
Deciphering Transcriptional Networks during Human Cardiac Development
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reprogramming and Maintenance of hiPSCs
2.2. Cardiac Differentiation of hiPSCs
2.3. Bulk Transcriptomics
2.3.1. RNA Extraction and Sequencing
2.3.2. Primary Analysis of Bulk Transcriptomic Data
2.3.3. PCA
2.3.4. Time-Course Gene Expression Analysis
2.3.5. Clustering and Heatmap
2.3.6. Gene Ontology Analyses
2.3.7. Network Construction and Analysis
2.4. Single-Cell Transcriptomic
2.4.1. Single-Cell RNA-Seq Data Generation
2.4.2. Primary Analysis of Single-Cell Transcriptomic Data
2.4.3. Secondary Analysis of Single-Cell Transcriptomic Data
2.5. Musclemotion
2.6. HEK293 Cell Culture and Transfection
2.7. Co-Immunoprecipitation
2.7.1. Protein Sample Extraction and Quantification
2.7.2. Bead-Antibody Complexes Preparation
2.7.3. Immunoprecipitation and Western Blotting Analysis
2.8. Luciferase Assay
2.9. Immunofluorescence
2.10. TF and Cardiac Phenotypes Association
2.11. Quantitative RT-PCR
3. Results
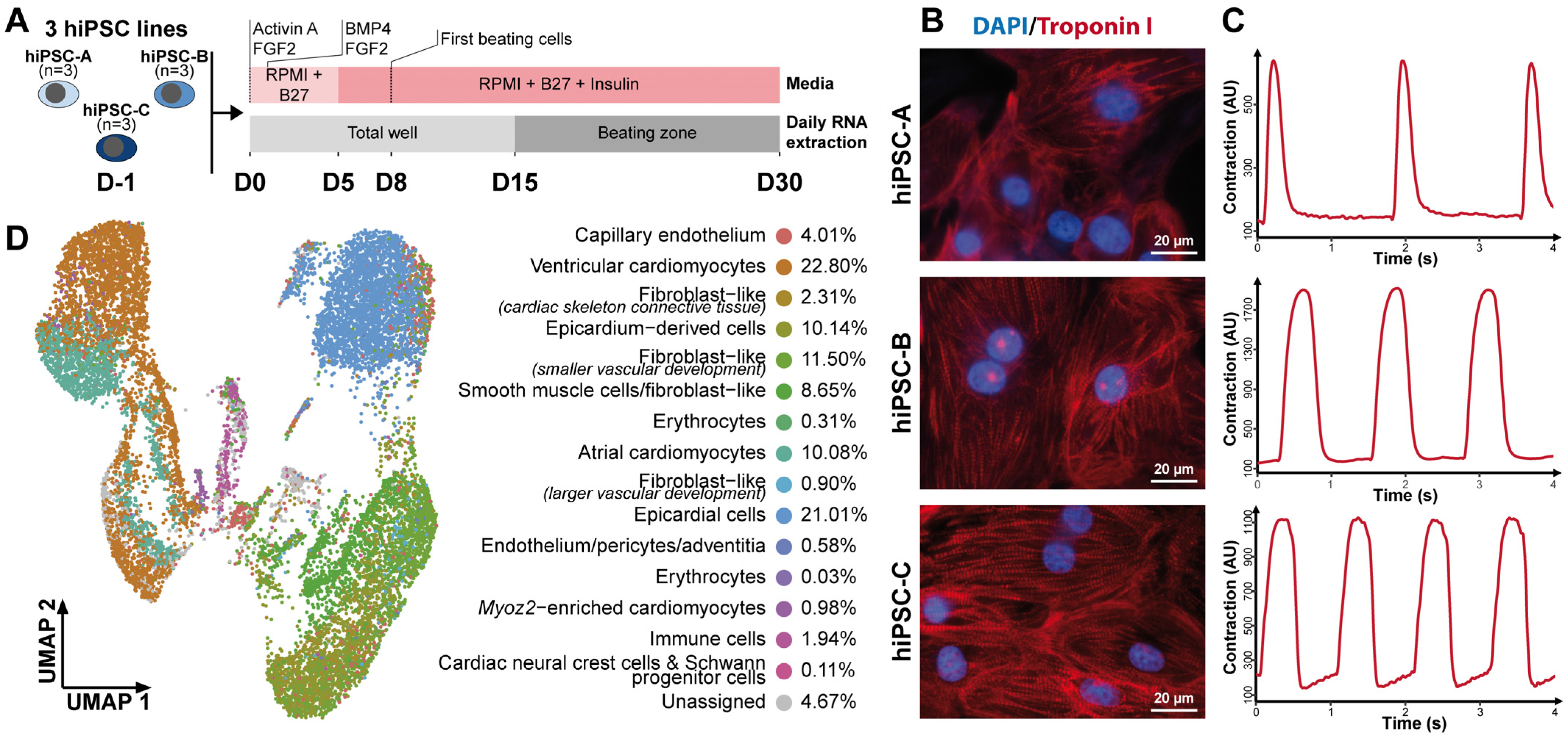
