
Loss of Pex1 in Inner Ear Hair Cells Contributes to Cochlear Synaptopathy and Hearing Loss
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Genotyping
2.3. Electrophysiology
2.4. Western Blots
2.5. Immunostaining and Synapse Analysis
2.6. Data Analysis and Statistics
2.7. Resources
3. Results
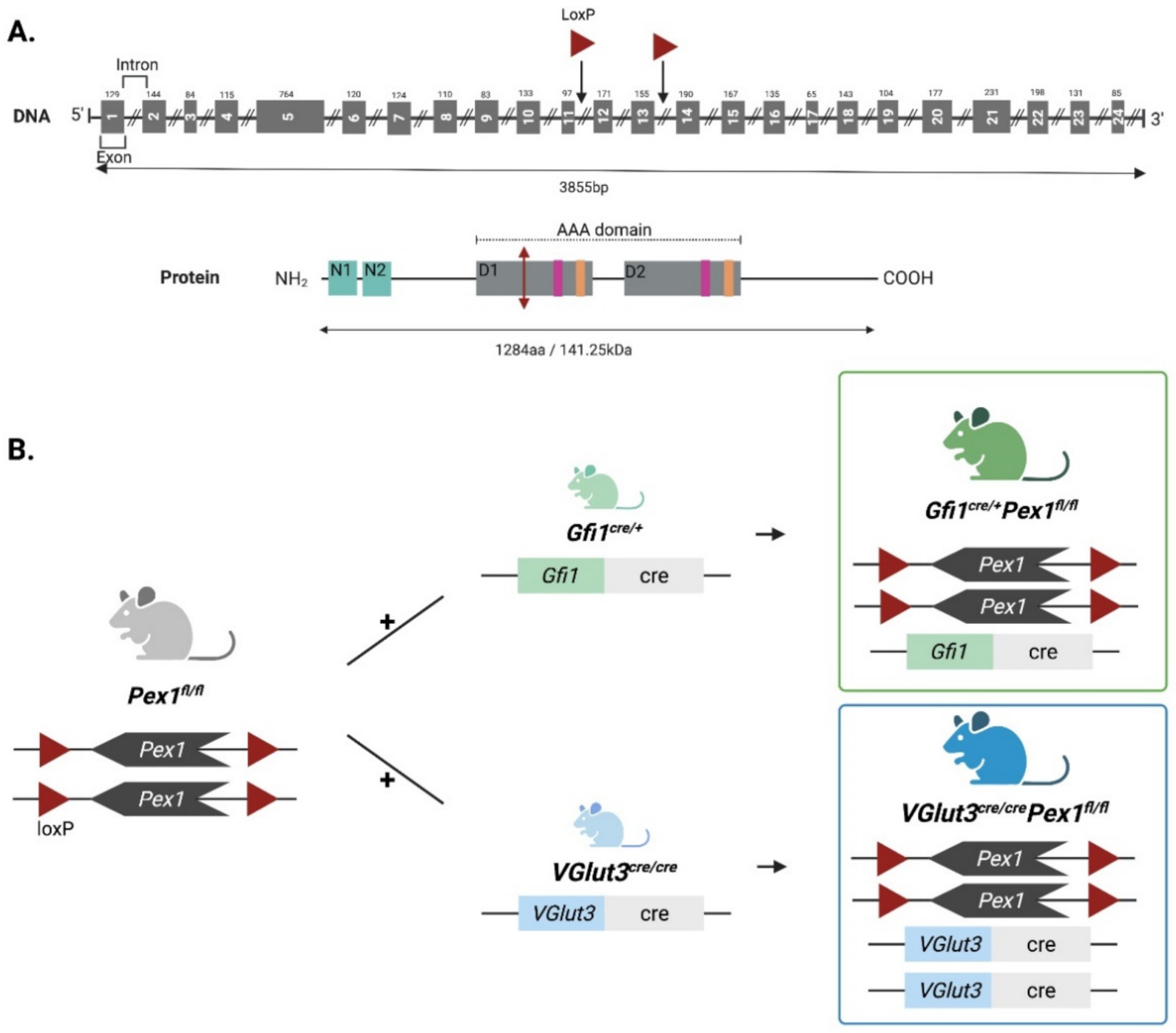
3.1. Generation and Characterization of Two New Organ-Specific Pex1 Knockout Mouse Lines
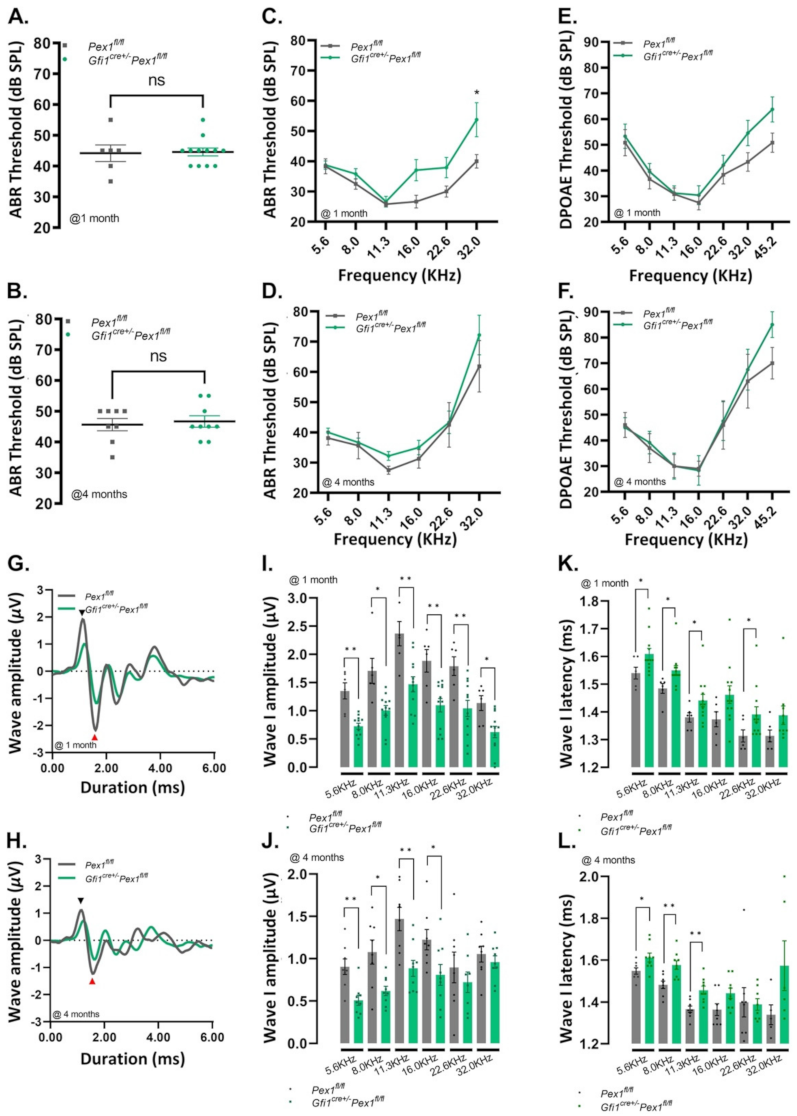
3.2. Gfi1Cre/+Pex1fl/fl Mice Show Normal Hearing but Reduced Wave I Amplitude
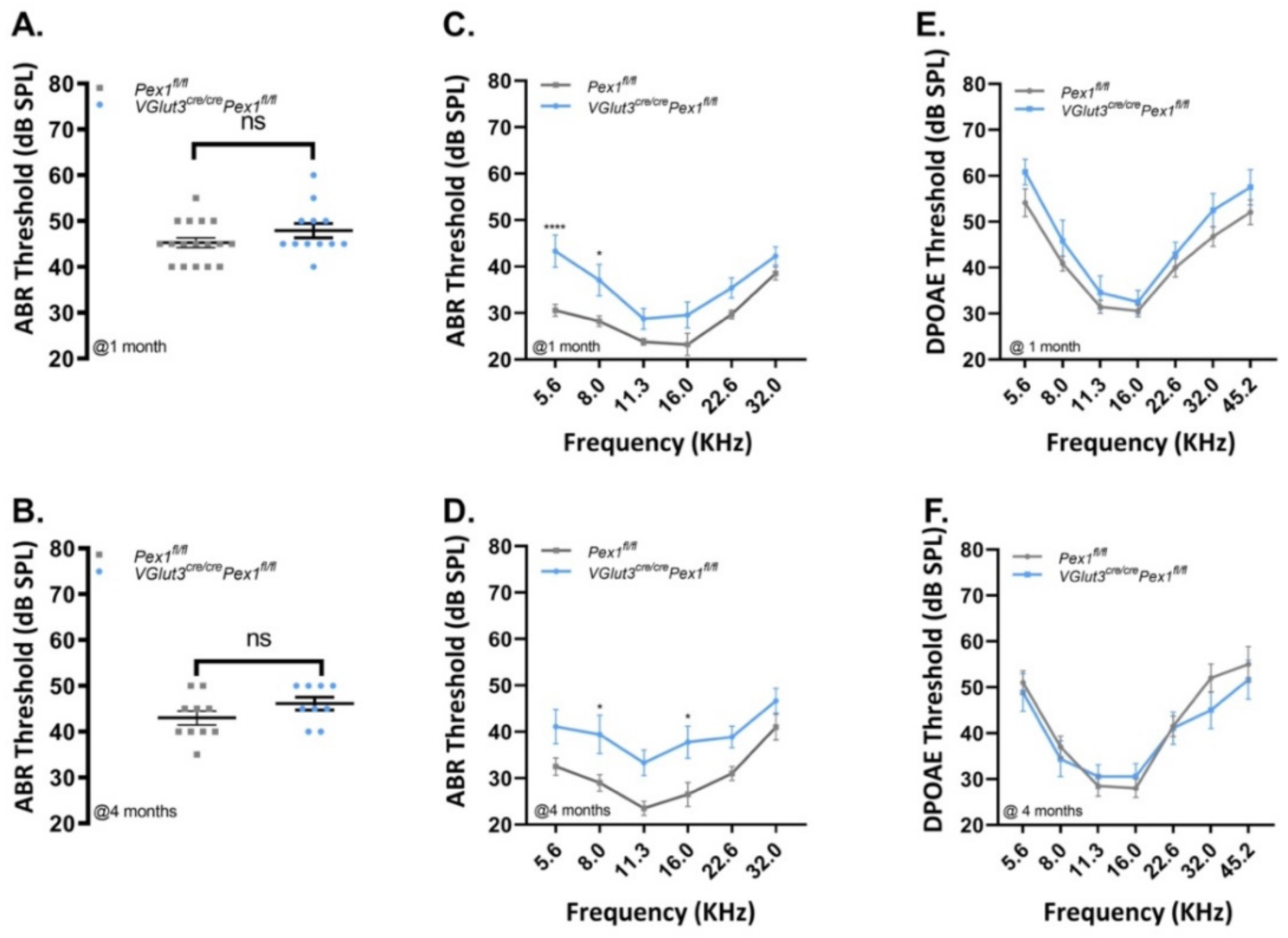
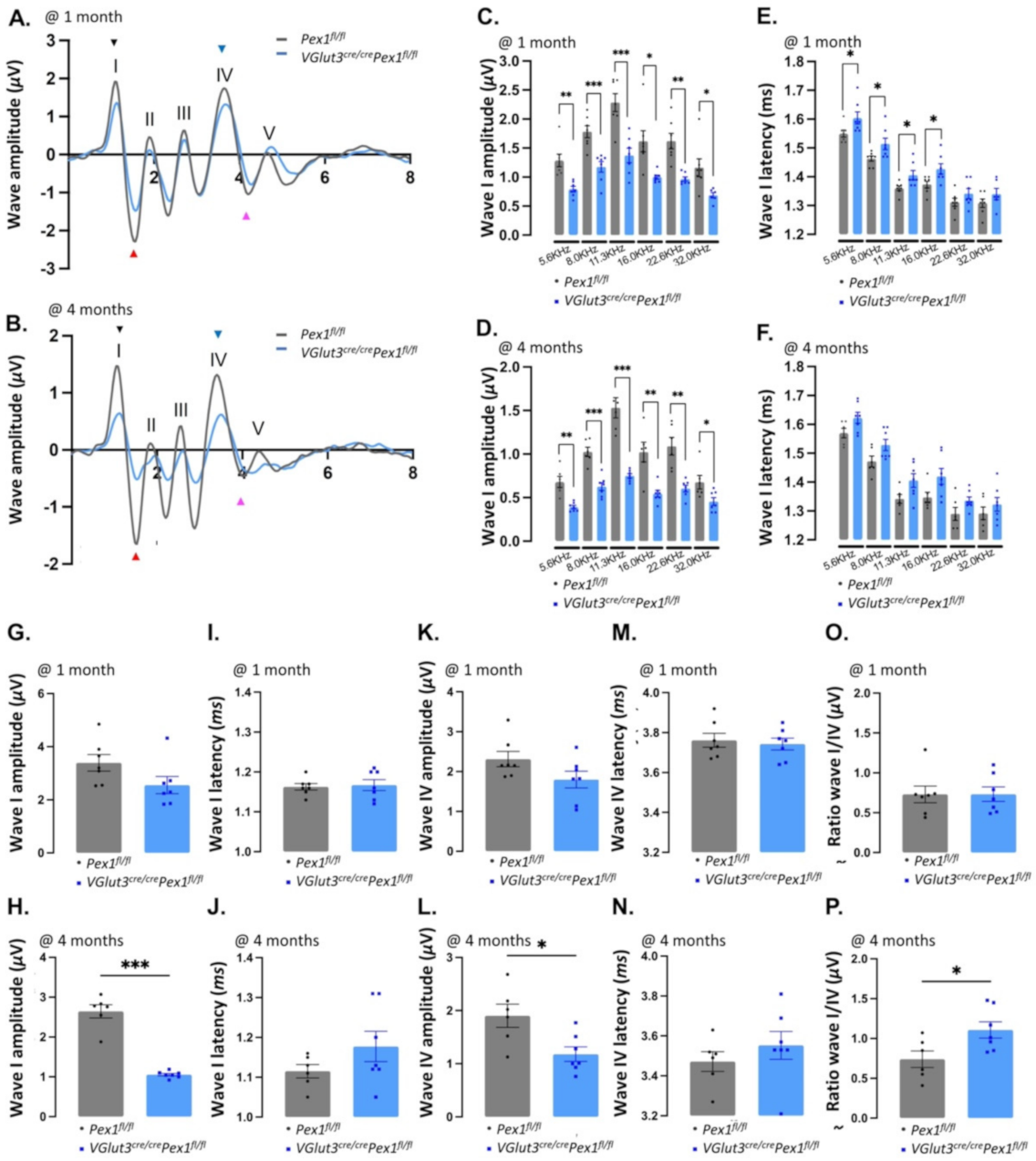
3.3. VGlut3Cre/CrePex1fl/fl Mice Exhibit Mild Hearing Loss and Preservation of Neural Central Gain
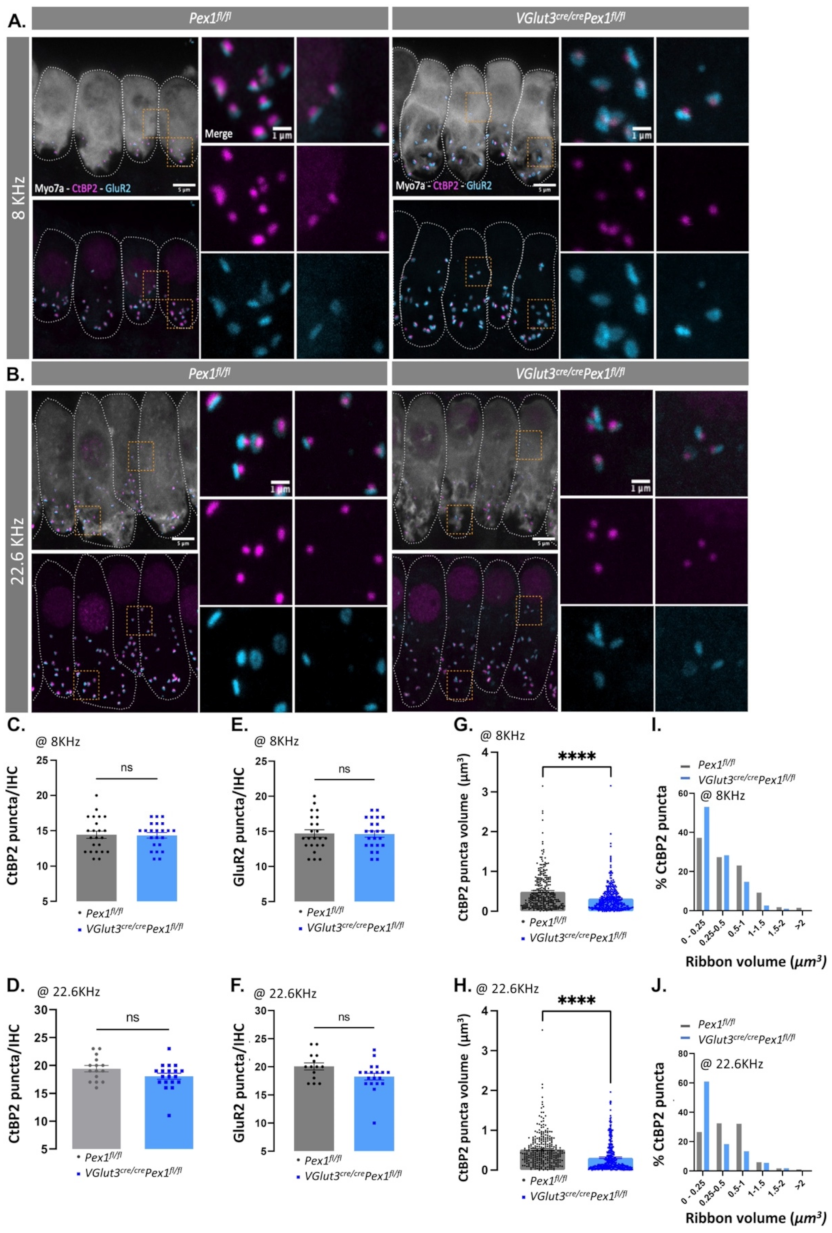
3.4. Synaptopathy Is Observed in VGlut3Cre/CrePex1fl/fl
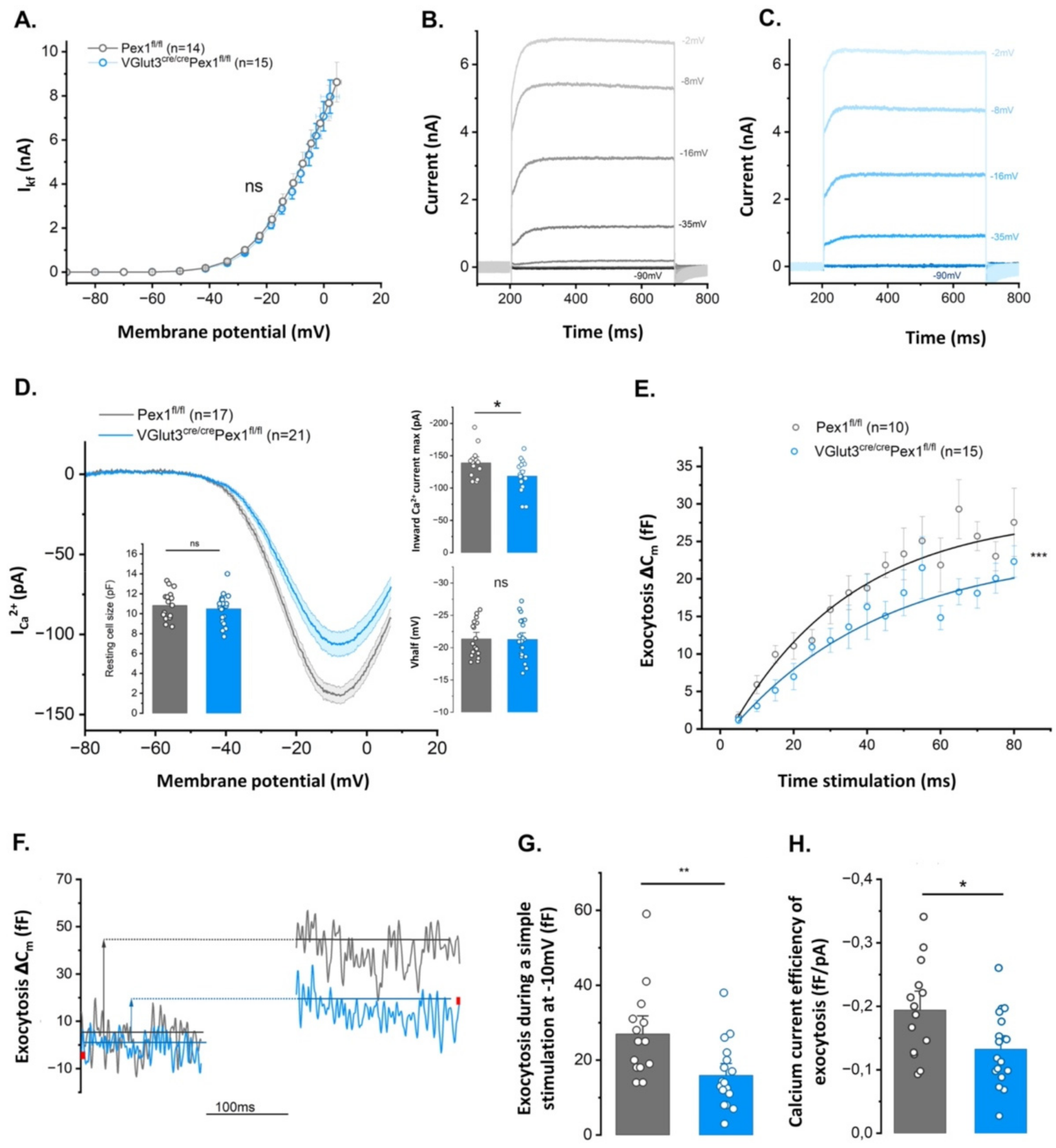
3.5. IHCs from VGlut3Cre/CrePex1fl/fl Mice Present Altered Exocystosis
3.6. Pex1 Deletion Affects Peroxisomal Number
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braverman, N.E.; D’Agostino, M.D.; Maclean, G.E. Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives. Dev. Disabil. Res. Rev. 2013, 17, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Barillari, M.R.; Karali, M.; Di Iorio, V.; Contaldo, M.; Piccolo, V.; Esposito, M.; Costa, G.; Argenziano, G.; Serpico, R.; Carotenuto, M.; et al. Mild form of Zellweger Spectrum Disorders (ZSD) due to variants in PEX1: Detailed clinical investigation in a 9-years-old female. Mol. Genet. Metab. Rep. 2020, 24, 100615. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yergeau, C.; Kawai, K.; Braverman, N.; Geleoc, G.S.G. A Retrospective Study of Hearing Loss in Patients Diagnosed with Peroxisome Biogenesis Disorders in the Zellweger Spectrum. Ear Hear. 2022, 43, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Braverman, N.E.; Raymond, G.V.; Rizzo, W.B.; Moser, A.B.; Wilkinson, M.E.; Stone, E.M.; Steinberg, S.J.; Wangler, M.F.; Rush, E.T.; Hacia, J.G.; et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 2016, 117, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enns, G.M.; Ammous, Z.; Himes, R.W.; Nogueira, J.; Palle, S.; Sullivan, M.; Ramirez, C. Diagnostic challenges and disease management in patients with a mild Zellweger spectrum disorder phenotype. Mol. Genet. Metab. 2021, 134, 217–222. [Google Scholar] [CrossRef]
