
Bioengineering Human Pluripotent Stem Cell-Derived Retinal Organoids and Optic Vesicle-Containing Brain Organoids for Ocular Diseases
 , ,
, , 
Abstract
:1. Introduction
2. Bioengineering Retinal Organoids from iPSCs
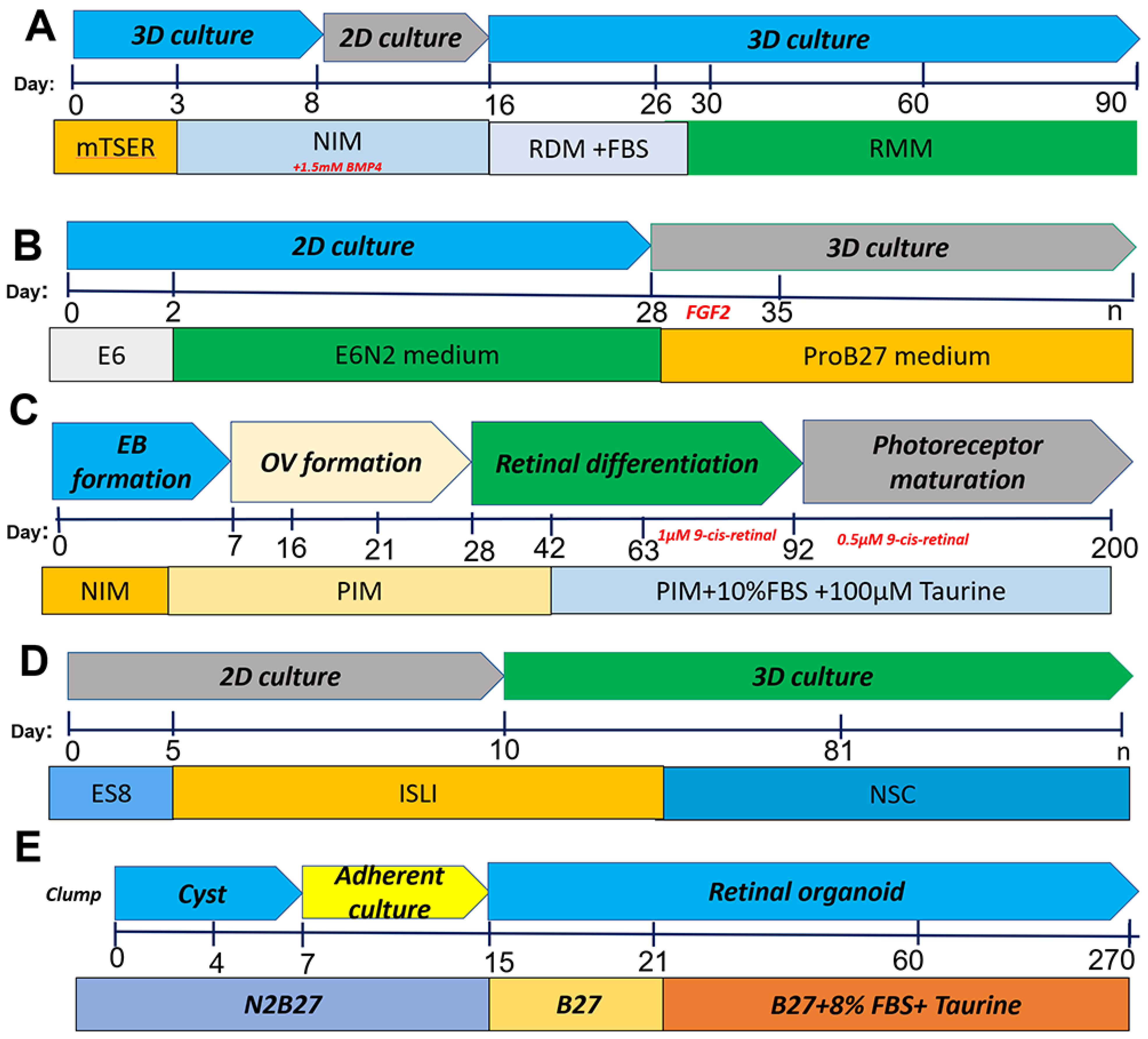
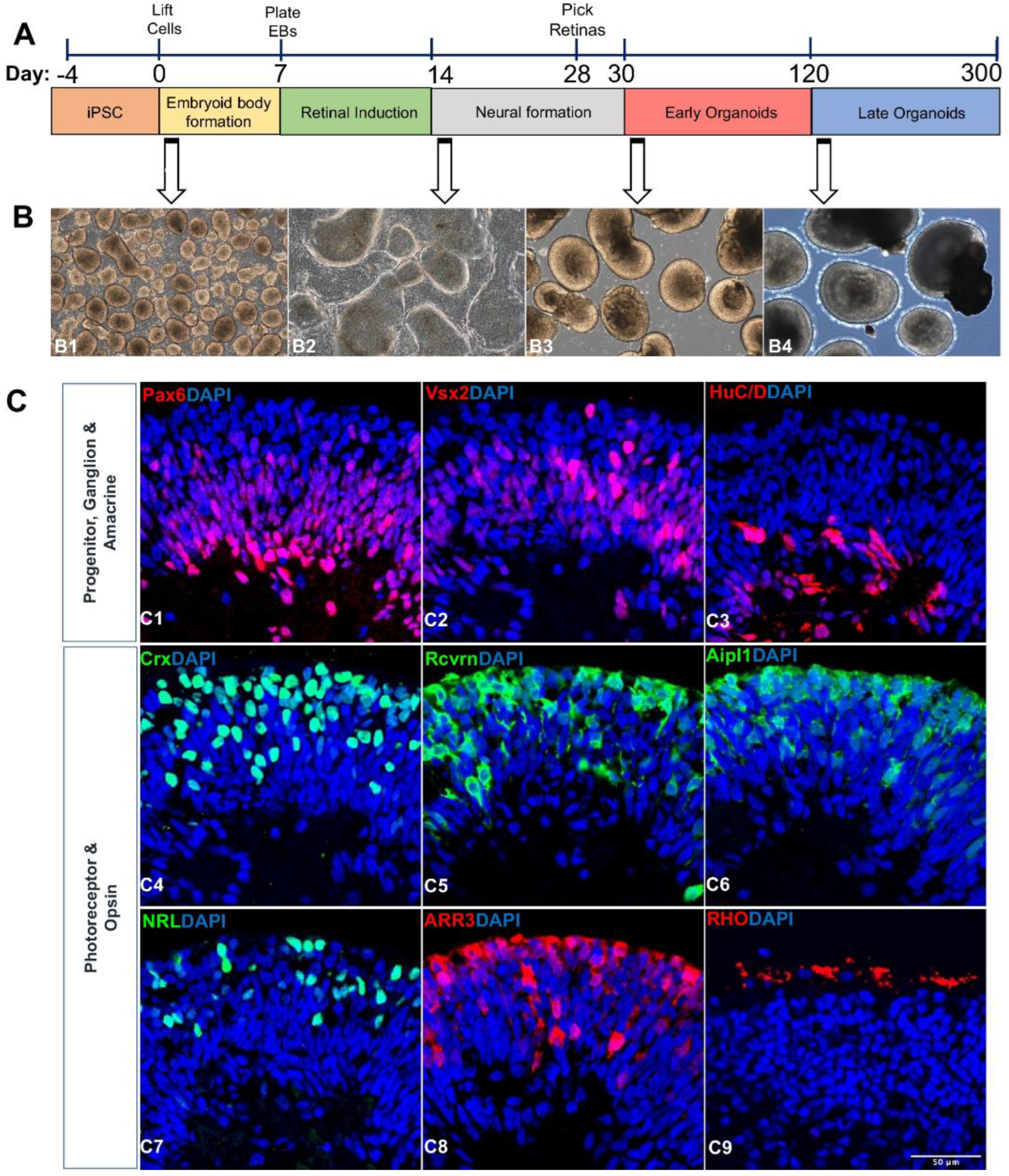
2.1. Differentiation of iPSCs into Retinal Organoids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Source | Differentiation Methods | Characterizations | Reference |
|---|---|---|---|
| IPSC (NCL1 from NxCell, Inc.) | Small molecule protocol: IWR1 (a Wnt signaling inhibitor) SB431542 (a TGFβ signaling inhibitor) LDN193189 (a BMP signaling inhibitor) IGF1; neural stem cell (NSC) medium | Pan-photoreceptor markers OTX2, CRX, and RECOVERIN in the outer layer of RO; retinal stem cell, ganglion cell and amacrine cell marker, PAX6; expressed markers of retinal ganglion cells, BRN3 After 12 weeks of differentiation and s expressed several pan photoreceptor markers, including BLIMP1, RECOVERIN, and AIPL1 | [46] |
| hiPSC lines PEN8E, 901, 902 | Scrapping method for RO generation; formation of EB from hPSC, neural induction medium (NIM),Scraping of adherent cells to form free floating OV, retinal induction medium (B27), 1% NEAA, 1% GlutaMAX | New scraping method displayed morphology similar to that of the dissected organoids and recapitulated the temporal development of the in vivo retina. Retinal ganglion cells (RGCs), horizontal or amacrine cells, ARL13B and basal body marker PCNT, photoreceptor cilia and basal bodies at day 200. | [48] |
| Three hiPSC lines, IMR90-437 (WiCell), CB-iPSC6.238 and KA.1 | Blebbistatin for aggregate formation induction (NIM), taurine and 2 mM GlutaMAX. Treatment with retinoic acid | Formation of optic cups; expression of a group of transcription factors including PAX6, RX, LHX2, SIX3, and SIX6, while the surrounding anterior neuroepithelial cells express PAX6 and SOX, | [49] |
| Human iPSCs | Based on the concept that IGF1 signaling, in conjunction with retinoic acid and triiodothyronine, crucial for retinal development NIM+ BMP4; RDM +FBS; RMM | Photoreceptors, bipolar, horizontal, amacrine, Müller, and retinal ganglion cells develop a thick layer of neuroepithelium in the organoids at 22 weeks | [43] |
| iPSCs from Nrl-GFP mouse | 10-day differentiation in static culture followed by maturation in RWV bioreactor (3D); RMM/N2; B-27-VitA, IGF1, NEAA | Transcriptome analysis shows ROs better mimic the spatiotemporal development observed in retinogenesis in vivo; when compared to static cultures, RWV organoids demonstrated rapid differentiation; expression of Müller glia cells and rod photoreceptor markers such as BRN3 and opsin 1 short-wavelength-sensitive | [50] |
2.2. Transcriptome Analysis of Retinal Organoids
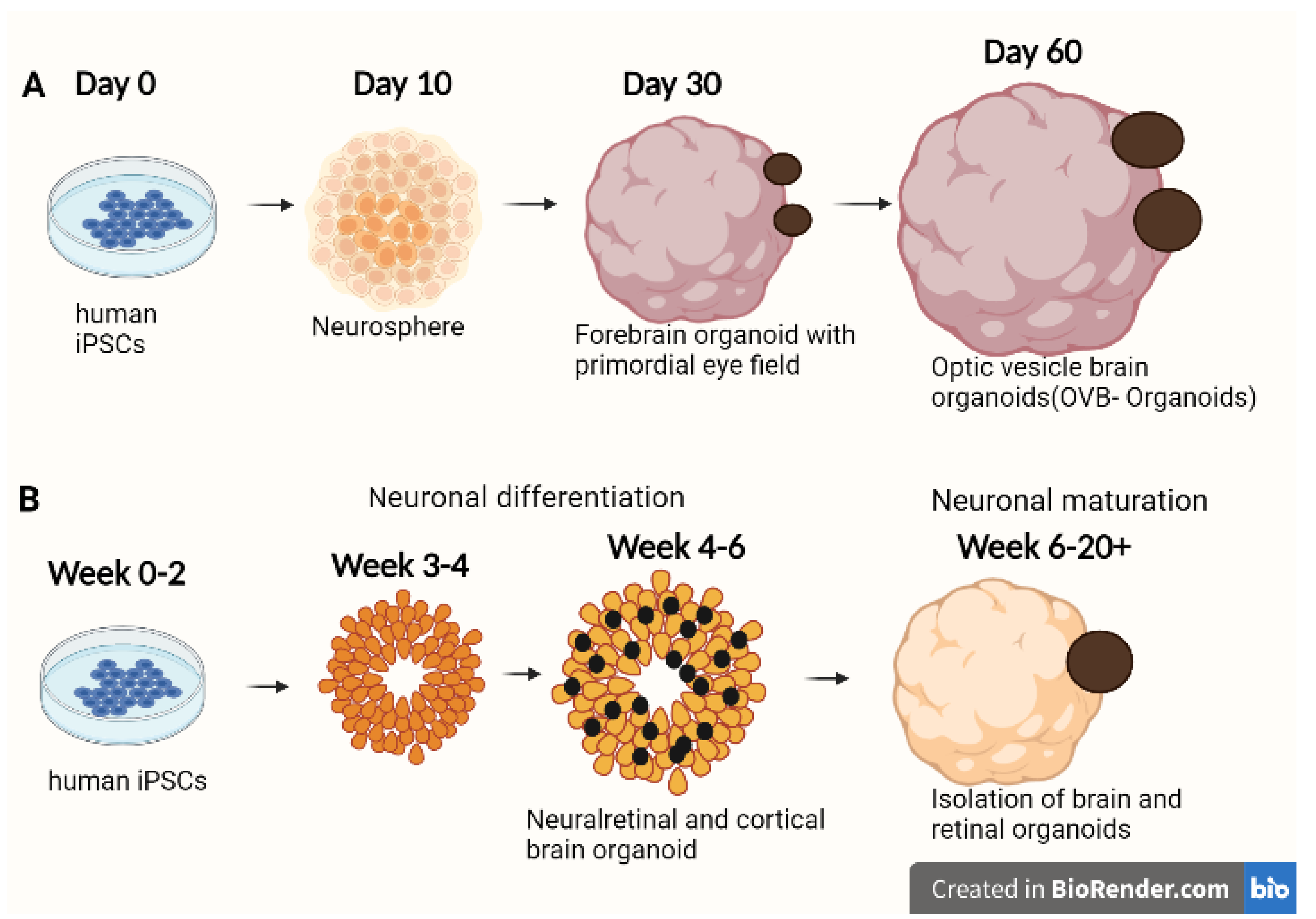
2.3. Bioengineering Optical Vesicle-Containing Brain Organoids from iPSCs
3. Applications of ROs in Various Ocular Diseases
| Eye Diseases | Disease Models | Characterizations of Disease Pathology, or Drug Screening | Reference |
|---|---|---|---|
| Glaucoma | ROs from hPSCs with an E50K mutation in the OPTN) gene | Optic nerve damage, high pressure in the eye causing pain and sudden visual disturbance; when derived from three-dimensional retinal organoids, retinal ganglion cells with a glaucoma OPTN gene mutation exhibit neurodegenerative phenotypes. | [89,90,91,92] |
| Leber congenital amaurosis | ROs from hiPSCs derived from LCA4 patient carrying a Cys89Arg mutation in AIPL1 | LCA is a retinopathy causing visual impairment. Patient ROs showed lower levels of AIPL1 with no retinal degeneration. ROs confirmed findings in animal models of the disease phenotype | [93,94,95] |
| Stargardt’ s disease | ROs from iPSCs of four STGD1 patients (homozygous for the p.Gly1961Glu variation, compound heterozygous, and two deleterious ABCA4 alleles) | Symptoms range from modest vision loss with less obvious fundus abnormalities to a more severe cone or cone-rod dystrophies accounting for 12% of IRD-related blindness | [96,97,98,99] |