3.1. Directed Cardiac Differentiation Robustly Generates Functional Cardiac Cells

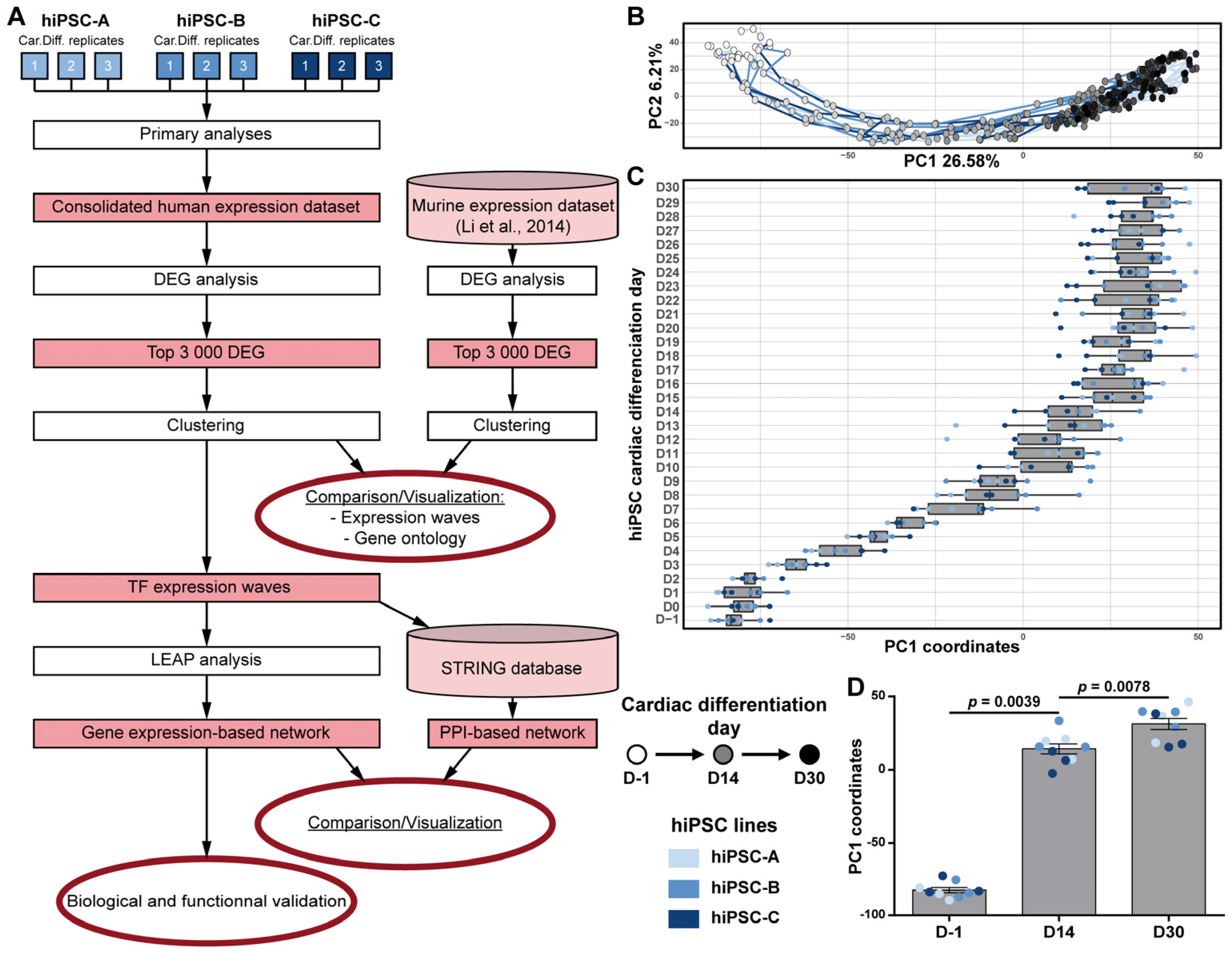
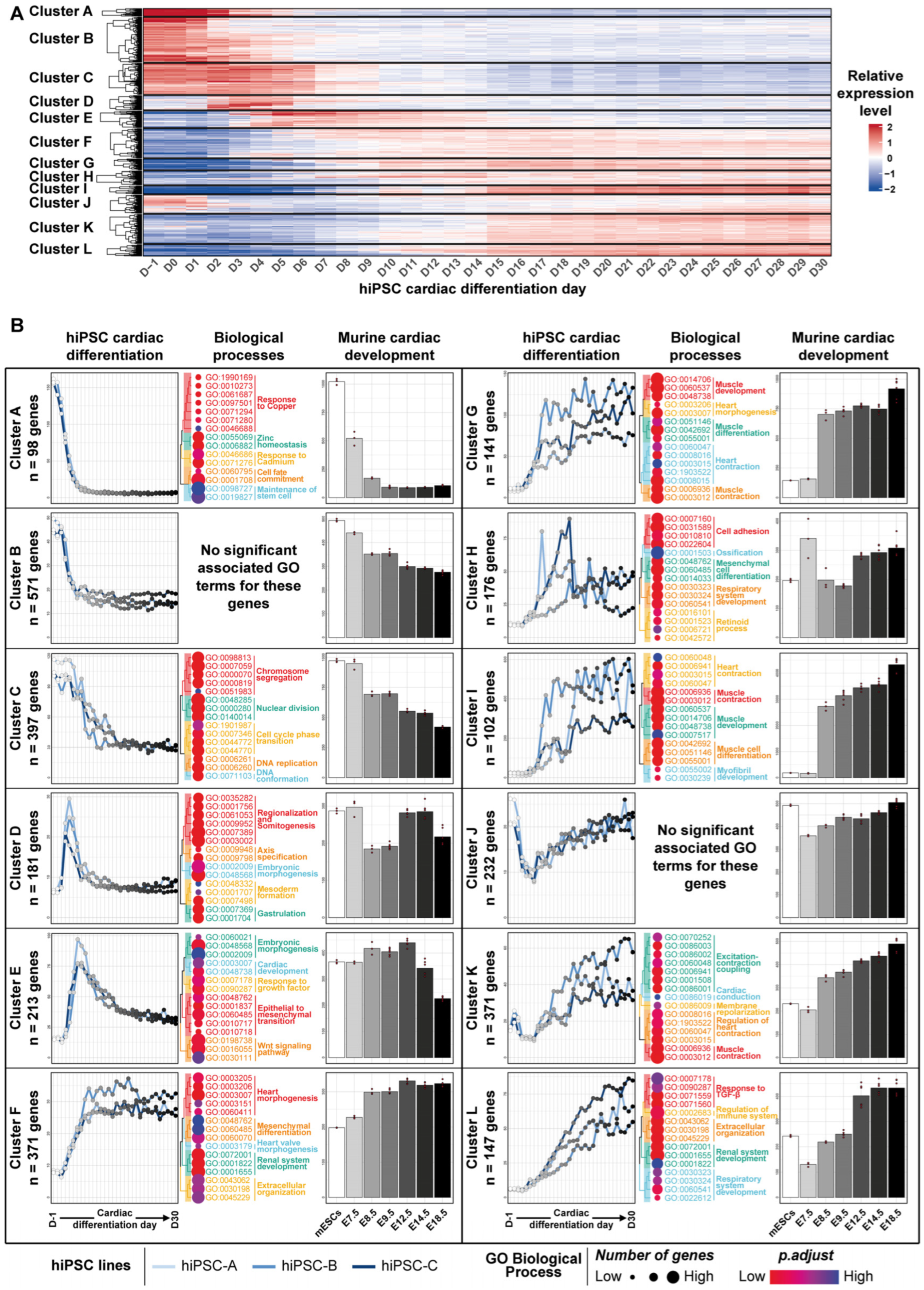
3.2. Transcriptomic Kinetics of hiPSC Cardiac Differentiation Unveiled Biological Processes Involved during Cardiac Development
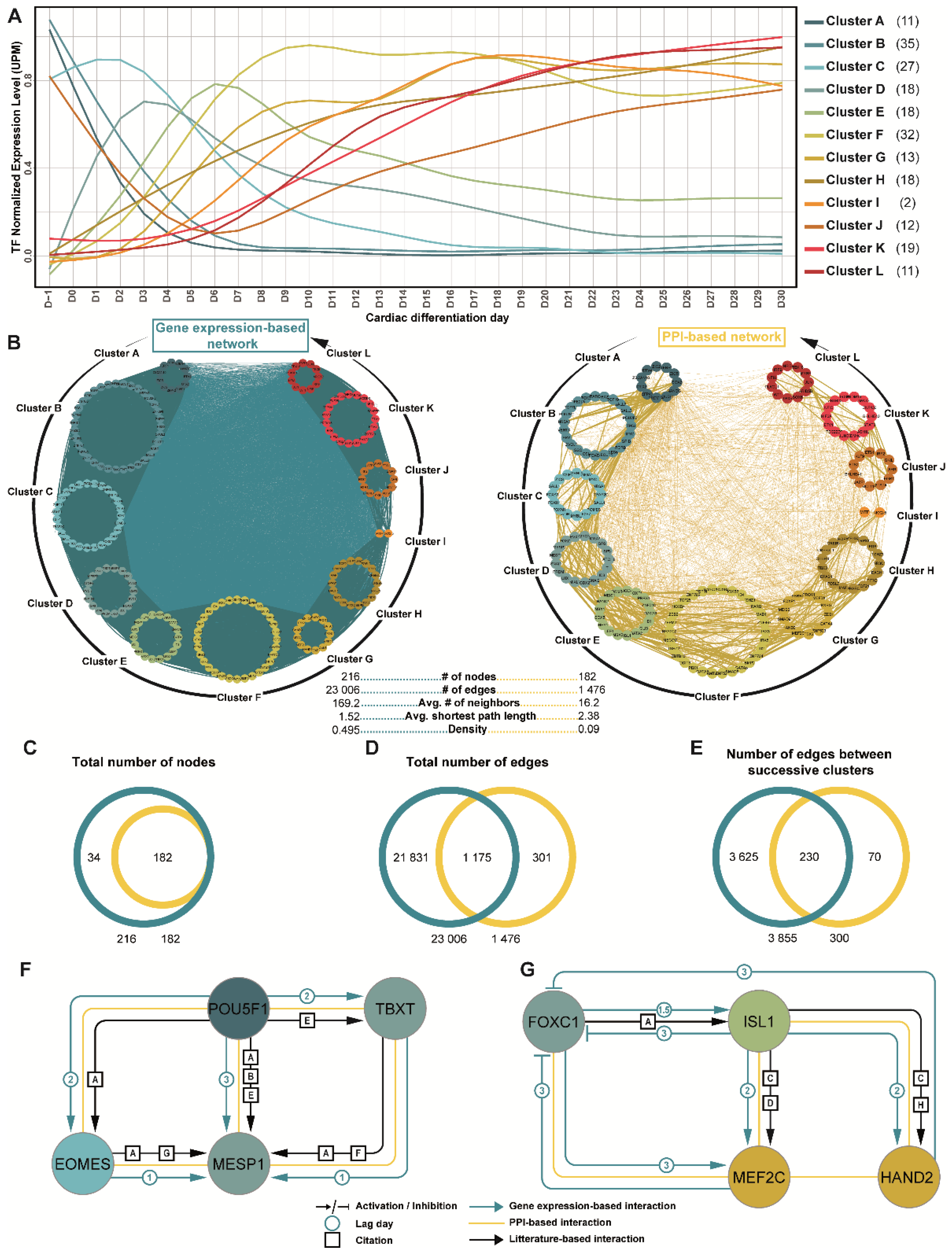
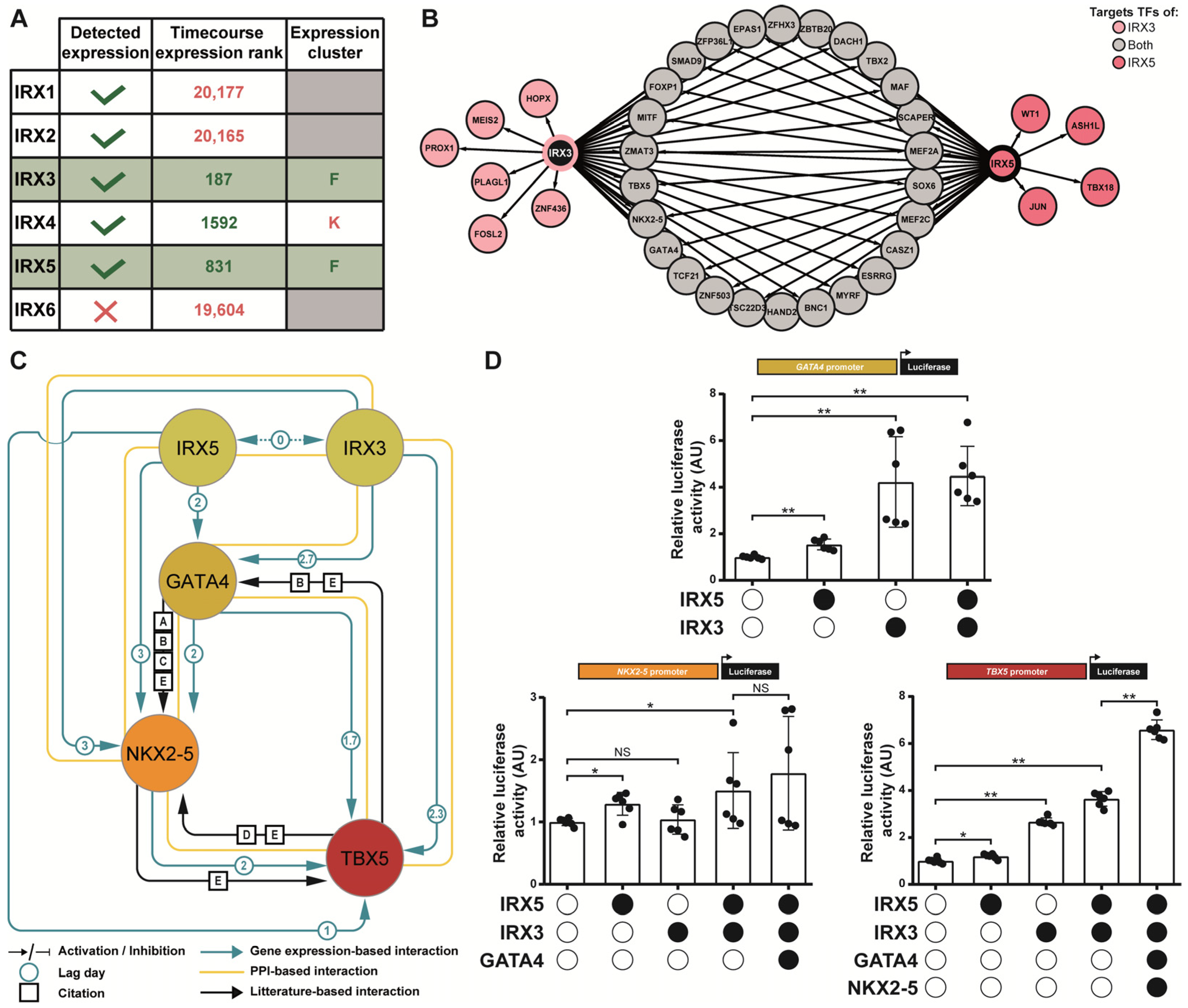
3.3. Prediction of Gene Regulatory Networks Governing hiPSC Cardiac Differentiation
3.4. IRX3 and IRX5 Are Involved in Triggering Expression of GATA4, NKX2-5, TBX5 Cardiac Transcription Factor Network
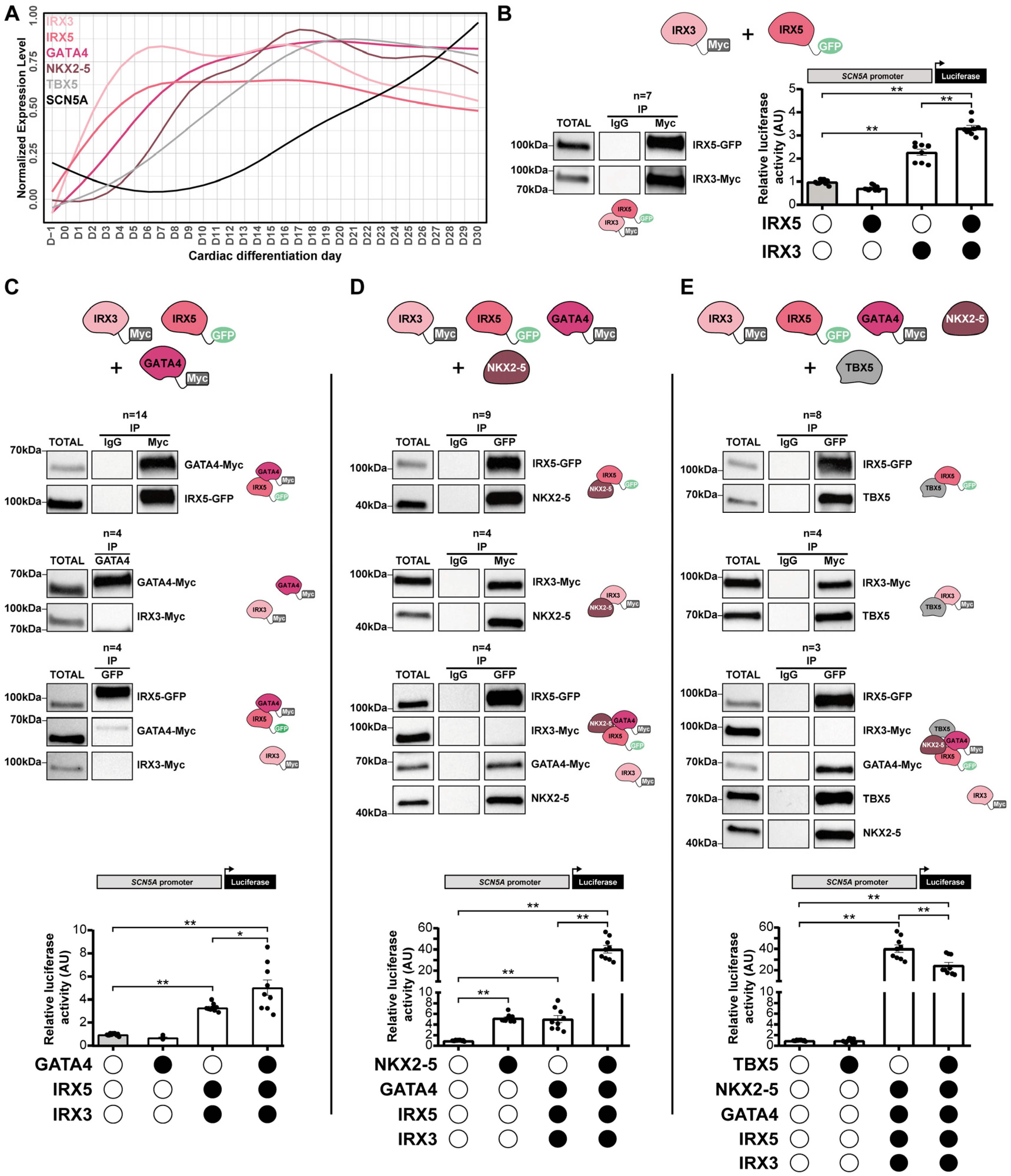
3.5. IRX3 and IRX5 Physically Interact with GATA4, NKX2-5 and TBX5 to Control SCN5A Expression
4. Discussion
4.1. In Vitro Modeling of Time in Cardiac Development
4.2. In Vitro Modeling of Cardiac Development-Associated Cellular Diversity
4.3. Uncovering New Regulatory Networks Using a Gene Expression Kinetics-Based Strategy
4.4. Activation Cascade of GATA4, NKX2-5, TBX5 Genes Triggered by IRX3 and IRX5
4.5. Exploring the Functional Interplay between IRX3/IRX5 and GATA4, NKX2-5, TBX5
