; Braca, A.; Trincavelli, M.L.; Martini, C. Carnosol controls the human glioblastoma stemness features through the epithelial-mesenchymal transition modulation and the induction of cancer stem cell apoptosis. Sci. Rep. 2017, 7, 15174. [Google Scholar] [CrossRef] [Green Version]
- Oblinger, J.L.; Burns, S.S.; Huang, J.; Pan, L.; Ren, Y.; Shen, R.; Kinghorn, A.D.; Welling, D.B.; Chang, L.S. Overexpression of eIF4F components in meningiomas and suppression of meningioma cell growth by inhibiting translation initiation. Exp. Neurol. 2018, 299, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Sufian, H.B.; Santos, J.M.; Khan, Z.S.; Halim, S.A.; Khan, A.; Munir, M.T.; Zahid, M.K.; Al-Harrasi, A.; Gollahon, L.S.; Hussain, F.; et al. Parthenolide reverses the epithelial to mesenchymal transition process in breast cancer by targeting TGFbeta1: In vitro and in silico studies. Life Sci. 2022, 301, 120610. [Google Scholar] [CrossRef]
- Galindo-Hernandez, O.; Cordova-Guerrero, I.; Diaz-Rubio, L.J.; Pulido-Capiz, A.; Diaz-Villanueva, J.F.; Castaneda-Sanchez, C.Y.; Serafin-Higuera, N.; Garcia-Gonzalez, V. Protein translation associated to PERK arm is a new target for regulation of metainflammation: A connection with hepatocyte cholesterol. J. Cell Biochem. 2019, 120, 4158–4171. [Google Scholar] [CrossRef]
- Chen, L.; Aktas, B.H.; Wang, Y.; He, X.; Sahoo, R.; Zhang, N.; Denoyelle, S.; Kabha, E.; Yang, H.; Freedman, R.Y.; et al. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget 2012, 3, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Ortiz, A.; Galindo-Hernandez, O.; Hernandez-Acevedo, G.N.; Hurtado-Ureta, G.; Garcia-Gonzalez, V. Impact of cholesterol-pathways on breast cancer development, a metabolic landscape. J. Cancer 2021, 12, 4307–4321. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, V.; Diaz-Villanueva, J.F.; Galindo-Hernandez, O.; Martinez-Navarro, I.; Hurtado-Ureta, G.; Perez-Arias, A.A. Ceramide Metabolism Balance, a Multifaceted Factor in Critical Steps of Breast Cancer Development. Int. J. Mol. Sci. 2018, 19, 2527. [Google Scholar] [CrossRef] [Green Version]
- Carlisi, D.; De Blasio, A.; Drago-Ferrante, R.; Di Fiore, R.; Buttitta, G.; Morreale, M.; Scerri, C.; Vento, R.; Tesoriere, G. Parthenolide prevents resistance of MDA-MB231 cells to doxorubicin and mitoxantrone: The role of Nrf2. Cell Death Discov. 2017, 3, 17078. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gonzalez, V.; Mas-Oliva, J. Amyloidogenic properties of a D/N mutated 12 amino acid fragment of the C-terminal domain of the Cholesteryl-Ester Transfer Protein (CETP). Int. J. Mol. Sci. 2011, 12, 2019–2035. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Gonzalez, V.; Mas-Oliva, J. A Novel beta-adaptin/c-Myc Complex Formation Modulated by Oxidative Stress in the Control of the Cell Cycle in Macrophages and its Implication in Atherogenesis. Sci. Rep. 2017, 7, 13442. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Chandra, A.; Kaur, A.; Sabnis, N.; Lacko, A.; Gryczynski, Z.; Fudala, R.; Gryczynski, I. Fluorescence properties of doxorubicin in PBS buffer and PVA films. J. Photochem. Photobiol. B 2017, 170, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Damian-Zamacona, S.; Garcia-Gonzalez, V.; Avila-Barrientos, L.P.; Delgado-Coello, B.; Reyes-Grajeda, J.P.; Mas-Oliva, J. Cell survival regulation during receptor-mediated endocytosis of chemically-modified lipoproteins associated to the formation of an Amphiphysin 2 (Bin1)/c-Myc complex. Biochem. Biophys. Res. Commun. 2018, 505, 365–371. [Google Scholar] [CrossRef]