| Retinitis pigmentosa | ROs with electrophysiological properties generated from iPSCs from RP patients with different mutations in the RPGR gene. | Loss of vision, complete blindness is uncommon. Pathogenesis of RPGR using patient-specific organoids. Significant abnormalities in photoreceptors were discovered, including decreased cilia length, photoreceptor cell quantity, and expression of photoreceptor-related genes. | [100,101,102] |
| RP11 (PRPF31-mutated) patient-derived ROs | In comparison to controls, TEM revealed that patient photoreceptors showed an increase in apoptotic nuclei and the presence of stress vacuoles, indicating progressive degenerative characteristics in ROs | [103] | |
| X-linked retinitis pigmentosa | Temporal maturation of CRISPR gene edited RP2 knockout Ros as well as ROs derived from patients with the same R120X nonsense mutation | Significant loss of vision, complete blindness is common in males. RP2 knockouts that were isogenic match the phenotype of RP2 patient-derived organoids. Rod photoreceptor cell death occurs in RP2 null ROs. | [104,105,106] |
| X-linked juvenile retinoschisis | hiPSCs from patients to study XLRS in a 3D retinal organoid in vitro differentiation system. | Damaged macula responsible for sharp central vision and visual acuity impairment in children. A retinal organoid model produced from hiPSCs recapitulates important XLRS characteristics. RS1 secretion and retinal development are normalized after CRISPR/Cas9 editing | [107,108,109,110] |
| Retinoblastoma | ROs generated from CRISPR/Cas9-derived RB1-null human embryonic stem cells (hESCs) | Eye cancer developed from immature cells of retina. The loss of RB1 enhanced apoptosis and reduced the number of photoreceptors in RB1-null ROs. The absence of RB1 did not, however, result in the formation of retinoblastoma in ROs | [109,111] |
3.1. Glaucoma
3.2. Leber Congenital Amaurosis (LCA)
3.3. Stargardt’s Disease (STGD1)
3.4. Retinitis Pigmentosa (RP)
3.5. Usher Syndrome
3.6. X-Linked Juvenile Retinoschisis (XLRS)
3.7. Retinoblastoma
3.8. Microphthalmia
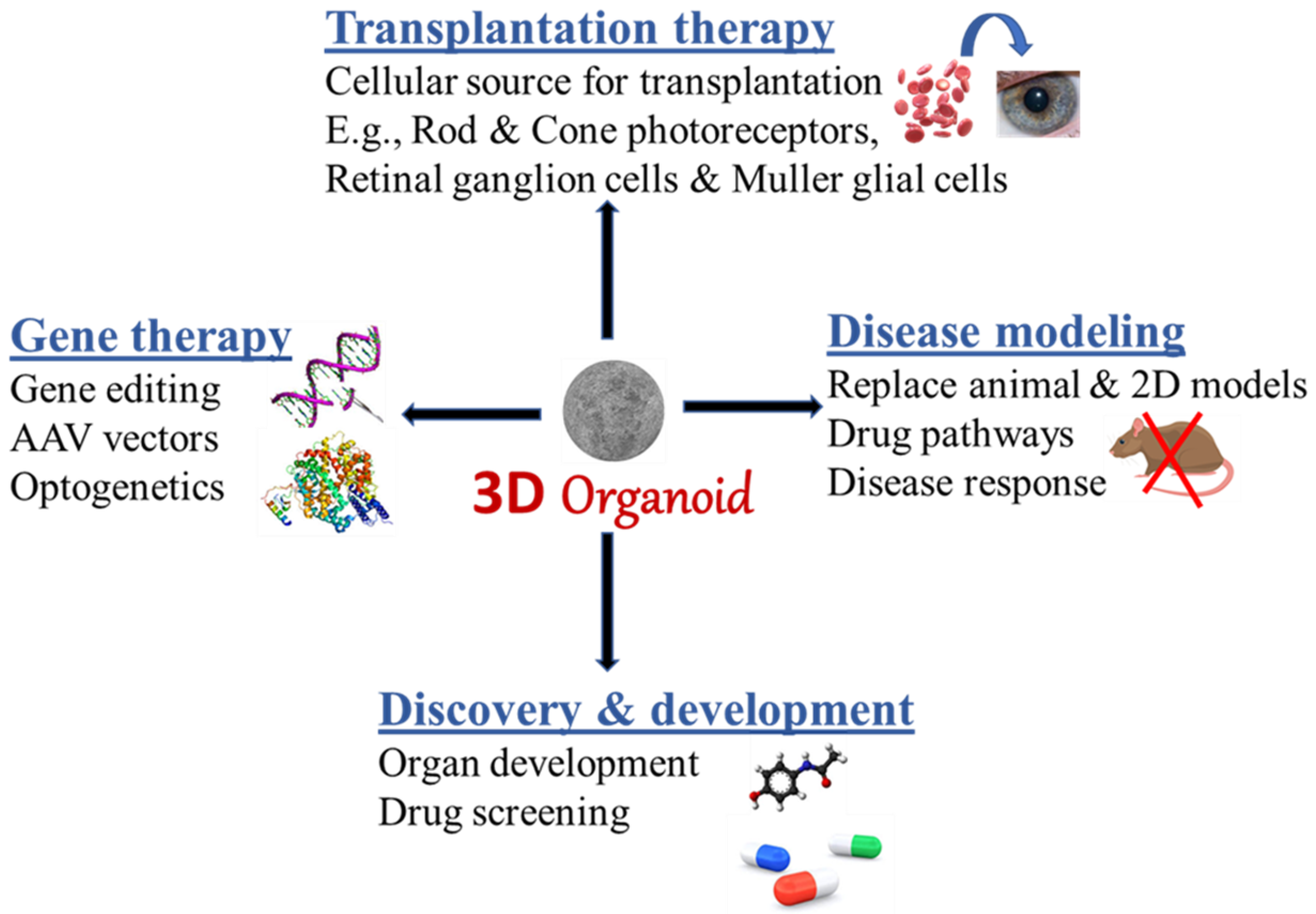
4. Platforms of Retinal and OVB Organoid Applications
4.1. A Source for Transplantation and Cell Replacement
4.2. A Platform for Drug Screening
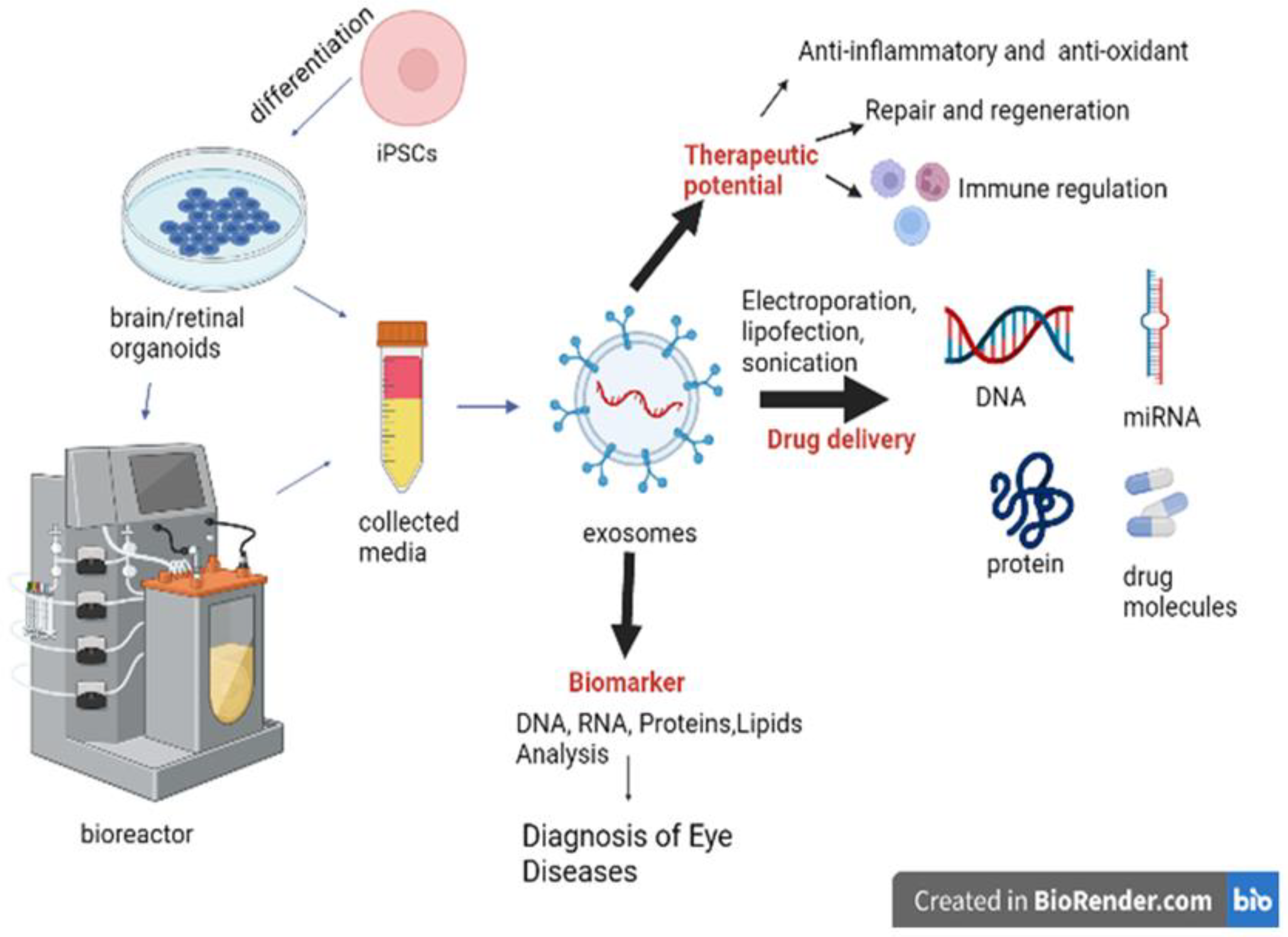
5. Importance of Extracellular Vesicles Secreted by Retinal and OVB Organoids
5.1. Characteristics of Extracellular Vesicles (EVs)
5.2. EVs as Drug Delivery Vehicles for Treating Ocular Diseases
| Source of EVs | Method of Isolation | Applications | Reference |
|---|---|---|---|
| Induced-primary RPE (ipRPE) from human retinal organoids | Iodixanol density gradient separation | EVs isolated from RPE derived from ROs contain proteins involved in AMD pathophysiology such as immune response, inflammation, oxidative stress, and drusen composition. This study shows that drusen-associated proteins are present in the RPE-derived EVs and play an active role in drusen growth which is relevant in AMD progression and other retinal diseases. | [204] |
| hiPSC-derived 3D retinal organoids | Differential centrifugation | The functional 3D RO-derived EVs contains contained small noncoding RNAs cargo including miRNA, tRNA, and piRNA which are released continuously and are related to post-translational modification and human retinal development regulation. The internalization of 3D RO EVs by hRPCs is necessary for the regulation of genes involved in retinal homeostasis and developmental processes, such as nuclear transport, transcription, GTPase regulation, and ganglion and photoreceptor cell differentiation. | [196] |
| hiPSC-derived 3D retinal organoids | Modified differential ultracentrifugation method with PEG precipitation | Bioreactors were used to upscale secretion of EVs by late time point ROs (>120 days). The EV protein profiling of conditioned medium across the complete development and maturation of retinogenesis can serve as a valuable biomarker for assessing the patient-specific retinal organoids with inherited retinal degenerations. Gene expression analysis by qPCR showed a high expression of exosome biogenesis genes in late retinal organoids derived EVs (>120 day). | [68] |