4.6. Perspectives
4.7. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meilhac, S.M.; Buckingham, M.E. The Deployment of Cell Lineages That Form the Mammalian Heart. Nat. Rev. Cardiol. 2018, 15, 705–724. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.D.; Buckingham, M.E. How to Make a Heart: The Origin and Regulation of Cardiac Progenitor Cells. Curr. Top. Dev. Biol. 2010, 90, 1–41. [Google Scholar] [CrossRef]
- Postma, A.V.; Christoffels, V.M.; Bezzina, C.R. Developmental Aspects of Cardiac Arrhythmogenesis. Cardiovasc. Res. 2011, 91, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660.e19. [Google Scholar] [CrossRef]
- Bruneau, B.G. Signaling and Transcriptional Networks in Heart Development and Regeneration. Cold Spring Harb. Perspect. Biol. 2013, 5, a008292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna-Zurita, L.; Stirnimann, C.U.; Glatt, S.; Kaynak, B.L.; Thomas, S.; Baudin, F.; Samee, M.A.H.; He, D.; Small, E.M.; Mileikovsky, M.; et al. Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell 2016, 164, 999–1014. [Google Scholar] [CrossRef] [Green Version]
- Al Sayed, Z.R.; Canac, R.; Cimarosti, B.; Bonnard, C.; Gourraud, J.-B.; Hamamy, H.; Kayserili, H.; Girardeau, A.; Jouni, M.; Jacob, N.; et al. Human Model of IRX5 Mutations Reveals Key Role for This Transcription Factor in Ventricular Conduction. Cardiovasc. Res. 2021, 117, 2092–2107. [Google Scholar] [CrossRef] [PubMed]
- Bruneau, B.G.; Bao, Z.Z.; Fatkin, D.; Xavier-Neto, J.; Georgakopoulos, D.; Maguire, C.T.; Berul, C.I.; Kass, D.A.; Kuroski-de Bold, M.L.; de Bold, A.J.; et al. Cardiomyopathy in Irx4-Deficient Mice Is Preceded by Abnormal Ventricular Gene Expression. Mol. Cell. Biol. 2001, 21, 1730–1736. [Google Scholar] [CrossRef] [Green Version]
- Costantini, D.L.; Arruda, E.P.; Agarwal, P.; Kim, K.-H.; Zhu, Y.; Zhu, W.; Lebel, M.; Cheng, C.W.; Park, C.Y.; Pierce, S.A.; et al. The Homeodomain Transcription Factor Irx5 Establishes the Mouse Cardiac Ventricular Repolarization Gradient. Cell 2005, 123, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Gaborit, N.; Sakuma, R.; Wylie, J.N.; Kim, K.-H.; Zhang, S.-S.; Hui, C.