- Waterham, H.R.; Ferdinandusse, S.; Wanders, R.J. Human disorders of peroxisome metabolism and biogenesis. Biochim. Biophys. Acta 2016, 1863, 922–933. [Google Scholar] [CrossRef]
- Wanders, R.J.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef] [Green Version]
- Farre, J.C.; Mahalingam, S.S.; Proietto, M.; Subramani, S. Peroxisome biogenesis, membrane contact sites, and quality control. EMBO Rep 2019, 20, e46864. [Google Scholar] [CrossRef]
- Raas-Rothschild, A.; Wanders, R.J.; Mooijer, P.A.; Gootjes, J.; Waterham, H.R.; Gutman, A.; Suzuki, Y.; Shimozawa, N.; Kondo, N.; Eshel, G.; et al. A PEX6-defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents. Am. J. Hum. Genet. 2002, 70, 1062–1068. [Google Scholar] [CrossRef] [Green Version]
- Poll-The, B.T.; Gootjes, J.; Duran, M.; De Klerk, J.B.; Wenniger-Prick, L.J.; Admiraal, R.J.; Waterham, H.R.; Wanders, R.J.; Barth, P.G. Peroxisome biogenesis disorders with prolonged survival: Phenotypic expression in a cohort of 31 patients. Am. J. Med. Genet. A 2004, 126, 333–338. [Google Scholar] [CrossRef]
- Berendse, K.; Engelen, M.; Ferdinandusse, S.; Majoie, C.B.; Waterham, H.R.; Vaz, F.M.; Koelman, J.H.; Barth, P.G.; Wanders, R.J.; Poll-The, B.T. Zellweger spectrum disorders: Clinical manifestations in patients surviving into adulthood. J. Inherit. Metab. Dis. 2016, 39, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Reuber, B.E.; Germain-Lee, E.; Collins, C.S.; Morrell, J.C.; Ameritunga, R.; Moser, H.W.; Valle, D.; Gould, S.J. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat. Genet. 1997, 17, 445–448. [Google Scholar] [CrossRef]
- Steinberg, S.J.; Dodt, G.; Raymond, G.V.; Braverman, N.E.; Moser, A.B.; Moser, H.W. Peroxisome biogenesis disorders. Biochim. Biophys. Acta 2006, 1763, 1733–1748. [Google Scholar] [CrossRef] [Green Version]
