
Generation of CD34+CD43+ Hematopoietic Progenitors to Induce Thymocytes from Human Pluripotent Stem Cells
 and
and
Abstract
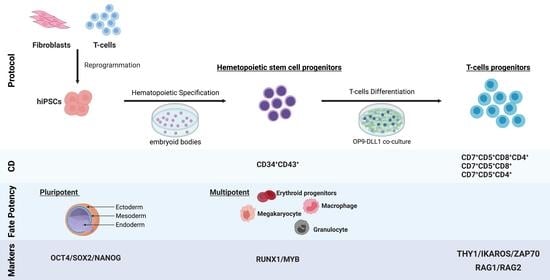
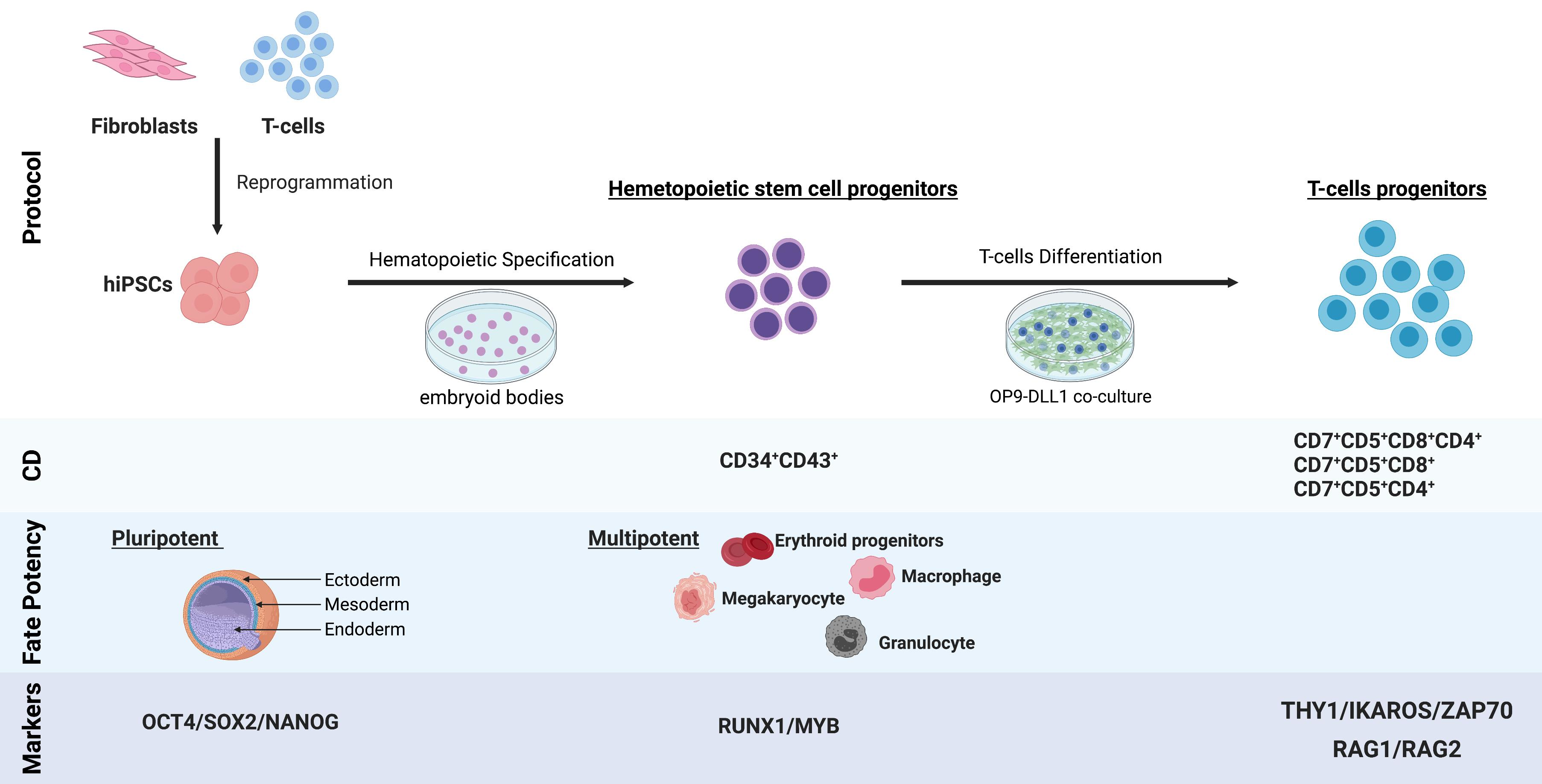
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
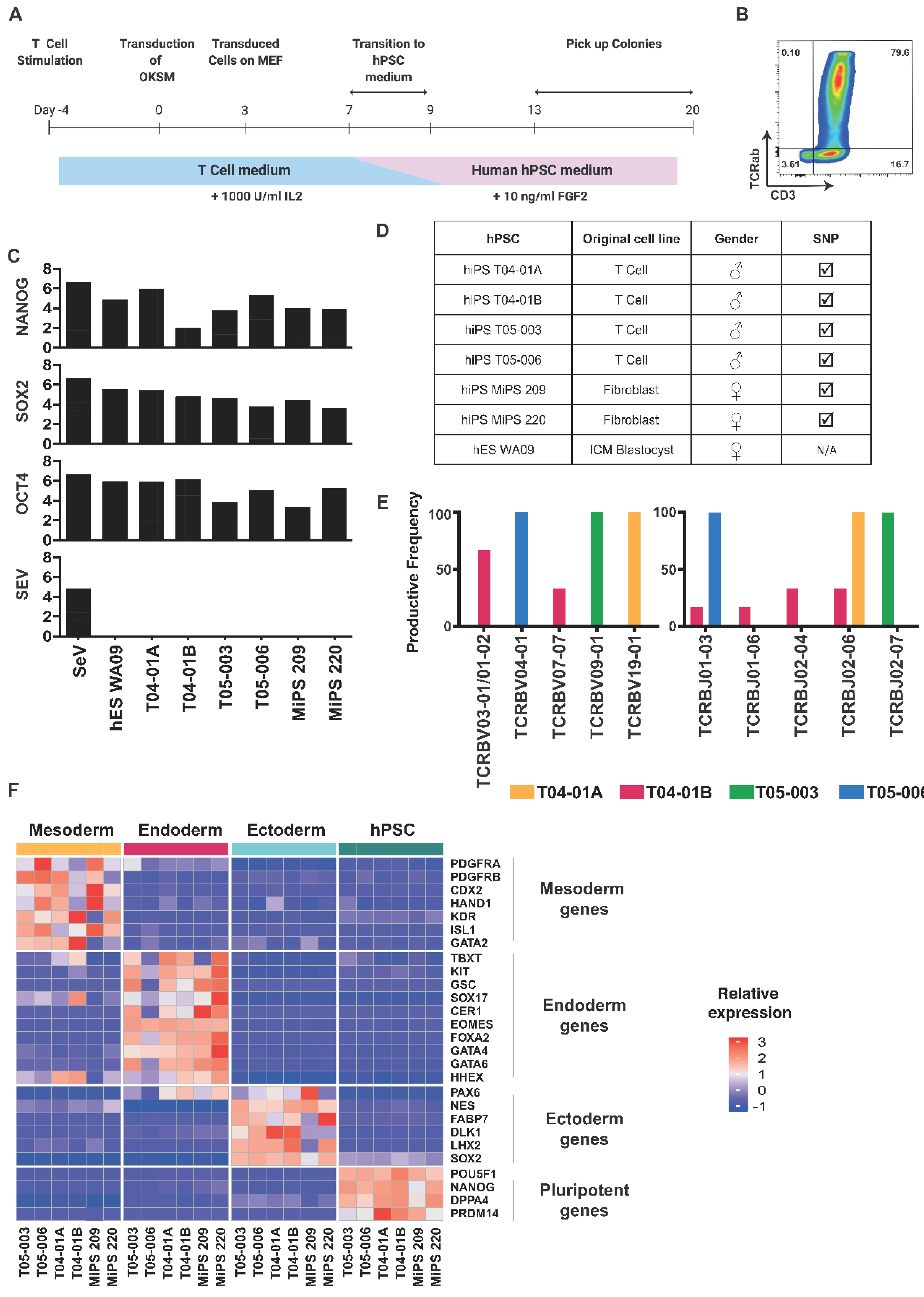
2.1. Efficient Establishment of hPSCs Clones from Blood T Cells
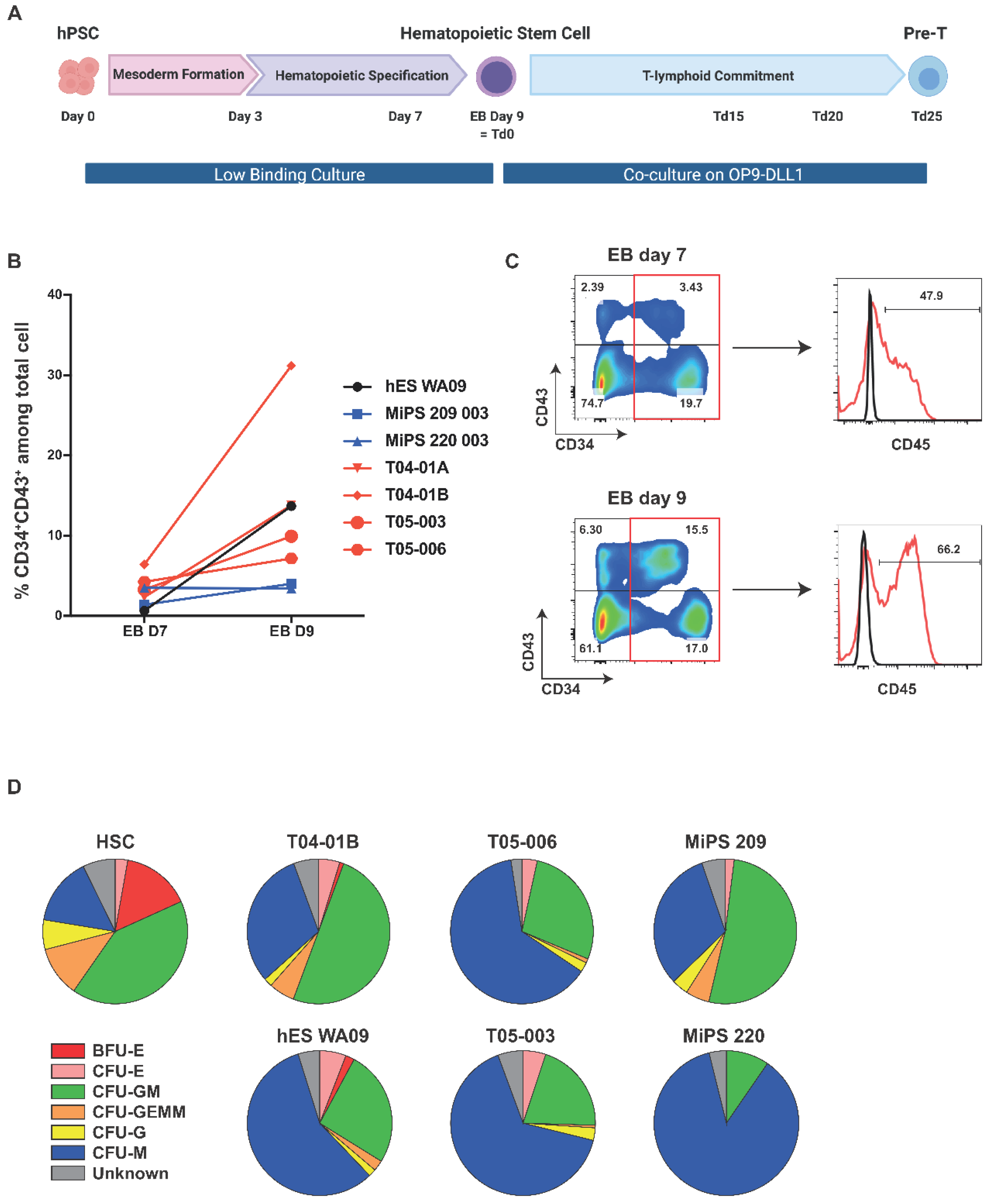
2.2. Functionally Competent Hematopoietic Progenitor Cell Differentiation from Reprogrammed T Cells
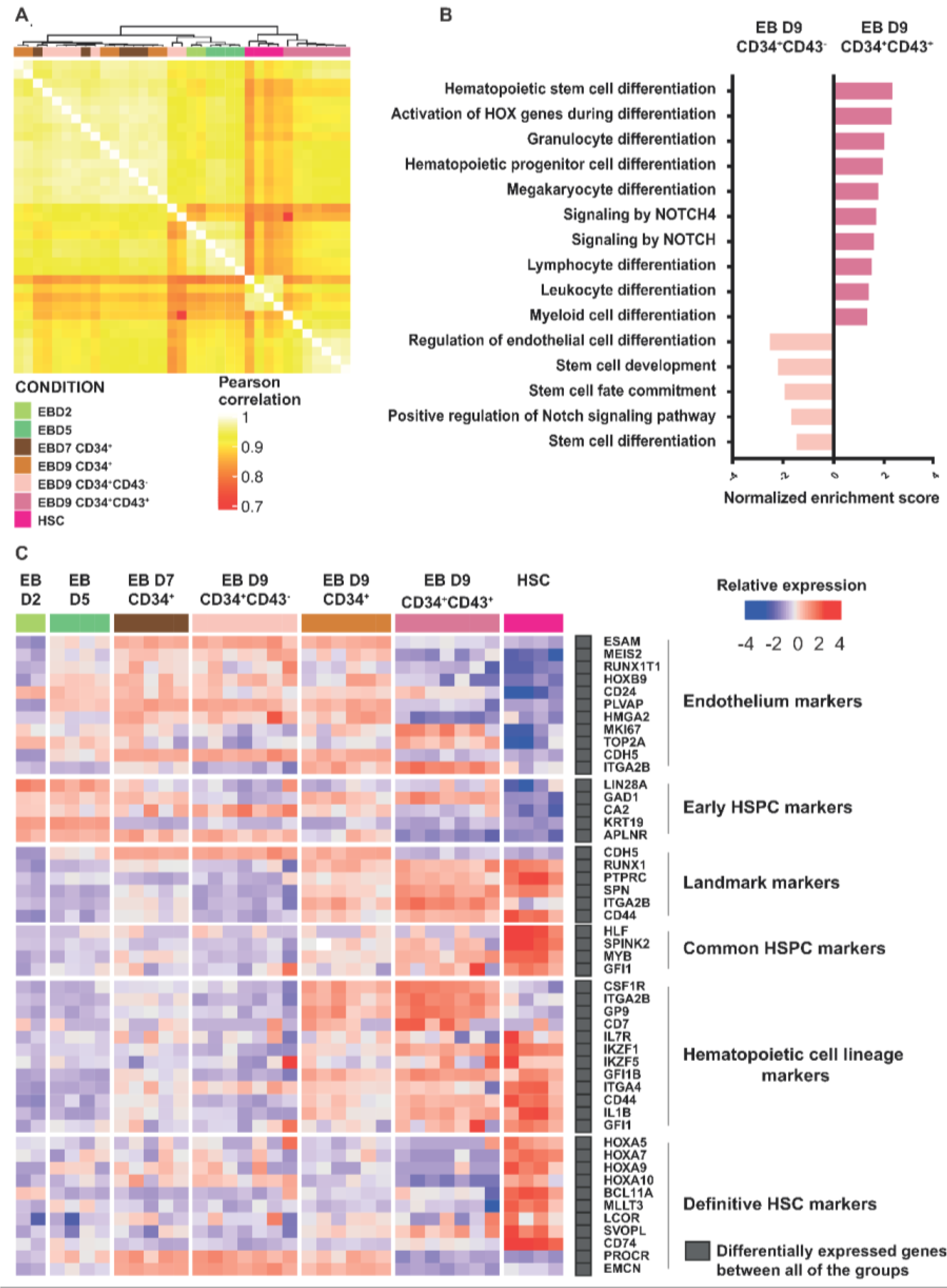
2.3. Transcriptomic Profiling Demonstrates That CD34+CD43+ Cells Derived from hPSCs Are the Most Similar to HSCs
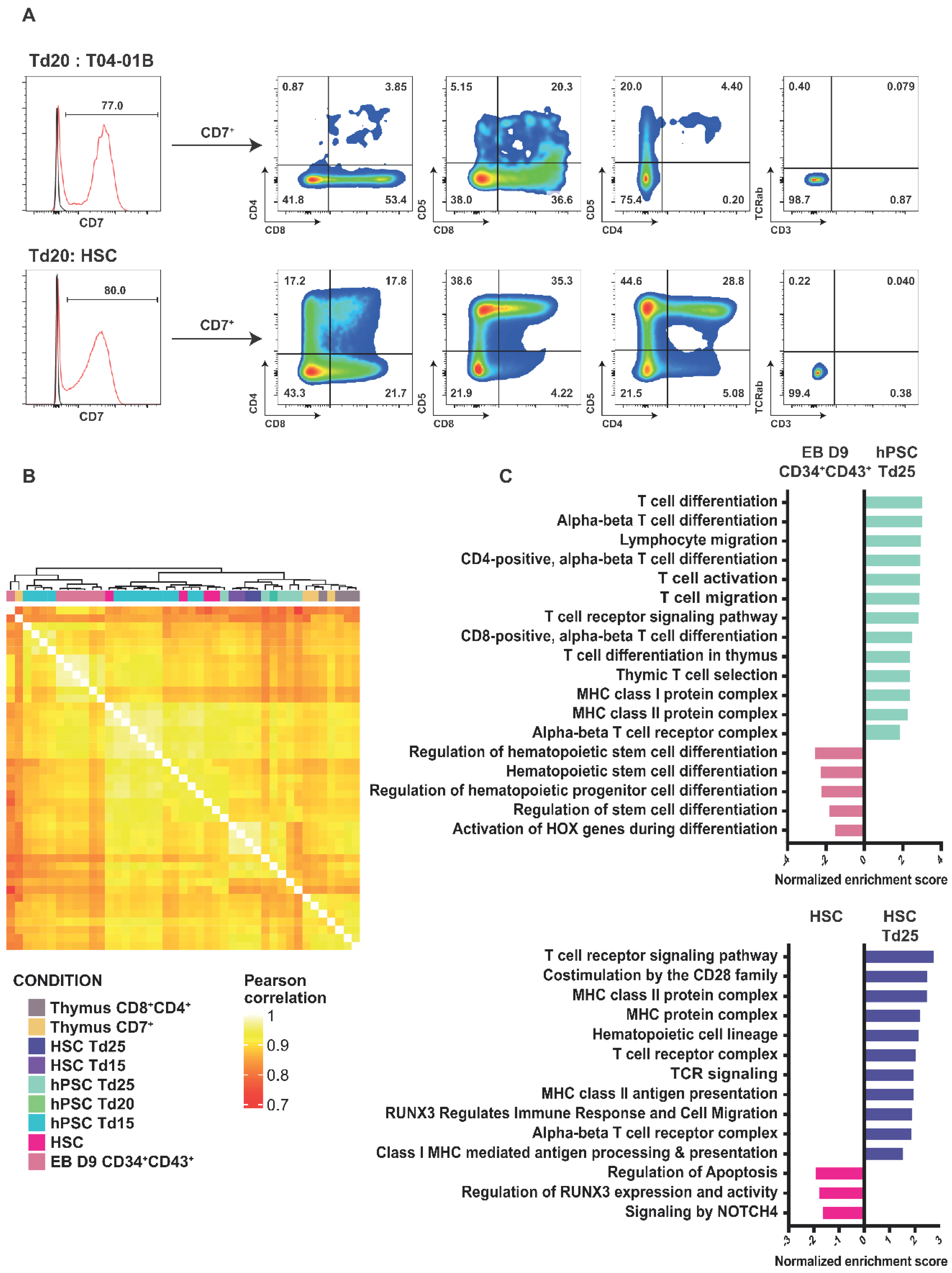
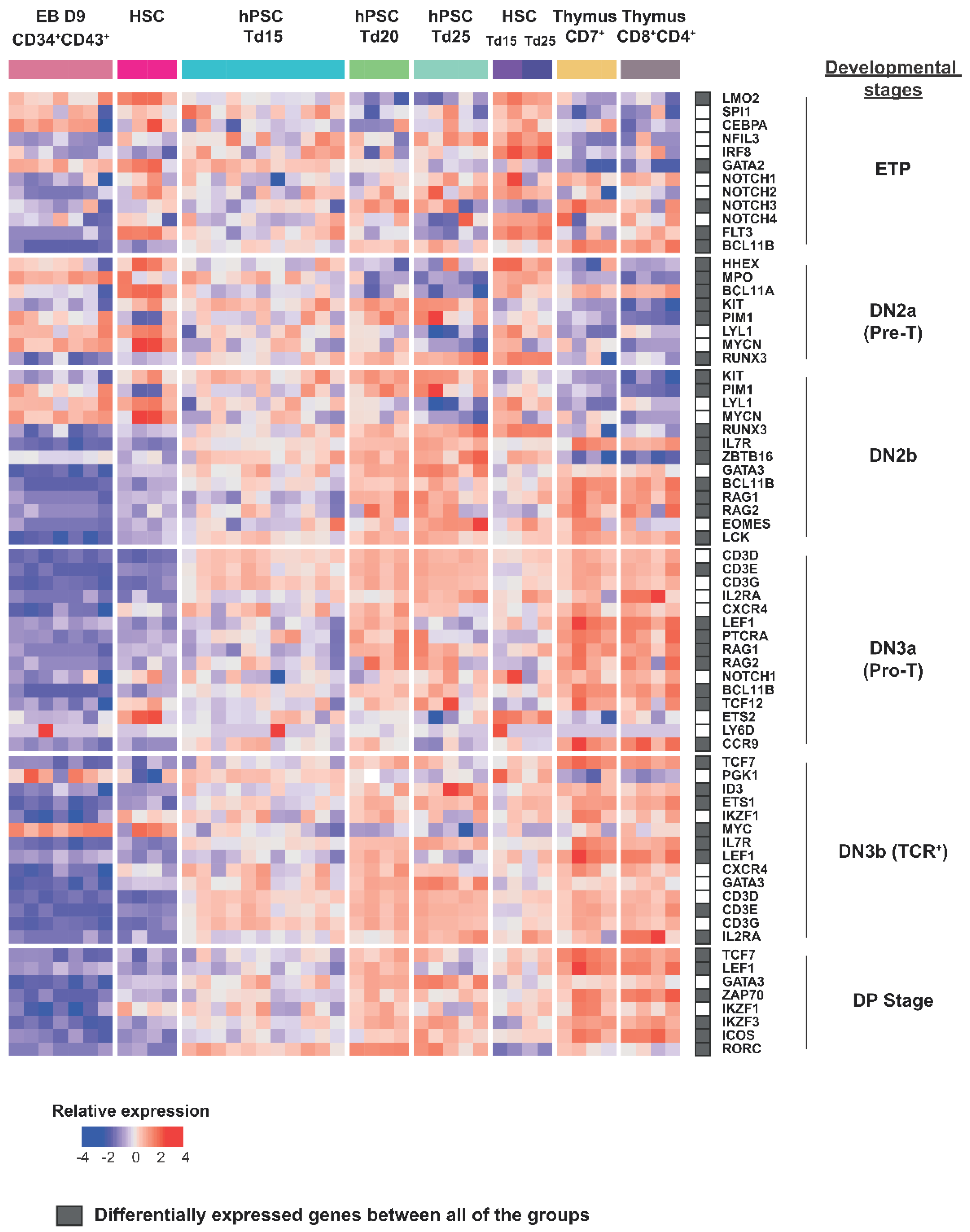
2.4. Efficient Lymphopoiesis Induction up to DN3a T-Cell Developmental Stage of HPSCs
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Tissue Culture
4.3. Reprogramming of Human T Cells into T-iPSCs
4.4. Early Germ Layer Differentiation
4.5. SNP Analysis