-C.; Bruneau, B.G. Cooperative and Antagonistic Roles for Irx3 and Irx5 in Cardiac Morphogenesis and Postnatal Physiology. Dev. Camb. Engl. 2012, 139, 4007–4019. [Google Scholar] [CrossRef]
- Zhang, S.-S.; Kim, K.-H.; Rosen, A.; Smyth, J.W.; Sakuma, R.; Delgado-Olguín, P.; Davis, M.; Chi, N.C.; Puviindran, V.; Gaborit, N.; et al. Iroquois Homeobox Gene 3 Establishes Fast Conduction in the Cardiac His–Purkinje Network. Proc. Natl. Acad. Sci. USA 2011, 108, 13576–13581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly Efficient Generation of Human Hepatocyte-like Cells from Induced Pluripotent Stem Cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canac, R.; Caillaud, A.; Cimarosti, B.; Girardeau, A.; Hamamy, H.; Reversade, B.; Bonnard, C.; Al Sayed, Z.R.; David, L.; Poschmann, J.; et al. Generation of Three Human Induced Pluripotent Stem Cell Lines with IRX5 Knockout and Knockin Genetic Editions Using CRISPR-Cas9 System. Stem Cell Res. 2022, 58, 102627. [Google Scholar] [CrossRef] [PubMed]