- Dubreuil, M.M.; Morgens, D.W.; Okumoto, K.; Honsho, M.; Contrepois, K.; Lee-McMullen, B.; Traber, G.M.; Sood, R.S.; Dixon, S.J.; Snyder, M.P.; et al. Systematic Identification of Regulators of Oxidative Stress Reveals Non-canonical Roles for Peroxisomal Import and the Pentose Phosphate Pathway. Cell Rep. 2020, 30, 1417–1433.e1417. [Google Scholar] [CrossRef]
- Heymans, H.S.; Schutgens, R.B.; Tan, R.; van den Bosch, H.; Borst, P. Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome). Nature 1983, 306, 69–70. [Google Scholar] [CrossRef]
- Yang, H.; Gan, J.; Xie, X.; Deng, M.; Feng, L.; Chen, X.; Gao, Z.; Gan, L. Gfi1-Cre knock-in mouse line: A tool for inner ear hair cell-specific gene deletion. Genesis 2010, 48, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Duan, B.; Huang, T.; Zhang, Y.; Chen, Y.; Britz, O.; Garcia-Campmany, L.; Ren, X.; Vong, L.; Lowell, B.B.; et al. Identification of spinal circuits involved in touch-evoked dynamic mechanical pain. Nat. Neurosci. 2017, 20, 804–814. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef]
- Peineau, T.; Belleudy, S.; Pietropaolo, S.; Bouleau, Y.; Dulon, D. Synaptic Release Potentiation at Aging Auditory Ribbon Synapses. Front. Aging Neurosci. 2021, 13, 756449. [Google Scholar] [CrossRef]
- Meyer, A.C.; Frank, T.; Khimich, D.; Hoch, G.; Riedel, D.; Chapochnikov, N.M.; Yarin, Y.M.; Harke, B.; Hell, S.W.; Egner, A.; et al. Tuning of synapse number, structure and function in the cochlea. Nat. Neurosci. 2009, 12, 444–453. [Google Scholar] [CrossRef]
- Michalski, N.; Michel, V.; Caberlotto, E.; Lefevre, G.M.; van Aken, A.F.; Tinevez, J.Y.; Bizard, E.; Houbron, C.; Weil, D.; Hardelin, J.P.; et al. Harmonin-b, an actin-binding scaffold protein, is involved in the adaptation of mechanoelectrical transduction by sensory hair cells. Pflugers Arch. 2009, 459, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, P.F.; Bouleau, Y.; Safieddine, S.; Petit, C.; Dulon, D. Exocytotic machineries of vestibular type I and cochlear ribbon synapses display similar intrinsic otoferlin-dependent Ca2+ sensitivity but a different coupling to Ca2+ channels. J. Neurosci. 