6. Limitations of Current Retinal and OVB Organoids
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Adeno-associated virus | AAV2/5 |
| ATP-binding cassette transporter subfamily A4 | ABCA4 |
| All-trans retinoic acid | ATRA |
| Aryl hydrocarbon receptor-interacting protein-like 1 | AIPL1 |
| Basic fibroblast growth factor | bFGF |
| Bone morphogenetic protein | BMP |
| 9-cis retinoic acid | 9CRA |
| CONE ARRESTIN | CAR |
| Cone-rod Homeobox | CRX |
| Current good manufacturing practices | cGMP |
| Embryoid body | EB |
| Embryonic stem cells | ESCs |
| Extracellular matrix | ECM |
| Extracellular vesicle | EV |
| Human neurodevelopmental diseases | NDDs |
| Human pluripotent stem cells | hPSCs |
| Human retinal progenitor cells | hRPCs |
| Human umbilical cord | hUC |
| Induced pluripotent stem cells | iPSCs |
| Inherited retinal diseases | IRDs |
| Insulin-like growth factor | IGF 1 |
| Intraocular pressure | IOP |
| a Wnt signaling inhibitor | IWR1 |
| Leber congenital amaurosis | LCA |
| Macular telangiectasia type 2 | MacTel |
| Mesenchymal stem cells | MSCs |
| Microphthalmia-associated transcription factor | MITF |
| Multivesicular body | MVB |
| Neurodevelopmental diseases | NDDs |
| Neural retina | NR |
| Neural retina progenitors | NRPCs |
| Outer nuclear layer | ONL |
| Optic nerve head | ONH |
| Optic vesicles | OVs |
| Optical vesicle-containing brain organoids | OVB-organoids |
| Optineurin | OPTN |
| Photoreceptor- induction medium | PIM |
| pre-mRNA processing factors | PRPFs |
| RECOVERIN | RCVRN |
| Retinal ganglion cells | RGC |
| Retinal nerve fiber layer | RNFL |
| Retinal organoids | ROs |
| Retinal pigment epithelium | RPE |
| Retinitis Pigmentosa | RP |
| Retinoblastoma | RB |
| Retinoic acid receptors | RARs |
| Retinoid X receptors | RXRs |
| Single cell RNA sequencing | scRNASeq |
| Sonic Hedgehog | SHH |
| Starburst amacrine cells | SACs |
| Stargardt’s disease | STGD1 |
| Three-dimensional | 3D |
| Transforming growth factor | TGF |
| Triiodothyronine | T3 |
| Visual System Homeobox 2 | VSX2 |
| X-linked juvenile retinoschisis | XLRS |
| X-linked RP | XLRP |
References
- Huch, M.; Koo, B.-K. Modeling mouse and human development using organoid cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Corrò, C.; Novellasdemunt, L.; Li, V.S. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef] [PubMed]
- Ootani, A.; Li, X.; Sangiorgi, E.; Ho, Q.T.; Ueno, H.; Toda, S.; Sugihara, H.; Fujimoto, K.; Weissman, I.L.; Capecchi, M.R.; et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat. Med. 2009, 15, 701–706. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Jiao, Y.; Qin, S.; Zhao, W.; Chu, Q.; Wu, K. Organoid technology in disease modelling, drug development, personalized treatment and regeneration medicine. Exp. Hematol. Oncol. 2018, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Kaplan, H.J.; Tezel, T.H.; Berger, A.S.; Wolf, M.L.; Del Priore, L.V. Human photoreceptor transplantation in retinitis pigmentosa: A safety study. Arch. Ophthalmol. 1997, 115, 1168–1172. [Google Scholar] [CrossRef]
- Radtke, N.D.; Aramant, R.B.; Seiler, M.; Petry, H.M. Preliminary report: Indications of improved visual function after retinal sheet transplantation in retinitis pigmentosa patients. Am. J. Ophthalmol. 1999, 128, 384–387. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-Formation of Optic Cups and Storable Stratified Neural Retina from Human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garreta, E.; Kamm, R.D.; Lopes, S.M.C.D.S.; Lancaster, M.A.; Weiss, R.; Trepat, X.; Hyun, I.; Montserrat, N. Rethinking organoid technology through bioengineering. Nat. Mater. 2020, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S.; Shearer, R.L.; Capowski, E.E.; Wright, L.S.; Wallace, K.A.; McMillan, E.L.; Zhang, S.-C.; Gamm, D.M. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16698–16703. [Google Scholar] [CrossRef] [Green Version]