- Girardeau, A.; Atticus, D.; Canac, R.; Cimarosti, B.; Caillaud, A.; Chariau, C.; Simonet, F.; Cariou, B.; Charpentier, F.; Gourraud, J.-B.; et al. Generation of Human Induced Pluripotent Stem Cell Lines from Four Unrelated Healthy Control Donors Carrying European Genetic Background. Stem Cell Res. 2021, 59, 102647. [Google Scholar] [CrossRef] [PubMed]
- Es-Salah-Lamoureux, Z.; Jouni, M.; Malak, O.A.; Belbachir, N.; Al Sayed, Z.R.; Gandon-Renard, M.; Lamirault, G.; Gauthier, C.; Baró, I.; Charpentier, F.; et al. HIV-Tat Induces a Decrease in IKr and IKsvia Reduction in Phosphatidylinositol-(4,5)-Bisphosphate Availability. J. Mol. Cell. Cardiol. 2016, 99, 1–13. [Google Scholar] [CrossRef]
- Charpentier, E.; Cornec, M.; Dumont, S.; Meistermann, D.; Bordron, P.; David, L.; Redon, R.; Bonnaud, S.; Bihouée, A. 3′RNA Sequencing for Robust and Low-Cost Gene Expression Profiling. Protoc. Exch. 2021. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.C. Timecourse: Statistical Analysis for Developmental Microarray Time Course Data. R package version 1.70.0. 2022. Available online: http://www.bioconductor.org (accessed on 3 November 2022).