2014, 34, 10853–10869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, J.Y.; Johnson, S.L.; Carlton, A.J.; De Tomasi, L.; Goodyear, R.J.; De Faveri, F.; Furness, D.N.; Wells, S.; Brown, S.D.M.; Holley, M.C.; et al. Age-related changes in the biophysical and morphological characteristics of mouse cochlear outer hair cells. J. Physiol. 2020, 598, 3891–3910. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kawai, K.; Holt, J.R.; Geleoc, G.S. Sensory transduction is required for normal development and maturation of cochlear inner hair cell synapses. Elife 2021, 10, e69433. [Google Scholar] [CrossRef] [PubMed]
- Kujawa, S.G.; Liberman, M.C. Adding insult to injury: Cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 2009, 29, 14077–14085. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, K.A.; Jeffers, P.W.; Lall, K.; Liberman, M.C.; Kujawa, S.G. Aging after noise exposure: Acceleration of cochlear synaptopathy in “recovered” ears. J. Neurosci. 2015, 35, 7509–7520. [Google Scholar] [CrossRef] [Green Version]
- Asai, Y.; Pan, B.; Nist-Lund, C.; Galvin, A.; Lukashkin, A.N.; Lukashkina, V.A.; Chen, T.; Zhou, W.; Zhu, H.; Russell, I.J.; et al. Transgenic Tmc2 expression preserves inner ear hair cells and vestibular function in mice lacking Tmc1. Sci. Rep. 2018, 8, 12124. [Google Scholar] [CrossRef] [Green Version]
- Matern, M.; Vijayakumar, S.; Margulies, Z.; Milon, B.; Song, Y.; Elkon, R.; Zhang, X.; Jones, S.M.; Hertzano, R. Gfi1(Cre) mice have early onset progressive hearing loss and induce recombination in numerous inner ear non-hair cells. Sci. Rep. 2017, 7, 42079. [Google Scholar] [CrossRef] [Green Version]
- Beer-Hammer, S.; Lee, S.C.; Mauriac, S.A.; Leiss, V.; Groh, I.A.M.; Novakovic, A.; Piekorz, R.P.; Bucher, K.; Chen, C.; Ni, K.; et al. Galphai Proteins are Indispensable for Hearing. Cell. Physiol. Biochem. 2018, 47, 1509–1532. [Google Scholar] [CrossRef]
- Mohrle, D.; Ni, K.; Varakina, K.; Bing, D.; Lee, S.C.; Zimmermann, U.; Knipper, M.; Ruttiger, L. Loss of auditory sensitivity from inner hair cell synaptopathy can be centrally compensated in the young but not old brain. Neurobiol. Aging 2016, 44, 173–184. [Google Scholar] [CrossRef]