- Reichman, S.; Terray, A.; Slembrouck, A.; Nanteau, C.; Orieux, G.; Habeler, W.; Nandrot, E.F.; Sahel, J.-A.; Monville, C.; Goureau, O. From confluent human iPS cells to self-forming neural retina and retinal pigmented epithelium. Proc. Natl. Acad. Sci. USA 2014, 111, 8518–8523. [Google Scholar] [CrossRef] [Green Version]
- Vugler, A.; Carr, A.-J.; Lawrence, J.; Chen, L.L.; Burrell, K.; Wright, A.; Lundh, P.; Semo, M.; Ahmado, A.; Gias, C.; et al. Elucidating the phenomenon of HESC-derived RPE: Anatomy of cell genesis, expansion and retinal transplantation. Exp. Neurol. 2008, 214, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.J.; Vugler, A.A.; Hikita, S.T.; Lawrence, J.M.; Gias, C.; Chen, L.L.; Buchholz, D.E.; Ahmado, A.; Semo, M.; Smart, M.J.K.; et al. Protective effects of human iPS-derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLoS ONE 2009, 4, e8152. [Google Scholar] [CrossRef] [PubMed]
- Ben M’Barek, K.; Habeler, W.; Plancheron, A.; Jarraya, M.; Regent, F.; Terray, A.; Yang, Y.; Chatrousse, L.; Domingues, S.; Masson, Y.; et al. Human ESC–derived retinal epithelial cell sheets potentiate rescue of photoreceptor cell loss in rats with retinal degeneration. Sci. Transl. Med. 2017, 9, eaai7471. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Muffat, J.; Li, Y.; Jaenisch, R. CNS disease models with human pluripotent stem cells in the CRISPR age. Curr. Opin. Cell Biol. 2016, 43, 96–103. [Google Scholar] [CrossRef]
- Poon, A.; Zhang, Y.; Chandrasekaran, A.; Phanthong, P.; Schmid, B.; Nielsen, T.T.; Freude, K.K. Modeling neurodegenerative diseases with patient-derived induced pluripotent cells: Possibilities and challenges. New Biotechnol. 2017, 39, 190–198. [Google Scholar] [CrossRef]
- Gabriel, E.; Wason, A.; Ramani, A.; Gooi, L.M.; Keller, P.; Pozniakovsky, A.; Poser, I.; Noack, F.; Telugu, N.S.; Calegari, F.; et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J. 2016, 35, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, M.; Lee, S.; Wark, J.R.; Xiao, D.; Lim, B.Y.; O’Hara-Wright, M.; Kim, H.J.; Smith, G.C.; Wong, T.; Teber, E.T.; et al. Differentiation of brain and retinal organoids from confluent cultures of pluripotent stem cells connected by nerve-like axonal projections of optic origin. Stem Cell Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- Collin, J.; Queen, R.; Zerti, D.; Dorgau, B.; Hussain, R.; Coxhead, J.; Cockell, S.; Lako, M. Deconstructing Retinal Organoids: Single Cell RNA-Seq Reveals the Cellular Components of Human Pluripotent Stem Cell-Derived Retina. Stem Cells 2018, 37, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron Cowan, A.S.; Renner, M.; De Gennaro, M.; Roma, G.; Nigsch, F.; Roska Correspondence, B.; Cowan, C.S.; Gross-Scherf, B.; Goldblum, D.; Hou, Y.; et al. Cell Types of the Human Retina and Its Organoids at Single-Cell Resolution. Cell 2020, 182, 1623–1640.e34. [Google Scholar] [CrossRef]
- Hoshino, A.; Ratnapriya, R.; Brooks, M.J.; Chaitankar, V.; Wilken, M.S.; Zhang, C.; Starostik, M.R.; Gieser, L.; La Torre, A.; Nishio, M.; et al. Molecular Anatomy of the Developing Human Retina. Dev. Cell 2017, 43, 763–779.e4. [Google Scholar] [CrossRef] [Green Version]
- Capowski, E.E.; Samimi, K.; Mayerl, S.J.; Phillips, M.J.; Pinilla, I.; Howden, S.E.; Saha, J.; Jansen, A.D.; Edwards, K.L.; Jager, L.D.; et al. Reproducibility and staging of 3D human retinal organoids across multiple pluripotent stem cell lines. Development 2018, 146, dev171686. [Google Scholar] [CrossRef] [Green Version]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Sasai, Y.; Eiraku, M.; Suga, H. In vitro organogenesis in three dimensions: Self-organising stem cells. Development 2012, 139, 4111–4121. [Google Scholar] [CrossRef] [Green Version]
- Centanin, L.; Wittbrodt, J. Retinal neurogenesis. Development 2014, 141, 241–244. [Google Scholar] [CrossRef]
- Sasai, Y. Grow your own eye. Sci. Am. 2012, 307, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, A.; Drucker, D. The development of parafoveal and mid-peripheral human retina. Behav. Brain Res. 1992, 49, 21–31. [Google Scholar] [CrossRef]
- Xiao, M.; Hendrickson, A. Spatial and temporal expression of short, long/medium, or both opsins in human fetal cones. J. Comp. Neurol. 2000, 425, 545–559. [Google Scholar] [CrossRef]