4.6. Hematopoietic T-Cell Differentiation from PSCs
4.7. CFU Assay
4.8. FACS Analysis
4.9. Extraction of RNA and DNA
4.10. 3′ Digital Gene Expression (3′ DGE) RNA Sequencing
4.11. High-Throughput Sequencing of the TCRβ Chain
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Themeli, M.; Rivière, I.; Sadelain, M. New Cell Sources for T Cell Engineering and Adoptive Immunotherapy. Cell Stem Cell 2015, 16, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bézie, S.; Meistermann, D.; Boucault, L.; Kilens, S.; Zoppi, J.; Autrusseau, E.; Donnart, A.; Nerriére-Daguin, V.; Bellier-Waast, F.; Charpentier, E.; et al. Ex Vivo Expanded Human Non-Cytotoxic CD8+CD45RClow/− Tregs Efficiently Delay Skin Graft Rejection and GVHD in Humanized Mice. Front. Immunol. 2018, 8, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bézie, S.; Charreau, B.; Vimond, N.; Lasselin, J.; Gérard, N.; Nerrière-Daguin, V.; Bellier-Waast, F.; Duteille, F.; Anegon, I.; Guillonneau, C. Human CD8+ Tregs expressing a MHC-specific CAR display enhanced suppression of human skin rejection and GVHD in NSG mice. Blood Adv. 2019, 3, 3522–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flippe, L.; Bézie, S.; Anegon, I.; Guillonneau, C. Future prospects for CD8 + regulatory T cells in immune tolerance. Immunol. Rev. 2019, 292, 209–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nianias, A.; Themeli, M. Induced Pluripotent Stem Cell (iPSC)–Derived Lymphocytes for Adoptive Cell Immunotherapy: Recent Advances and Challenges. Curr. Hematol. Malig. Rep. 2019, 14, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-E.; Botting, R.A.; Conde, C.D.; Popescu, D.-M.; Lavaert, M.; Kunz, D.J.; Goh, I.; Stephenson, E.; Ragazzini, R.; Tuck, E.; et al. A cell atlas of human thymic development defines T cell repertoire formation. Science 2020, 367, eaay3224. [Google Scholar] [CrossRef] [Green Version]