- Li, X.; Martinez-Fernandez, A.; Hartjes, K.A.; Kocher, J.-P.A.; Olson, T.M.; Terzic, A.; Nelson, T.J. Transcriptional Atlas of Cardiogenesis Maps Congenital Heart Disease Interactome. Physiol. Genom. 2014, 46, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping Identifiers for the Integration of Genomic Datasets with the R/Bioconductor Package BiomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinforma. Oxf. Engl. 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. The Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef] [PubMed]
- Cortal, A.; Martignetti, L.; Six, E.; Rausell, A. Gene Signature Extraction and Cell Identity Recognition at the Single-Cell Level with Cell-ID. Nat. Biotechnol. 2021, 39, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; van Meer, B.J.; Tertoolen, L.G.J.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.; Dieben, M.A.E.; Eschenhagen, T.; Giacomelli, E.; et al. MUSCLEMOTION: A Versatile Open Software Tool to Quantify Cardiomyocyte and Cardiac Muscle Contraction In Vitro and In Vivo. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of Published Genome-Wide Association Studies, Targeted Arrays and Summary Statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the Adult Human Heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Meilhac, S.M. Cardiac Growth I: Cardiomyocyte Proliferation. ESC Textb. Cardiovasc. Dev. 2018, 75–85. [Google Scholar]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, A.H.M.; Khoshakhlagh, P.; Rojo Arias, J.E.; Pasquini, G.; Wang, K.; Swiersy, A.; Shipman, S.L.; Appleton, E.; Kiaee, K.; Kohman, R.E.; et al. A Comprehensive Library of Human Transcription Factors for Cell Fate Engineering. Nat. Biotechnol. 2021, 39, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Ang, Y.-S.; Rivas, R.N.; Ribeiro, A.J.S.; Srivas, R.; Rivera, J.; Stone, N.R.; Pratt, K.; Mohamed, T.M.A.; Fu, J.-D.; Spencer, C.I.; et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell 2016, 167, 1734–1749.e22. [Google Scholar] [CrossRef] [Green Version]
- Durocher, D.; Charron, F.; Warren, R.; Schwartz, R.J.; Nemer, M. The Cardiac Transcription Factors Nkx2-5 and GATA-4 Are Mutual Cofactors. EMBO J. 1997, 16, 5687–5696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulley, D.J.; Black, B.L. Transcription Factor Pathways and Congenital Heart Disease. Curr. Top. Dev. Biol. 2012, 100, 253–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemer, G.; Nemer, M. GATA4 in Heart Development and Disease. Heart Dev. Regen. 2010, 2, 599–616. [Google Scholar] [CrossRef]
- Waardenberg, A.J.; Ramialison, M.; Bouveret, R.; Harvey, R.P. Genetic Networks Governing Heart Development. Cold Spring Harb. Perspect. Med. 2014, 4, a013839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnolds, D.E.; Liu, F.; Fahrenbach, J.P.; Kim, G.H.; Schillinger, K.J.; Smemo, S.; McNally, E.M.; Nobrega, M.A.; Patel, V.V.; Moskowitz, I.P. TBX5 Drives Scn5a Expression to Regulate Cardiac Conduction System Function. J. Clin. Investig. 2012, 122, 2509–2518. [Google Scholar] [CrossRef] [Green Version]
- Briggs, L.E.; Takeda, M.; Cuadra, A.E.; Wakimoto, H.; Marks, M.H.; Walker, A.J.; Seki, T.; Oh, S.P.; Lu, J.T.; Sumners, C.; et al. Perinatal Loss of Nkx2-5 Results in Rapid Conduction and Contraction Defects. Circ. Res. 2008, 103, 580–590. [Google Scholar] [CrossRef] [Green Version]
- Koizumi, A.; Sasano, T.; Kimura, W.; Miyamoto, Y.; Aiba, T.; Ishikawa, T.; Nogami, A.; Fukamizu, S.; Sakurada, H.; Takahashi, Y.; et al. Genetic Defects in a His-Purkinje System Transcription Factor, IRX3, Cause Lethal Cardiac Arrhythmias. Eur. Heart J. 2016, 37, 1469–1475. [Google Scholar] [CrossRef] [PubMed]
- Tarradas, A.; Pinsach-Abuin, M.L.; Mackintosh, C.; Llorà-Batlle, O.; Pérez-Serra, A.; Batlle, M.; Pérez-Villa, F.; Zimmer, T.; Garcia-Bassets, I.; Brugada, R.; et al. Transcriptional Regulation of the Sodium Channel Gene (SCN5A) by GATA4 in Human Heart. J. Mol. Cell. Cardiol. 2017, 102, 74–82. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Jia, Y.; Takimoto, K. Interaction between Transcription Factors Iroquois Proteins 4 and 5 Controls Cardiac Potassium Channel Kv4.2 Gene Transcription. Cardiovasc. Res. 2009, 81, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, A.; Samtani, R.; Dhanantwari, P.; Lee, E.; Yamada, S.; Shiota, K.; Donofrio, M.T.; Leatherbury, L.; Lo, C.W. A Detailed Comparison of Mouse and Human Cardiac Development. Pediatr. Res. 2014, 76, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Anzai, T.; Yamagata, T.; Uosaki, H. Comparative Transcriptome Landscape of Mouse and Human Hearts. Front. Cell Dev. Biol. 2020, 8, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, E.N.; Srivastava, D. Molecular Pathways Controlling Heart Development. Science 1996, 272, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, I.S.; Rao, K.S.; Iacono, G.; Devine, W.P.; Blair, A.P.; Hota, S.K.; Lai, M.H.; Garay, B.I.; Thomas, R.; Gong, H.Z.; et al. Modeling Human TBX5 Haploinsufficiency Predicts Regulatory Networks for Congenital Heart Disease. Dev. Cell 2021, 56, 292–309.e9. [Google Scholar] [CrossRef] [PubMed]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and Coordinated Epigenetic Regulation of Developmental Transitions in the Cardiac Lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Yamanaka, S. Induced Pluripotent Stem Cells 10 Years Later: For Cardiac Applications. Circ. Res. 2017, 120, 1958–1968. [Google Scholar] [CrossRef]