- Hendrickson, A. Development of Retinal Layers in Prenatal Human Retina. Am. J. Ophthalmol. 2015, 161, 29–35.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, M.J.; Wallace, K.A.; Dickerson, S.J.; Miller, M.J.; Verhoeven, A.D.; Martin, J.M.; Wright, L.S.; Shen, W.; Capowski, E.E.; Percin, E.F.; et al. Blood-Derived Human iPS Cells Generate Optic Vesicle–Like Structures with the Capacity to Form Retinal Laminae and Develop Synapses. Investig. Opthalmology Vis. Sci. 2012, 53, 2007–2019. [Google Scholar] [CrossRef] [PubMed]
- Osakada, F.; Ikeda, H.; Mandai, M.; Wataya, T.; Watanabe, K.; Yoshimura, N.; Akaike, A.; Sasai, Y.; Takahashi, M. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat. Biotechnol. 2008, 26, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Osakada, F.; Jin, Z.-B.; Hirami, Y.; Ikeda, H.; Danjyo, T.; Watanabe, K.; Sasai, Y.; Takahashi, M. In vitro differentiation of retinal cells from human pluripotent stem cells by small-molecule induction. J. Cell Sci. 2009, 122, 3169–3179. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, H.; Osakada, F.; Watanabe, K.; Mizuseki, K.; Haraguchi, T.; Miyoshi, H.; Kamiya, D.; Honda, Y.; Sasai, N.; Yoshimura, N.; et al. Generation of Rx+/Pax6+ neural retinal precursors from embryonic stem cells. Proc. Natl. Acad. Sci. USA 2005, 102, 11331–11336. [Google Scholar] [CrossRef] [Green Version]
- Hirami, Y.; Osakada, F.; Takahashi, K.; Okita, K.; Yamanaka, S.; Ikeda, H.; Yoshimura, N.; Takahashi, M. Generation of retinal cells from mouse and human induced pluripotent stem cells. Neurosci. Lett. 2009, 458, 126–131. [Google Scholar] [CrossRef] [Green Version]
- Tucker, B.A.; Park, I.-H.; Qi, S.D.; Klassen, H.J.; Jiang, C.; Yao, J.; Redenti, S.; Daley, G.Q.; Young, M.J. Transplantation of Adult Mouse iPS Cell-Derived Photoreceptor Precursors Restores Retinal Structure and Function in Degenerative Mice. PLoS ONE 2011, 6, e18992. [Google Scholar] [CrossRef]
- Lamba, D.A.; McUsic, A.; Hirata, R.K.; Wang, P.-R.; Russell, D.; Reh, T.A. Generation, Purification and Transplantation of Photoreceptors Derived from Human Induced Pluripotent Stem Cells. PLoS ONE 2010, 5, e8763. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.A.; Mullins, R.F.; Streb, L.M.; Anfinson, K.; Eyestone, M.E.; Kaalberg, E.; Riker, M.J.; Drack, A.V.; Braun, T.A.; Stone, E.M. Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. Elife 2013, 2, e00824. [Google Scholar] [CrossRef] [PubMed]
- Chichagova, V.; Dorgau, B.; Felemban, M.; Georgiou, M.; Armstrong, L.; Lako, M. Differentiation of Retinal Organoids from Human Pluripotent Stem Cells. Curr. Protoc. Stem Cell Biol. 2019, 50, e95. [Google Scholar] [CrossRef] [PubMed]
- Reichman, S.; Slembrouck, A.; Gagliardi, G.; Chaffiol, A.; Terray, A.; Nanteau, C.; Potey, A.; Belle, M.; Rabesandratana, O.; Duebel, J.; et al. Generation of Storable Retinal Organoids and Retinal Pigmented Epithelium from Adherent Human iPS Cells in Xeno-Free and Feeder-Free Conditions. Stem Cells 2017, 35, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Kaya, K.D.; Chen, H.Y.; Brooks, M.J.; Kelley, R.A.; Shimada, H.; Nagashima, K.; De Val, N.; Drinnan, C.T.; Gieser, L.; Kruczek, K.; et al. Transcriptome-based molecular staging of human stem cell-derived retinal organoids uncovers accelerated photoreceptor differentiation by 9-cis retinal. Mol. Vis. 2019, 25, 663–678. [Google Scholar]
- Zhu, J.; Reynolds, J.; Garcia, T.; Cifuentes, H.; Chew, S.; Zeng, X.; Lamba, D.A. Generation of Transplantable Retinal Photoreceptors from a Current Good Manufacturing Practice-Manufactured Human Induced Pluripotent Stem Cell Line. Stem Cells Transl. Med. 2018, 7, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lowe, A.; Dharmat, R.; Lee, S.; Owen, L.A.; Wang, J.; Shakoor, A.; Li, Y.; Morgan, D.J.; Hejazi, A.A.; et al. Generation, transcriptome profiling, and functional validation of cone-rich human retinal organoids. Proc. Natl. Acad. Sci. USA 2019, 116, 10824–10833. [Google Scholar] [CrossRef] [Green Version]