- Awong, G.; Herer, E.; La Motte-Mohs, R.N.; Zúñiga-Pflücker, J.C. Human CD8 T cells generated in vitro from hematopoietic stem cells are functionally mature. BMC Immunol. 2011, 12, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smedt, M.; Hoebeke, I.; Plum, J. Human bone marrow CD34+ progenitor cells mature to T cells on OP9-DL1 stromal cell line without thymus microenvironment. Blood Cells Mol. Dis. 2004, 33, 227–232. [Google Scholar] [CrossRef]
- La Motte-Mohs, R.N.; Herer, E.; Zúñiga-Pflücker, J.C. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood 2005, 105, 1431–1439. [Google Scholar] [CrossRef]
- Schmitt, T.M.; Zúñiga-Pflücker, J.C. Induction of T Cell Development from Hematopoietic Progenitor Cells by Delta-like-1 In Vitro. Immunity 2002, 17, 749–756. [Google Scholar] [CrossRef]
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of CD8αβ T Cells from T-cell–Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016, 76, 6839–6850. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, H.; Masuda, K.; Nagano, S.; Maeda, T. Cloning and expansion of antigen-specific T cells using iPS cell technology: Development of “off-the-shelf” T cells for the use in allogeneic transfusion settings. Int. J. Hematol. 2018, 107, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, M.; Lei, F.; Xiong, X.; Das, J.K.; Ren, X.; Fang, D.; Salek-Ardakani, S.; Yang, J.-M.; Song, J. Stem cell–derived tissue-associated regulatory T cells suppress the activity of pathogenic cells in autoimmune diabetes. J. Clin. Investig. 2019, 4, e126471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plath, K.; Lowry, W.E. Progress in understanding reprogramming to the induced pluripotent state. Nat. Rev. Genet. 2011, 12, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eric Charpentier; Marine Cornec; Solenne Dumont; Dimitri Meistermann; Philippe Bordron; Laurent David; Richard Redon; Stéphanie Bonnaud; Audrey Bihouée 3’ RNA Sequencing for Robust and Low-Cost Gene Expression Profiling. Available online: https://www.researchsquare.com (accessed on 9 November 2022).