- Hofbauer, P.; Jahnel, S.M.; Papai, N.; Giesshammer, M.; Deyett, A.; Schmidt, C.; Penc, M.; Tavernini, K.; Grdseloff, N.; Meledeth, C.; et al. Cardioids Reveal Self-Organizing Principles of Human Cardiogenesis. Cell 2021, 184, 3299–3317.e22. [Google Scholar] [CrossRef]
- Specht, A.T.; Li, J. LEAP: Constructing Gene Co-Expression Networks for Single-Cell RNA-Sequencing Data Using Pseudotime Ordering. Bioinforma. Oxf. Engl. 2017, 33, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yi, N.; Hu, Y.; Zhou, X.; Jiang, H.; Lin, Q.; Chen, R.; Liu, H.; Gu, Y.; Tong, C.; et al. Molecular Signatures and Networks of Cardiomyocyte Differentiation in Humans and Mice. Mol. Ther. Nucleic Acids 2020, 21, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Teran, B.; Pittman, M.; Felix, F.; Thomas, R.; Richmond-Buccola, D.; Hüttenhain, R.; Choudhary, K.; Moroni, E.; Costa, M.W.; Huang, Y.; et al. Transcription Factor Protein Interactomes Reveal Genetic Determinants in Heart Disease. Cell 2022, 185, 794–814.e30. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, C.; Strobl, A.C.; Shboul, M.; Lee, H.; Merriman, B.; Nelson, S.F.; Ababneh, O.H.; Uz, E.; Güran, T.; Kayserili, H.; et al. Mutations in IRX5 Impair Craniofacial Development and Germ Cell Migration via SDF1. Nat. Genet. 2012, 44, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Aiba, T.; Sasano, T.; Furukawa, T.; Kusano, K.; Shimizu, W. IRX3 Variant as a Modifier of Brugada Syndrome with Frequent Ventricular Fibrillation. Hear. Case Rep. 2016, 2, 465–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowton, M.; Guzzetta, A.; Rydeen, A.B.; Moskowitz, I.P. Control of Cardiomyocyte Differentiation Timing by Intercellular Signaling Pathways. Semin. Cell Dev. Biol. 2021, 118, 94–106. [Google Scholar] [CrossRef]
- Barc, J.; Tadros, R.; Glinge, C.; Chiang, D.Y.; Jouni, M.; Simonet, F.; Jurgens, S.J.; Baudic, M.; Nicastro, M.; Potet, F.; et al. Genome-Wide Association Analyses Identify New Brugada Syndrome Risk Loci and Highlight a New Mechanism of Sodium Channel Regulation in Disease Susceptibility. Nat. Genet. 2022, 54, 232–239. [Google Scholar] [CrossRef]
- Roselli, C.; Chaffin, M.D.; Weng, L.-C.; Aeschbacher, S.; Ahlberg, G.; Albert, C.M.; Almgren, P.; Alonso, A.; Anderson, C.D.; Aragam, K.G.; et al. Multi-Ethnic Genome-Wide Association Study for Atrial Fibrillation. Nat. Genet. 2018, 50, 1225–1233. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid Name | Sequence/Reference | Supplier | |
|---|---|---|---|
| NKX2.5 promoter–FireflyLuc | -2000 bp_Start codon | Vectorbuilder | |
| GATA4 promoter–FireflyLuc | -1800_TSS_+200 | Vectorbuilder | |
| TBX5 promoter–FireflyLuc | -1800_TSS_+200 | Vectorbuilder | |
| SCN5A promoter–FireflyLuc | -2109_TSS_+1072 | Adapted from [7] | |
| pGL2 Renilla luciferase | Promega | ||
| IRX5 | RG234228 | Origene | |
| IRX3 | RG205722 | Origene | |
| GATA4 | RC210945 | Origene | |
| TBX5 | SC120046 | Origene | |
| NKX2.5 | SC122678 | Origene | |
| pcDNA3.1 | Invitrogen | ||
| Antibody | Reference | RRID | Supplier |
| anti-GFP | TA150041 | AB_2622256 | Origene |
| anti-Myc Tag | 05-724 | AB_309938 | Merck Millipore |
| anti-IRX5 | sc-81102 | AB_1124818 | Santa Cruz |
| anti-IRX3 | sc-166877 | AB_10609525 | Santa Cruz |
| anti-GATA4 | sc-25310 | AB_627667 | Santa Cruz |
| anti-TBX5 | sc-515536 | Santa Cruz | |
| anti-NKX2.5 | sc-8697 | AB_650280 | Santa Cruz |
| anti-Troponin I | sc-15368 | AB_793465 | Santa Cruz |
| Mouse IgG Isotype Control | 02-6502 | AB_2532951 | Thermo Fisher Scientific |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canac, R.; Cimarosti, B.; Girardeau, A.; Forest, V.; Olchesqui, P.; Poschmann, J.; Redon, R.; Lemarchand, P.; Gaborit, N.; Lamirault, G. Deciphering Transcriptional Networks during Human Cardiac Development. Cells 2022, 11, 3915. https://doi.org/10.3390/cells11233915
Canac R, Cimarosti B, Girardeau A, Forest V, Olchesqui P, Poschmann J, Redon R, Lemarchand P, Gaborit N, Lamirault G. Deciphering Transcriptional Networks during Human Cardiac Development. Cells. 2022; 11(23):3915. https://doi.org/10.3390/cells11233915
Chicago/Turabian StyleCanac, Robin, Bastien Cimarosti, Aurore Girardeau, Virginie Forest, Pierre Olchesqui, Jeremie Poschmann, Richard Redon, Patricia Lemarchand, Nathalie Gaborit, and Guillaume Lamirault. 2022. "Deciphering Transcriptional Networks during Human Cardiac Development" Cells 11, no. 23: 3915. https://doi.org/10.3390/cells11233915