- Regent, F.; Chen, H.Y.; Kelley, R.A.; Qu, Z.; Swaroop, A.; Li, T. A simple and efficient method for generating human retinal organoids. Mol. Vis. 2020, 26, 97–105. [Google Scholar]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.-H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [Green Version]
- DiStefano, T.; Chen, H.Y.; Panebianco, C.; Kaya, K.D.; Brooks, M.J.; Gieser, L.; Morgan, N.Y.; Pohida, T.; Swaroop, A. Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors. Stem Cell Rep. 2017, 10, 300–313. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Xie, T. Stem Cell Niche: Structure and Function. Annu. Rev. Cell Dev. Biol. 2005, 21, 605–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, S.J.; Spradling, A.C. Stem Cells and Niches: Mechanisms That Promote Stem Cell Maintenance throughout Life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Adamson, P.C.; Widemann, B.C.; Reaman, G.H.; Seibel, N.L.; Murphy, R.F.; Gillespie, A.F.; Balis, F.M. A phase I trial and pharmacokinetic study of 9-cis-retinoic acid (ALRT1057) in pediatric patients with refractory cancer: A joint Pediatric Oncology Branch, National Cancer Institute, and Children’s Cancer Group study. Clin. Cancer Res. 2001, 7, 3034–3039. [Google Scholar] [PubMed]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Hyatt, G.A.; Schmitt, E.A.; Fadool, J.M.; Dowling, J.E. Retinoic acid alters photoreceptor development in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 13298–13303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, M.W.; Williams, R.C.; Turner, J.K.; Creech-Kraft, J.M.; Reh, T.A. Retinoic acid promotes rod photoreceptor differentiation in rat retina in vivo. NeuroReport 1999, 10, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; Wang, W.-L. Retinoic acid signaling in mammalian eye development. Exp. Eye Res. 2009, 89, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Khanna, H.; Akimoto, M.; Siffroi-Fernandez, S.; Friedman, J.S.; Hicks, D.; Swaroop, A. Retinoic Acid Regulates the Expression of Photoreceptor Transcription Factor NRL. J. Biol. Chem. 2006, 281, 27327–27334. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures in Vitro without a Mesenchymal Niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Sasai, Y. Next-Generation Regenerative Medicine: Organogenesis from Stem Cells in 3D Culture. Cell Stem Cell 2013, 12, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Ader, M.; Tanaka, E.M. Modeling human development in 3D culture. Curr. Opin. Cell Biol. 2014, 31, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [PubMed]
- Barishak, Y. Embryology of the Eye and Its Adnexa, 2nd ed.; Karger: New York, NY, USA, 2001. [Google Scholar] [CrossRef]
- Finlay, B.L. The developing and evolving retina: Using time to organize form. Brain Res. 2008, 1192, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Li, H.S.; Tierney, C.; Wen, L.; Wu, J.Y.; Rao, Y. A single morphogenetic field gives rise to two retina primordia under the influence of the prechordal plate. Development 1997, 124, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Mathers, P.H.; Jamrich, M. Regulation of eye formation by the Rx and pax6 homeobox genes. Experientia 2000, 57, 186–194. [Google Scholar] [CrossRef]
- Bailey, T.J. Regulation of Rx Genes in Vertebrate Eye Development: Baylor College of Medicine. Program Dev. Biol. 2005. [Google Scholar]
- Arthur, P.; Kandoi, S.; Sun, L.; Kalvala, A.; Kutlehria, S.; Bhattacharya, S.; Kulkarni, T.; Nimma, R.; Li, Y.; Lamba, D.A.; et al. Biophysical, Molecular and Proteomic Profiling of Human Retinal Organoid-Derived Exosomes. Pharm. Res. 2022, 1–16. [Google Scholar] [CrossRef]