- Brandt, A.; Striessnig, J.; Moser, T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J. Neurosci. 2003, 23, 10832–10840. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.B.; Rutherford, M.A.; Gabrielaitis, M.; Pangrsic, T.; Gottfert, F.; Frank, T.; Michanski, S.; Hell, S.; Wolf, F.; Wichmann, C.; et al. Developmental refinement of hair cell synapses tightens the coupling of Ca2+ influx to exocytosis. EMBO J. 2014, 33, 247–264. [Google Scholar] [CrossRef]
- Frank, T.; Rutherford, M.A.; Strenzke, N.; Neef, A.; Pangrsic, T.; Khimich, D.; Fejtova, A.; Gundelfinger, E.D.; Liberman, M.C.; Harke, B.; et al. Bassoon and the synaptic ribbon organize Ca(2)+ channels and vesicles to add release sites and promote refilling. Neuron 2010, 68, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Graydon, C.W.; Cho, S.; Li, G.L.; Kachar, B.; von Gersdorff, H. Sharp Ca(2)(+) nanodomains beneath the ribbon promote highly synchronous multivesicular release at hair cell synapses. J. Neurosci. 2011, 31, 16637–16650. [Google Scholar] [CrossRef] [Green Version]
- Graydon, C.W.; Zhang, J.; Oesch, N.W.; Sousa, A.A.; Leapman, R.D.; Diamond, J.S. Passive diffusion as a mechanism underlying ribbon synapse vesicle release and resupply. J. Neurosci. 2014, 34, 8948–8962. [Google Scholar] [CrossRef] [Green Version]
- Nazarko, T.Y. Pexophagy is responsible for 65% of cases of peroxisome biogenesis disorders. Autophagy 2017, 13, 991–994. [Google Scholar] [CrossRef] [Green Version]
- Hiebler, S.; Masuda, T.; Hacia, J.G.; Moser, A.B.; Faust, P.L.; Liu, A.; Chowdhury, N.; Huang, N.; Lauer, A.; Bennett, J.; et al. The Pex1-G844D mouse: A model for mild human Zellweger spectrum disorder. Mol. Genet. Metab. 2014, 111, 522–532. [Google Scholar] [CrossRef] [Green Version]
- Argyriou, C.; Polosa, A.; Cecyre, B.; Hsieh, M.; Di Pietro, E.; Cui, W.; Bouchard, J.F.; Lachapelle, P.; Braverman, N. A longitudinal study of retinopathy in the PEX1-Gly844Asp mouse model for mild Zellweger Spectrum Disorder. Exp. Eye Res. 2019, 186, 107713. [Google Scholar] [CrossRef]
- Argyriou, C.; Polosa, A.; Song, J.Y.; Omri, S.; Steele, B.; Cecyre, B.; McDougald, D.S.; Di Pietro, E.; Bouchard, J.F.; Bennett, J.; et al. AAV-mediated PEX1 gene augmentation improves visual function in the PEX1-Gly844Asp mouse model for mild Zellweger spectrum disorder. Mol. Ther. Methods Clin. Dev. 2021, 23, 225–240. [Google Scholar] [CrossRef]