- Acosta-Montano, P.; Rodriguez-Velazquez, E.; Ibarra-Lopez, E.; Frayde-Gomez, H.; Mas-Oliva, J.; Delgado-Coello, B.; Rivero, I.A.; Alatorre-Meda, M.; Aguilera, J.; Guevara-Olaya, L.; et al. Fatty Acid and Lipopolysaccharide Effect on Beta Cells Proteostasis and its Impact on Insulin Secretion. Cells 2019, 8, 884. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [Green Version]
- Soga, S.; Shirai, H.; Kobori, M.; Hirayama, N. Use of Amino Acid Composition to Predict Ligand-Binding Sites. J. Chem. Inf. Model. 2007, 47, 400–406. [Google Scholar] [CrossRef]
- Clark, A.M.; Labute, P. 2D depiction of protein-ligand complexes. J. Chem. Inf. Model. 2007, 47, 1933–1944. [Google Scholar] [CrossRef]
- Kumar, D.; Haldar, S.; Gorain, M.; Kumar, S.; Mulani, F.A.; Yadav, A.S.; Miele, L.; Thulasiram, H.V.; Kundu, G.C. Epoxyazadiradione suppresses breast tumor growth through mitochondrial depolarization and caspase-dependent apoptosis by targeting PI3K/Akt pathway. BMC Cancer 2018, 18, 52. [Google Scholar] [CrossRef]
- He, Y.; Sun, S.; Sha, H.; Liu, Z.; Yang, L.; Xue, Z.; Chen, H.; Qi, L. Emerging roles for XBP1, a sUPeR transcription factor. Gene Expr. 2010, 15, 13–25. [Google Scholar] [CrossRef]
- Martinez-Navarro, I.; Diaz-Molina, R.; Pulido-Capiz, A.; Mas-Oliva, J.; Luna-Reyes, I.; Rodriguez-Velazquez, E.; Rivero, I.A.; Ramos-Ibarra, M.A.; Alatorre-Meda, M.; Garcia-Gonzalez, V. Lipid Modulation in the Formation of beta-Sheet Structures. Implications for De Novo Design of Human Islet Amyloid Polypeptide and the Impact on beta-Cell Homeostasis. Biomolecules 2020, 10, 1201. [Google Scholar] [CrossRef]
- Saini, J.; Sharma, P.K. Clinical, Prognostic and Therapeutic Significance of Heat Shock Proteins in Cancer. Curr. Drug Targets 2018, 19, 1478–1490. [Google Scholar] [CrossRef]
- Uluckan, O.; Becker, S.N.; Deng, H.; Zou, W.; Prior, J.L.; Piwnica-Worms, D.; Frazier, W.A.; Weilbaecher, K.N. CD47 regulates bone mass and tumor metastasis to bone. Cancer Res. 2009, 69, 3196–3204. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, C.; Rani, S.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; Crown, J.; et al. Docetaxel-resistance in prostate cancer: Evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackwood, E.A.; Thuerauf, D.J.; Stastna, M.; Stephens, H.; Sand, Z.; Pentoney, A.; Azizi, K.; Jakobi, T.; Van Eyk, J.E.; Katus, H.A.; et al. Proteomic analysis of the cardiac myocyte secretome reveals extracellular protective functions for the ER stress response. J. Mol. Cell Cardiol. 2020, 143, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Manie, S.N.; Lebeau, J.; Chevet, E. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 3. Orchestrating the unfolded protein response in oncogenesis: An update. Am. J. Physiol. Cell Physiol. 2014, 307, C901–C907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liwak, U.; Jordan, L.E.; Von-Holt, S.D.; Singh, P.; Hanson, J.E.; Lorimer, I.A.; Roncaroli, F.; Holcik, M. Loss of PDCD4 contributes to enhanced chemoresistance in Glioblastoma multiforme through de-repression of Bcl-xL translation. Oncotarget 2013, 4, 1365–1372. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Yang, H.S.; Li, Y.C.; Zhu, J. Dissecting the Roles of PDCD4 in Breast Cancer. Front. Oncol. 2022, 12, 855807. [Google Scholar] [CrossRef]