- Welby, E.; Lakowski, J.; Di Foggia, V.; Budinger, D.; Gonzalez-Cordero, A.; Lun, A.T.; Epstein, M.; Patel, A.; Cuevas, E.; Kruczek, K.; et al. Isolation and comparative transcriptome analysis of human fetal and iPSC-derived cone photoreceptor cells. Stem Cell Rep. 2017, 9, 1898–1915. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.J.; Jiang, P.; Howden, S.; Barney, P.; Min, J.; York, N.W.; Chu, L.-F.; Capowski, E.E.; Cash, A.; Jain, S.; et al. A Novel Approach to Single Cell RNA-Sequence Analysis Facilitates In Silico Gene Reporting of Human Pluripotent Stem Cell-Derived Retinal Cell Types. Stem Cells 2017, 36, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Langer, K.B.; Ohlemacher, S.K.; Phillips, M.J.; Fligor, C.M.; Jiang, P.; Gamm, D.M.; Meyer, J.S. Retinal Ganglion Cell Diversity and Subtype Specification from Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Guo, Y.; Zhou, Y.; Mao, S.; Yan, X.; Zeng, Y.; Ding, C.; Chan, H.F.; Tang, S.; Tang, L.; et al. Transcriptomic Analysis of the Developmental Similarities and Differences Between the Native Retina and Retinal Organoids. Investig. Opthalmology Vis. Sci. 2020, 61, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.; Kim, H.J.; Yang, J.; Chae, S.; Lee, W.; Chung, S.; Kim, J.; Choi, H.; Song, H.; Lee, C.K.; et al. Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 2019, 19, e13081. [Google Scholar] [CrossRef] [PubMed]
- Osaki, T.; Uzel, S.G.; Kamm, R.D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 2018, 4, eaat5847. [Google Scholar] [CrossRef] [Green Version]
- Fereshtehnejad, S.-M.; Zeighami, Y.; Dagher, A.; Postuma, R.B. Clinical criteria for subtyping Parkinson’s disease: Biomarkers and longitudinal progression. Brain 2017, 140, 1959–1976. [Google Scholar] [CrossRef] [PubMed]
- Brookmeyer, R.; Abdalla, N.; Kawas, C.H.; Corrada, M.M. Forecasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimer’s Dement. 2018, 14, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, K.; Feneberg, E.; Scaber, J.; Thompson, A.G.; Turner, M.R. Amyotrophic lateral sclerosis: The complex path to precision medicine. J. Neurol. 2018, 265, 2454–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.-Y. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Pander, C.H. Beiträge zur Entwickelungsgeschichte des Hühnchens im Eye; Bayerische Julius-Maximilians-Universität Würzburg: Wurzburg, Germany, 1817. [Google Scholar]
- Huschke, E. Über einige Streitpunkte aus der Anatomie des Auges. Z Ophthalmol. 1835, 4, 273–295. [Google Scholar]
- Adelmann, H.B.; Malpighi, M. The Evolution of Embryology; Cornell UP: Ithaca, NY, USA, 1966. [Google Scholar]
- Gabriel, E.; Albanna, W.; Pasquini, G.; Ramani, A.; Josipovic, N.; Mariappan, A.; Schinzel, F.; Karch, C.M.; Bao, G.; Gottardo, M.; et al. Human brain organoids assemble functionally integrated bilateral optic vesicles. Cell Stem Cell 2021, 28, 1740–1757.e8. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 2020, 183, 1913–1929.e26. [Google Scholar] [CrossRef]
- Archibald, N.K.; Clarke, M.P.; Mosimann, U.P.; Burn, D.J. The retina in Parkinson’s disease. Brain 2009, 132, 1128–1145. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Singh, N.; Chaudhary, S.; Bellamkonda, V.; Kritikos, A.E.; Wise, A.S.; Rana, N.; McDonald, D.; Ayyagari, R. Retinal Degeneration and Alzheimer’s Disease: An Evolving Link. Int. J. Mol. Sci. 2020, 21, 7290. [Google Scholar] [CrossRef] [PubMed]
- Kruczek, K.; Swaroop, A. Pluripotent stem cell-derived retinal organoids for disease modeling and development of therapies. Stem Cells 2020, 38, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A. Open-angle glaucoma. N. Engl. J. Med. 1993, 328, 1097–1106. [Google Scholar] [CrossRef]
- Quigley, H.A.; Nickells, R.W.; Kerrigan, L.A.; Pease, M.E.; Thibault, D.J.; Zack, D.J. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Investig. Ophthalmol. Vis. Sci. 1995, 36, 774–786. [Google Scholar]
- Boland, M.V.; Quigley, H.A. Risk factors and open-angle glaucoma: Classification and application. J. Glaucoma 2007, 16, 408–418. [Google Scholar] [CrossRef]