- Demaret, T.; Roumain, M.; Ambroise, J.; Evraerts, J.; Ravau, J.; Bouzin, C.; Bearzatto, B.; Gala, J.L.; Stepman, H.; Marie, S.; et al. Longitudinal study of Pex1-G844D NMRI mouse model: A robust pre-clinical model for mild Zellweger spectrum disorder. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165900. [Google Scholar] [CrossRef]
- Liberman, M.C. Noise-Induced Hearing Loss: Permanent Versus Temporary Threshold Shifts and the Effects of Hair Cell Versus Neuronal Degeneration. Adv. Exp. Med. Biol. 2016, 875, 1–7. [Google Scholar] [PubMed]
- Liberman, M.C.; Kujawa, S.G. Cochlear synaptopathy in acquired sensorineural hearing loss: Manifestations and mechanisms. Hear. Res. 2017, 349, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, K.A.; Guo, D.; Micucci, S.; De Gruttola, V.; Liberman, M.C.; Kujawa, S.G. Noise-induced Cochlear Synaptopathy with and Without Sensory Cell Loss. Neuroscience 2020, 427, 43–57. [Google Scholar] [CrossRef]
- Kujawa, S.G.; Liberman, M.C. Translating animal models to human therapeutics in noise-induced and age-related hearing loss. Hear. Res. 2019, 377, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Paquette, S.T.; Gilels, F.; White, P.M. Noise exposure modulates cochlear inner hair cell ribbon volumes, correlating with changes in auditory measures in the FVB/nJ mouse. Sci. Rep. 2016, 6, 25056. [Google Scholar] [CrossRef] [Green Version]
- Defourny, J.; Aghaie, A.; Perfettini, I.; Avan, P.; Delmaghani, S.; Petit, C. Pejvakin-mediated pexophagy protects auditory hair cells against noise-induced damage. Proc. Natl. Acad. Sci. USA 2019, 116, 8010–8017. [Google Scholar] [CrossRef] [Green Version]
- Delmaghani, S.; Defourny, J.; Aghaie, A.; Beurg, M.; Dulon, D.; Thelen, N.; Perfettini, I.; Zelles, T.; Aller, M.; Meyer, A.; et al. Hypervulnerability to Sound Exposure through Impaired Adaptive Proliferation of Peroxisomes. Cell 2015, 163, 894–906. [Google Scholar] [CrossRef] [Green Version]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [Green Version]