- Kyttälä, A.; Moraghebi, R.; Valensisi, C.; Kettunen, J.; Andrus, C.; Pasumarthy, K.K.; Nakanishi, M.; Nishimura, K.; Ohtaka, M.; Weltner, J.; et al. Genetic Variability Overrides the Impact of Parental Cell Type and Determines iPSC Differentiation Potential. Stem Cell Rep. 2016, 6, 200–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015, 33, 1173–1181. [Google Scholar] [CrossRef]
- Nishizawa, M.; Chonabayashi, K.; Nomura, M.; Tanaka, A.; Nakamura, M.; Inagaki, A.; Nishikawa, M.; Takei, I.; Oishi, A.; Tanabe, K.; et al. Epigenetic Variation between Human Induced Pluripotent Stem Cell Lines Is an Indicator of Differentiation Capacity. Cell Stem Cell 2016, 19, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Van Lochem, E.; van der Velden, V.; Wind, H.; Marvelde, J.T.; Westerdaal, N.; van Dongen, J. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: Reference patterns for age-related changes and disease-induced shifts. Cytometry 2004, 60B, 1–13. [Google Scholar] [CrossRef]
- Loh, Y.-H.; Hartung, O.; Li, H.; Guo, C.; Sahalie, J.M.; Manos, P.D.; Urbach, A.; Heffner, G.C.; Grskovic, M.; Vigneault, F.; et al. Reprogramming of T Cells from Human Peripheral Blood. Cell Stem Cell 2010, 7, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.E.; Rondon, E.; Rajesh, D.; Mack, A.; Lewis, R.; Feng, X.; Zitur, L.; Learish, R.D.; Nuwaysir, E.F. Derivation of Induced Pluripotent Stem Cells from Human Peripheral Blood T Lymphocytes. PLoS ONE 2010, 5, e11373. [Google Scholar] [CrossRef]
- Husa, A.-M.; Strobl, M.R.; Strajeriu, A.; Wieser, M.; Strehl, S.; Fortschegger, K. Generation of CD34 Fluorescent Reporter Human Induced Pluripotent Stem Cells for Monitoring Hematopoietic Differentiation. Stem Cells Dev. 2018, 27, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Netsrithong, R.; Suwanpitak, S.; Boonkaew, B.; Trakarnsanga, K.; Chang, L.-J.; Tipgomut, C.; Vatanashevanopakorn, C.; Pattanapanyasat, K.; Wattanapanitch, M. Multilineage differentiation potential of hematoendothelial progenitors derived from human induced pluripotent stem cells. Stem Cell Res. Ther. 2020, 11, 481. [Google Scholar] [CrossRef] [PubMed]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A clinically applicable and scalable method to regenerate T-cells from iPSCs for off-the-shelf T-cell immunotherapy. Nat. Commun. 2021, 12, 430. [Google Scholar] [CrossRef]
- Vodyanik, M.A.; Thomson, J.A.; Slukvin, I.I. Leukosialin (CD43) defines hematopoietic progenitors in human embryonic stem cell differentiation cultures. Blood 2006, 108, 2095–2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wang, H.; Wen, Y.; Chen, X.; Liu, X.; Gao, J.; Su, P.; Xu, Y.; Zhou, W.; Shi, L.; et al. MEIS2 regulates endothelial to hematopoietic transition of human embryonic stem cells by targeting TAL1. Stem Cell Res. Ther. 2018, 9, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimura, R.; Jha, D.K.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.-F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, L.T.; Kinney, M.A.; Liu, X.; Zhang, Y.; Barragan, J.; Sousa, P.M.; Jha, D.K.; Han, A.; Cesana, M.; Shao, Z.; et al. Regulation of embryonic haematopoietic multipotency by EZH1. Nature 2018, 553, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Calvanese, V.; Nguyen, A.T.; Bolan, T.J.; Vavilina, A.; Su, T.; Lee, L.K.; Wang, Y.; Lay, F.D.; Magnusson, M.; Crooks, G.M.; et al. MLLT3 governs human haematopoietic stem-cell self-renewal and engraftment. Nature 2019, 576, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Flippe, L.; Gaignerie, A.; Sérazin, C.; Baron, O.; Saulquin, X.; Themeli, M.; Guillonneau, C.; David, L. Rapid and Reproducible Differentiation of Hematopoietic and T Cell Progenitors From Pluripotent Stem Cells. Front. Cell Dev. Biol. 2020, 8, 577464. [Google Scholar] [CrossRef]
- Rothenberg, E.V.; Kueh, H.Y.; Yui, M.A.; Zhang, J.A. Hematopoiesis and T-cell specification as a model developmental system. Immunol. Rev. 2016, 271, 72–97. [Google Scholar] [CrossRef]