- Vikhreva, P.N.; Kalinichenko, S.V.; Korobko, I.V. Programmed cell death 4 mechanism of action: The model to be updated? Cell Cycle 2017, 16, 1761–1764. [Google Scholar] [CrossRef] [Green Version]
- Aman, S.; Li, Y.; Cheng, Y.; Yang, Y.; Lv, L.; Li, B.; Xia, K.; Li, S.; Wu, H. DACH1 inhibits breast cancer cell invasion and metastasis by down-regulating the transcription of matrix metalloproteinase 9. Cell Death Discov 2021, 7, 351. [Google Scholar] [CrossRef]
- Yen, Y.T.; Yang, J.C.; Chang, J.B.; Tsai, S.C. Down-Regulation of miR-194-5p for Predicting Metastasis in Breast Cancer Cells. Int. J. Mol. Sci. 2021, 23, 325. [Google Scholar] [CrossRef]
- Hong, O.-Y.; Jang, H.-Y.; Lee, Y.-R.; Jung, S.H.; Youn, H.J.; Kim, J.-S. Inhibition of cell invasion and migration by targeting matrix metalloproteinase-9 expression via sirtuin 6 silencing in human breast cancer cells. Sci. Rep. 2022, 12, 12125. [Google Scholar] [CrossRef]
- Fan, B.; Jin, Y.; Zhang, H.; Zhao, R.; Sun, M.; Sun, M.; Yuan, X.; Wang, W.; Wang, X.; Chen, Z.; et al. MicroRNA-21 contributes to renal cell carcinoma cell invasiveness and angiogenesis via the PDCD4/c-Jun (AP-1) signalling pathway. Int. J. Oncol. 2020, 56, 178–192. [Google Scholar] [CrossRef]
- Saeki, T.; Nomizu, T.; Toi, M.; Ito, Y.; Noguchi, S.; Kobayashi, T.; Asaga, T.; Minami, H.; Yamamoto, N.; Aogi, K.; et al. Dofequidar fumarate (MS-209) in combination with cyclophosphamide, doxorubicin, and fluorouracil for patients with advanced or recurrent breast cancer. J. Clin. Oncol. 2007, 25, 411–417. [Google Scholar] [CrossRef] [PubMed]
- de Cabo, R.; Mattson, M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019, 381, 2541–2551. [Google Scholar] [CrossRef] [PubMed]
- Samec, M.; Liskova, A.; Koklesova, L.; Samuel, S.M.; Zhai, K.; Buhrmann, C.; Varghese, E.; Abotaleb, M.; Qaradakhi, T.; Zulli, A.; et al. Flavonoids against the Warburg phenotype-concepts of predictive, preventive and personalised medicine to cut the Gordian knot of cancer cell metabolism. EPMA J. 2020, 11, 377–398. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Iwasaki, W.; Takahashi, M.; Sakamoto, A.; Watanabe, C.; Shichino, Y.; Floor, S.N.; Fujiwara, K.; Mito, M.; Dodo, K.; et al. The Translation Inhibitor Rocaglamide Targets a Bimolecular Cavity between eIF4A and Polypurine RNA. Mol. Cell 2019, 73, 738–748.e739. [Google Scholar] [CrossRef] [Green Version]
- Khaled, J.; Kopsida, M.; Lennernas, H.; Heindryckx, F. Drug Resistance and Endoplasmic Reticulum Stress in Hepatocellular Carcinoma. Cells 2022, 11, 632. [Google Scholar] [CrossRef]
- Gupta, A.; Hossain, M.M.; Miller, N.; Kerin, M.; Callagy, G.; Gupta, S. NCOA3 coactivator is a transcriptional target of XBP1 and regulates PERK-eIF2alpha-ATF4 signalling in breast cancer. Oncogene 2016, 35, 5860–5871. [Google Scholar] [CrossRef] [Green Version]