- Germain, K.; Kim, P.K. Pexophagy: A Model for Selective Autophagy. Int. J. Mol. Sci. 2020, 21, 578. [Google Scholar] [CrossRef] [Green Version]
- Walter, C.; Gootjes, J.; Mooijer, P.A.; Portsteffen, H.; Klein, C.; Waterham, H.R.; Barth, P.G.; Epplen, J.T.; Kunau, W.H.; Wanders, R.J.; et al. Disorders of peroxisome biogenesis due to mutations in PEX1: Phenotypes and PEX1 protein levels. Am J Hum. Genet. 2001, 69, 35–48. [Google Scholar] [CrossRef]
- Seal, R.P.; Akil, O.; Yi, E.; Weber, C.M.; Grant, L.; Yoo, J.; Clause, A.; Kandler, K.; Noebels, J.L.; Glowatzki, E.; et al. Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron 2008, 57, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Weisz, C.J.C.; Williams, S.G.; Eckard, C.S.; Divito, C.B.; Ferreira, D.W.; Fantetti, K.N.; Dettwyler, S.A.; Cai, H.M.; Rubio, M.E.; Kandler, K.; et al. Outer Hair Cell Glutamate Signaling through Type II Spiral Ganglion Afferents Activates Neurons in the Cochlear Nucleus in Response to Nondamaging Sounds. J. Neurosci. 2021, 41, 2930–2943. [Google Scholar] [CrossRef]
- Orvis, J.; Gottfried, B.; Kancherla, J.; Adkins, R.S.; Song, Y.; Dror, A.A.; Olley, D.; Rose, K.; Chrysostomou, E.; Kelly, M.C.; et al. gEAR: Gene Expression Analysis Resource portal for community-driven, multi-omic data exploration. Nat. Methods 2021, 18, 843–844. [Google Scholar] [CrossRef]
- Wangemann, P. Adrenergic and muscarinic control of cochlear endolymph production. Adv. Otorhinolaryngol. 2002, 59, 42–50. [Google Scholar]
- Wangemann, P. Supporting sensory transduction: Cochlear fluid homeostasis and the endocochlear potential. J. Physiol. 2006, 576, 11–21. [Google Scholar] [CrossRef]
- Spector, G.J.; Carr, C. The ultrastructural cytochemistry of peroxisomes in the guinea pig cochlea: A metabolic hypothesis for the stria vascularis. Laryngoscope 1979, 89, 1–38. [Google Scholar] [CrossRef]
- Kochaj, R.M.; Martelletti, E.; Ingham, N.J.; Buniello, A.; Sousa, B.C.; Wakelam, M.J.O.; Lopez-Clavijo, A.F.; Steel, K.P. The Effect of a Pex3 Mutation on Hearing and Lipid Content of the Inner Ear. Cells 2022, 11, 3206. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Sequence | Product | ||