- Zhou, W.; Yui, M.A.; Williams, B.A.; Yun, J.; Wold, B.J.; Cai, L.; Rothenberg, E.V. Single-Cell Analysis Reveals Regulatory Gene Expression Dynamics Leading to Lineage Commitment in Early T Cell Development. Cell Syst. 2019, 9, 321–337.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberg, E.; Moore, J.; Yui, M. Launching the T-cell-lineage developmental programme. Nat. Rev. Immunol. 2008, 8, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Yui, M.; Rothenberg, E. Developmental gene networks: A triathlon on the course to T cell identity. Nat. Rev. Immunol. 2014, 14, 529–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buono, M.; Facchini, R.; Matsuoka, S.; Thongjuea, S.; Waithe, D.; Luis, T.C.; Giustacchini, A.; Besmer, P.; Mead, A.J.; Jacobsen, S.E.W.; et al. A dynamic niche provides Kit ligand in a stage-specific manner to the earliest thymocyte progenitors. Nature 2016, 18, 157–167. [Google Scholar] [CrossRef]
- Desanti, G.E.; Jenkinson, W.E.; Parnell, S.M.; Boudil, A.; Gautreau-Rolland, L.; Eksteen, B.; Ezine, S.; Lane, P.J.L.; Jenkinson, E.J.; Anderson, G. Clonal Analysis Reveals Uniformity in the Molecular Profile and Lineage Potential of CCR9+and CCR9−Thymus-Settling Progenitors. J. Immunol. 2011, 186, 5227–5235. [Google Scholar] [CrossRef] [Green Version]
- Canté-Barrett, K.; Mendes, R.D.; Li, Y.; Vroegindeweij, E.; Pike-Overzet, K.; Wabeke, T.; Langerak, A.W.; Pieters, R.; Staal, F.J.T.; Meijerink, J.P.P. Loss of CD44dim Expression from Early Progenitor Cells Marks T-Cell Lineage Commitment in the Human Thymus. Front. Immunol. 2017, 8, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Perez, L.; Famili, F.; Cordes, M.; Brugman, M.; van Eggermond, M.; Wu, H.; Chouaref, J.; Granado, D.S.L.; Tiemessen, M.M.; Pike-Overzet, K.; et al. Functional definition of a transcription factor hierarchy regulating T cell lineage commitment. Sci. Adv. 2020, 6, eaaw7313. [Google Scholar] [CrossRef]
- Lavaert, M.; Liang, K.L.; Vandamme, N.; Park, J.-E.; Roels, J.; Kowalczyk, M.S.; Li, B.; Ashenberg, O.; Tabaka, M.; Dionne, D.; et al. Integrated scRNA-Seq Identifies Human Postnatal Thymus Seeding Progenitors and Regulatory Dynamics of Differentiating Immature Thymocytes. Immunity 2020, 52, 1088–1104.e6. [Google Scholar] [CrossRef] [PubMed]
- Dik, W.A.; Pike-Overzet, K.; Weerkamp, F.; de Ridder, D.; de Haas, E.F.; Baert, M.R.; van der Spek, P.; Koster, E.E.; Reinders, M.J.; van Dongen, J.J.; et al. New insights on human T cell development by quantitative T cell receptor gene rearrangement studies and gene expression profiling. J. Exp. Med. 2005, 201, 1715–1723. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Juedes, A.E.; Temann, U.-A.; Shresta, S.; Allison, J.P.; Ruddle, N.H.; Flavell, R.A. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001, 409, 97–101. [Google Scholar] [CrossRef]
- Georgopoulos, K. The making of a lymphocyte: The choice among disparate cell fates and the IKAROS enigma. Genes Dev. 2017, 31, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgopoulos, K.; Winandy, S.; Avitahl, N. The role of the Ikaros gene in lymphocyte development and homeostasis. Annu. Rev. Immunol. 1997, 15, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kadlecek, T.; Au-Yeung, B.; Goodfellow, H.E.S.; Hsu, L.-Y.; Freedman, T.; Weiss, A. ZAP-70: An Essential Kinase in T-cell Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [Green Version]
- Outters, P.; Jaeger, S.; Zaarour, N.; Ferrier, P. Long-Range Control of V(D)J Recombination & Allelic Exclusion: Modeling Views. Adv. Immunol. 2015, 128, 363–413. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of Rejuvenated Antigen-Specific T Cells by Reprogramming to Pluripotency and Redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Paes, B.C.M.F.; Moço, P.D.; Pereira, C.G.; Porto, G.S.; Russo, E.M.D.S.; Reis, L.C.J.; Covas, D.T.; Picanço-Castro, V. Ten years of iPSC: Clinical potential and advances in vitro hematopoietic differentiation. Cell Biol. Toxicol. 2016, 33, 233–250. [Google Scholar] [CrossRef] [PubMed]
- Demirci, S.; Leonard, A.; Tisdale, J.F. Hematopoietic stem cells from pluripotent stem cells: Clinical potential, challenges, and future perspectives. Stem Cells Transl. Med. 2020, 9, 1549–1557. [Google Scholar] [CrossRef]