- Banach, A.; Jiang, Y.P.; Roth, E.; Kuscu, C.; Cao, J.; Lin, R.Z. CEMIP upregulates BiP to promote breast cancer cell survival in hypoxia. Oncotarget 2019, 10, 4307–4320. [Google Scholar] [CrossRef] [Green Version]
- Dauer, P.; Sharma, N.S.; Gupta, V.K.; Nomura, A.; Dudeja, V.; Saluja, A.; Banerjee, S. GRP78-mediated antioxidant response and ABC transporter activity confers chemoresistance to pancreatic cancer cells. Mol. Oncol. 2018, 12, 1498–1512. [Google Scholar] [CrossRef] [Green Version]
- Bennett, R.L.; Carruthers, A.L.; Hui, T.; Kerney, K.R.; Liu, X.; May, W.S., Jr. Increased expression of the dsRNA-activated protein kinase PKR in breast cancer promotes sensitivity to doxorubicin. PLoS ONE 2012, 7, e46040. [Google Scholar] [CrossRef] [Green Version]
- Tuval-Kochen, L.; Paglin, S.; Keshet, G.; Lerenthal, Y.; Nakar, C.; Golani, T.; Toren, A.; Yahalom, J.; Pfeffer, R.; Lawrence, Y. Eukaryotic Initiation Factor 2α-a Downstream Effector of Mammalian Target of Rapamycin-Modulates DNA Repair and Cancer Response to Treatment. PLoS ONE 2013, 8, e77260. [Google Scholar] [CrossRef]
- Alsterda, A.; Asha, K.; Powrozek, O.; Repak, M.; Goswami, S.; Dunn, A.M.; Memmel, H.C.; Sharma-Walia, N. Salubrinal Exposes Anticancer Properties in Inflammatory Breast Cancer Cells by Manipulating the Endoplasmic Reticulum Stress Pathway. Front. Oncol. 2021, 11, 654940. [Google Scholar] [CrossRef] [PubMed]
- Gafar, A.A.; Draz, H.M.; Goldberg, A.A.; Bashandy, M.A.; Bakry, S.; Khalifa, M.A.; AbuShair, W.; Titorenko, V.I.; Sanderson, J.T. Lithocholic acid induces endoplasmic reticulum stress, autophagy and mitochondrial dysfunction in human prostate cancer cells. PeerJ 2016, 4, e2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.T.; Chen, R.Q.; Lin, G.B.; Fang, X.L.; Yu, S.J.; Liang, X.H.; Zhang, R. Defining the regulatory role of programmed cell death 4 in laryngeal squamous cell carcinoma. Biochem. Cell Biol. 2018, 96, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Steinberger, J.; Shen, L.; Kiniry, S.J.; Naineni, S.K.; Cencic, R.; Amiri, M.; Aboushawareb, S.A.E.; Chu, J.; Maiga, R.I.; Yachnin, B.J.; et al. Identification and characterization of hippuristanol-resistant mutants reveals eIF4A1 dependencies within mRNA 5’ leader regions. Nucleic Acids Res. 2020, 48, 9521–9537. [Google Scholar] [CrossRef]
- Howard, C.M.; Bearss, N.; Subramaniyan, B.; Tilley, A.; Sridharan, S.; Villa, N.; Fraser, C.S.; Raman, D. The CXCR4-LASP1-eIF4F Axis Promotes Translation of Oncogenic Proteins in Triple-Negative Breast Cancer Cells. Front. Oncol. 2019, 9, 284. [Google Scholar] [CrossRef]
- Xue, C.; Gu, X.; Li, G.; Bao, Z.; Li, L. Expression and Functional Roles of Eukaryotic Initiation Factor 4A Family Proteins in Human Cancers. Front. Cell Dev. Biol 2021, 9, 711965. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Hou, J.; Tan, Y.; Su, C.; Wang, T.; Gao, Z.; Song, D.; Zhao, J.; Liao, Y.; Liu, X.; Jiang, Y.; et al. Inhibition of protein FAK enhances 5-FU chemosensitivity to gastric carcinoma via p53 signaling pathways. Comput. Struct. Biotechnol. J. 2020, 18, 125–136. [Google Scholar] [CrossRef]