|---|---|---|---|
| Pex1fl/fl | Forward | 5′-GAAGCATCCCTGCTCACTTC-3′ | WT = 249 bp Floxed = 430 bp |
| Reverse | 5′-CCCTTCCACATACTAGGCAAGC-3′ | ||
| Gfi1Cre | Cre | 5′-GCCCAAATGTTGCTGGATAGT-3′ | WT = 600 bp Cre = 700 bp |
| Forward | 5′-GGGATAACGGACCAGTTG-3′ | ||
| Reverse | 5′-CCGAGGGGCGTTAGGATA-3′ | ||
| VGlut3Cre | Cre | 5′-ATCGACCGGTAATGCAGGCAA-3′ | WT = 300 bp Cre = 800 bp |
| Forward | 5′-GATGTCTTATGGAGCCACCACCCAG-3′ | ||
| Reverse | 5′-CGTAGACCAAGGTCCATATTCCCA-3′ |
| Software | Source | More Information |
|---|---|---|
| Imaris Cell Imaging 9.6.1 | Oxford Instruments (Abingdon, UK) | https://imaris.oxinst.com/ (accessed on 1 November 2022) |
| ImageJ- Fiji | NIH (Bethesda, MD, USA) | https://imagej.nih.gov/ij/index.html (accessed on 1 November 2022) |
| GraphPad Prism 9.1 | California USA | https://www.graphpad.com/ (accessed on 1 November 2022) |
| OriginPro 2022b | Northampton, MA, USA | https://www.originlab.com/2022 (accessed on 1 November 2022) |
| ZEN 2.3 (blue edition) | Carl Zeiss (Jena, Germany) | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html#zenversions (accessed on 1 November 2022) |
| EPL Cochlear Function Test Suite | EPL Engineering, Boston, MA, USA | https://www.masseyeandear.org/research/otolaryngology/eaton-peabody-laboratories/engineering-core (accessed on 1 November 2022) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauriac, S.A.; Peineau, T.; Zuberi, A.; Lutz, C.; Géléoc, G.S.G. Loss of Pex1 in Inner Ear Hair Cells Contributes to Cochlear Synaptopathy and Hearing Loss. Cells 2022, 11, 3982. https://doi.org/10.3390/cells11243982
Mauriac SA, Peineau T, Zuberi A, Lutz C, Géléoc GSG. Loss of Pex1 in Inner Ear Hair Cells Contributes to Cochlear Synaptopathy and Hearing Loss. Cells. 2022; 11(24):3982. https://doi.org/10.3390/cells11243982
Chicago/Turabian StyleMauriac, Stephanie A., Thibault Peineau, Aamir Zuberi, Cathleen Lutz, and Gwénaëlle S. G. Géléoc. 2022. "Loss of Pex1 in Inner Ear Hair Cells Contributes to Cochlear Synaptopathy and Hearing Loss" Cells 11, no. 24: 3982. https://doi.org/10.3390/cells11243982