- Doulatov, S.; Vo, L.T.; Chou, S.S.; Kim, P.G.; Arora, N.; Li, H.; Hadland, B.K.; Bernstein, I.D.; Collins, J.J.; Zon, L.I.; et al. Induction of Multipotential Hematopoietic Progenitors from Human Pluripotent Stem Cells via Respecification of Lineage-Restricted Precursors. Cell Stem Cell 2013, 13, 459–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyonneau-Harmand, L.; L’Homme, B.; Birebent, B.; Desterke, C.; Chevallier, N.; Garcon, L.; Lapillonne, H.; Benderitter, M.; Delhommeau, F.; Jaffredo, T.; et al. Transgene-Free Hematopoietic Stem and Progenitor Cells from Human Induced Pluripotent Stem Cells. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Kawai, Y.; Kawana-Tachikawa, A.; Kitayama, S.; Ueda, T.; Miki, S.; Watanabe, A.; Kaneko, S. Generation of highly proliferative, rejuvenated cytotoxic T cell clones through pluripotency reprogramming for adoptive immunotherapy. Mol. Ther. 2021, 29, 3027–3041. [Google Scholar] [CrossRef]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, J.L.; A Kochman, A.; Lu, S.; Terwey, T.H.; Kim, T.D.; Hubbard, V.M.; Muriglan, S.J.; Suh, D.; Smith, O.M.; Grubin, J.; et al. Adoptive transfer of T-cell precursors enhances T-cell reconstitution after allogeneic hematopoietic stem cell transplantation. Nat. Med. 2006, 12, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Awong, G.; Herer, E.; Surh, C.D.; Dick, J.E.; La Motte-Mohs, R.N.; Zúñiga-Pflücker, J.C. Characterization in vitro and engraftment potential in vivo of human progenitor T cells generated from hematopoietic stem cells. Blood 2009, 114, 972–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Six, E.; Benjelloun, F.; Garrigue, A.; Bonhomme, D.; Morillon, E.; Rouiller, J.; Cacavelli, L.; Blondeau, J.; Beldjord, K.; Hacein-Bey-Abina, S.; et al. Cytokines and culture medium have a major impact on human in vitro T-cell differentiation. Blood Cells Mol. Dis. 2011, 47, 72–78. [Google Scholar] [CrossRef]
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell 2018, 23, 850–858.e4. [Google Scholar] [CrossRef] [Green Version]
- Nezhad, M.S.; Abdollahpour-Alitappeh, M.; Rezaei, B.; Yazdanifar, M.; Seifalian, A.M. Induced Pluripotent Stem Cells (iPSCs) Provide a Potentially Unlimited T Cell Source for CAR-T Cell Development and Off-the-Shelf Products. Pharm. Res. 2021, 38, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Amini, L.; Greig, J.; Schmueck-Henneresse, M.; Volk, H.-D.; Bézie, S.; Reinke, P.; Guillonneau, C.; Wagner, D.L.; Anegon, I. Super-Treg: Toward a New Era of Adoptive Treg Therapy Enabled by Genetic Modifications. Front. Immunol. 2021, 11, 611638. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Song, J.; Fino, K.; Sandhu, P.; Song, X.; Lei, F.; Zheng, S.; Ni, B.; Fang, D.; Song, J. Stem cell-derived tissue-associated regulatory T cells ameliorate the development of autoimmunity. Sci. Rep. 2016, 6, 20588. [Google Scholar] [CrossRef] [Green Version]
- Haque, R.; Lei, F.; Xiong, X.; Bian, Y.; Zhao, B.; Wu, Y.; Song, J. Programming of Regulatory T Cells from Pluripotent Stem Cells and Prevention of Autoimmunity. J. Immunol. 2012, 189, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Kilens, S.; Meistermann, D.; Moreno, D.; Chariau, C.; Gaignerie, A.; Reignier, A.; Lelièvre, Y.; Casanova, M.; Vallot, C.; Nedellec, S.; et al. Parallel derivation of isogenic human primed and naive induced pluripotent stem cells. Nat. Commun. 2018, 9, 360. [Google Scholar] [CrossRef]
- Gaignerie, A.; Lefort, N.; Rousselle, M.; Forest-Choquet, V.; Flippe, L.; Francois–Campion, V.; Girardeau, A.; Caillaud, A.; Chariau, C.; Francheteau, Q.; et al. Urine-derived cells provide a readily accessible cell type for feeder-free mRNA reprogramming. Sci. Rep. 2018, 8, 14363. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flippe, L.; Gaignerie, A.; Sérazin, C.; Baron, O.; Saulquin, X.; Anegon, I.; David, L.; Guillonneau, C. Generation of CD34+CD43+ Hematopoietic Progenitors to Induce Thymocytes from Human Pluripotent Stem Cells. Cells 2022, 11, 4046. https://doi.org/10.3390/cells11244046
Flippe L, Gaignerie A, Sérazin C, Baron O, Saulquin X, Anegon I, David L, Guillonneau C. Generation of CD34+CD43+ Hematopoietic Progenitors to Induce Thymocytes from Human Pluripotent Stem Cells. Cells. 2022; 11(24):4046. https://doi.org/10.3390/cells11244046
Chicago/Turabian StyleFlippe, Léa, Anne Gaignerie, Céline Sérazin, Olivier Baron, Xavier Saulquin, Ignacio Anegon, Laurent David, and Carole Guillonneau. 2022. "Generation of CD34+CD43+ Hematopoietic Progenitors to Induce Thymocytes from Human Pluripotent Stem Cells" Cells 11, no. 24: 4046. https://doi.org/10.3390/cells11244046