- Huanwen, W.; Zhiyong, L.; Xiaohua, S.; Xinyu, R.; Kai, W.; Tonghua, L. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol. Cancer 2009, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Ohta, Y.; Fujii, M.; Takahashi, S.; Takano, A.; Nanki, K.; Matano, M.; Hanyu, H.; Saito, M.; Shimokawa, M.; Nishikori, S.; et al. Cell-matrix interface regulates dormancy in human colon cancer stem cells. Nature 2022, 608, 784–794. [Google Scholar] [CrossRef]
- Wolfe, A.L.; Singh, K.; Zhong, Y.; Drewe, P.; Rajasekhar, V.K.; Sanghvi, V.R.; Mavrakis, K.J.; Jiang, M.; Roderick, J.E.; Van der Meulen, J.; et al. RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 2014, 513, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Naineni, S.K.; Itoua Maiga, R.; Cencic, R.; Putnam, A.A.; Amador, L.A.; Rodriguez, A.D.; Jankowsky, E.; Pelletier, J. A comparative study of small molecules targeting eIF4A. RNA 2020, 26, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Zhao, R.; Heese, L.E.; Akiyama, H.; Patel, S.; Jaeger, A.M.; Jacamo, R.O.; Kojima, K.; Ma, M.C.J.; Ruvolo, V.R.; et al. Inhibition of translation initiation factor eIF4a inactivates heat shock factor 1 (HSF1) and exerts anti-leukemia activity in AML. Leukemia 2021, 35, 2469–2481. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhu, M.; Chaudhari, R.R.; Robles, O.; Chen, Y.; Skillern, W.; Qin, Q.; Wierda, W.G.; Zhang, S.; Hull, K.G.; et al. Creating novel translation inhibitors to target pro-survival proteins in chronic lymphocytic leukemia. Leukemia 2019, 33, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.; Foster, F.; Wood, A.; Owens, T.; Brennan, K.; Streuli, C.H.; Gilmore, A.P. Oncogenic activation of FAK drives apoptosis suppression in a 3D-culture model of breast cancer initiation. Oncotarget 2016, 7, 70336–70352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo-Sanchez, R.; Villegas-Comonfort, S.; Galindo-Hernandez, O.; Gomez, R.; Salazar, E.P. Benzo-[a]-pyrene induces FAK activation and cell migration in MDA-MB-231 breast cancer cells. Cell Biol. Toxicol. 2013, 29, 303–319. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Ortiz, A.; Pulido-Capiz, A.; Castañeda-Sánchez, C.Y.; Ibarra-López, E.; Galindo-Hernández, O.; Calderón-Fernández, M.A.; López-Cossio, L.Y.; Díaz-Molina, R.; Chimal-Vega, B.; Serafín-Higuera, N.; et al. eIF4A/PDCD4 Pathway, a Factor for Doxorubicin Chemoresistance in a Triple-Negative Breast Cancer Cell Model. Cells 2022, 11, 4069. https://doi.org/10.3390/cells11244069
González-Ortiz A, Pulido-Capiz A, Castañeda-Sánchez CY, Ibarra-López E, Galindo-Hernández O, Calderón-Fernández MA, López-Cossio LY, Díaz-Molina R, Chimal-Vega B, Serafín-Higuera N, et al. eIF4A/PDCD4 Pathway, a Factor for Doxorubicin Chemoresistance in a Triple-Negative Breast Cancer Cell Model. Cells. 2022; 11(24):4069. https://doi.org/10.3390/cells11244069
Chicago/Turabian StyleGonzález-Ortiz, Alina, Angel Pulido-Capiz, César Y. Castañeda-Sánchez, Esmeralda Ibarra-López, Octavio Galindo-Hernández, Maritza Anahí Calderón-Fernández, Leslie Y. López-Cossio, Raul Díaz-Molina, Brenda Chimal-Vega, Nicolás Serafín-Higuera, and et al. 2022. "eIF4A/PDCD4 Pathway, a Factor for Doxorubicin Chemoresistance in a Triple-Negative Breast Cancer Cell Model" Cells 11, no. 24: 4069. https://doi.org/10.3390/cells11244069