- VanderWall, K.B.; Huang, K.-C.; Pan, Y.; Lavekar, S.S.; Fligor, C.M.; Allsop, A.R.; Lentsch, K.A.; Dang, P.; Zhang, C.; Tseng, H.C.; et al. Retinal Ganglion Cells with a Glaucoma OPTN(E50K) Mutation Exhibit Neurodegenerative Phenotypes when Derived from Three-Dimensional Retinal Organoids. Stem Cell Rep. 2020, 15, 52–66. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Veleri, S.; Lazar, C.H.; Chang, B.; Sieving, P.A.; Banin, E.; Swaroop, A. Biology and therapy of inherited retinal degenerative disease: Insights from mouse models. Dis. Model. Mech. 2015, 8, 109–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukovic, D.; Castro, A.A.; Kaya, K.D.; Munezero, D.; Gieser, L.; Davó-Martínez, C.; Corton, M.; Cuenca, N.; Swaroop, A.; Ramamurthy, V.; et al. Retinal Organoids derived from hiPSCs of an AIPL1-LCA Patient Maintain Cytoarchitecture despite Reduced levels of Mutant AIPL1. Sci. Rep. 2020, 10, 5426. [Google Scholar] [CrossRef] [Green Version]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Starqardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef]
- Anderson, K.L.; Baird, L.; Lewis, R.A.; Chinault, A.C.; Otterud, B.; Leppert, M.; Lupski, J.R. A YAC contig encompassing the recessive Stargardt disease gene (STGD) on chromosome 1p. Am. J. Hum. Genet. 1995, 57, 1351–1363. [Google Scholar] [PubMed]
- Westeneng-van Haaften, S.C.; Boon, C.J.; Cremers, F.P.; Hoefsloot, L.H.; den Hollander, A.I.; Hoyng, C.B. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 2012, 119, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Su, P.Y.; Chuang, J.Z.; Lee, W.; Zernant, J.; Tsang, S.H.; Nagasaki, T.; Sung, C.H.; Allikmets, R. Characterization of Stargardt disease patient-derived human retinal organoids harboring the p. Gly1961Glu mutation in the ABCA4 gene. Investig. Ophthalmol. Vis. Sci. 2021, 62, 2595. [Google Scholar]
- Pagon, R.A. Retinitis pigmentosa. Surv. Ophthalmol. 1988, 33, 137–177. [Google Scholar] [CrossRef]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.-L.; Lei, X.-L.; Han, F.; He, K.-W.; Jin, S.-Q.; Zhang, Y.-Y.; Jin, Z.-B. Patient-Specific Retinal Organoids Recapitulate Disease Features of Late-Onset Retinitis Pigmentosa. Front. Cell Dev. Biol. 2020, 8, 128. [Google Scholar] [CrossRef]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun. 2018, 9, 4234. [Google Scholar] [CrossRef] [Green Version]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and Rescue of RP2 Retinitis Pigmentosa Using iPSC-Derived Retinal Organoids. Stem Cell Rep. 2020, 15, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez De La Camara, C.; Nanda, A.; Salvetti, A.P.; Fischer, M.D.; MacLaren, R.E. Gene therapy for the treatment of X-linked retinitis pigmentosa. Expert Opin Orphan Drugs 2018, 6, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neidhardt, J.; Glaus, E.; Lorenz, B.; Netzer, C.; Li, Y.; Schambeck, M.; Wittmer, M.; Feil, S.; Kirschner-Schwabe, R.; Rosenberg, T.; et al. Identification of novel mutations in X-linked retinitis pigmentosa families and implications for diagnostic testing. Mol. Vis. 2008, 14, 1081–1093. [Google Scholar]
- Sauer, C.G.; Gehrig, A.; Warneke-Wittstock, R.; Marquardt, A.; Ewing, C.C.; Gibson, A.; Lorenz, B.; Jurklies, B.; Weber, B.H. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat. Genet. 1997, 17, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-C.; Wang, M.-L.; Chen, S.-J.; Kuo, J.-C.; Wang, W.-J.; Nguyen, P.N.N.; Wahlin, K.J.; Lu, J.-F.; Tran, A.A.; Shi, M.; et al. Morphological and Molecular Defects in Human Three-Dimensional Retinal Organoid Model of X-Linked Juvenile Retinoschisis. Stem Cell Rep. 2019, 13, 906–923. [Google Scholar] [CrossRef] [PubMed]
- Saengwimol, D.; Rojanaporn, D.; Chaitankar, V.; Chittavanich, P.; Aroonroch, R.; Boontawon, T.; Thammachote, W.; Jinawath, N.; Hongeng, S.; Kaewkhaw, R. A three-dimensional organoid model recapitulates tumorigenic aspects and drug responses of advanced human retinoblastoma. Sci. Rep. 2018, 8, 15664. [Google Scholar] [CrossRef]
- Tantri, A.; Vrabec, T.R.; Cu-Unjieng, A.; Frost, A.; Annesley, W.H., Jr.; Donoso, L.A. X-linked retinoschisis: A clinical and molecular genetic review. Surv. Ophthalmol. 2004, 49, 214–230. [Google Scholar] [CrossRef]
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A.; Zhao, J.; Munier, F.L.; Abramson, D.H.; Shields, C.L.; Chantada, G.L.; Njuguna, F.; et al. Retinoblastoma. Nat. Rev. Dis. Primers 2015, 1, 15021. [Google Scholar] [CrossRef] [Green Version]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quigley, H.A.; Dunkelberger, G.R.; Green, W.R. Retinal Ganglion Cell Atrophy Correlated with Automated Perimetry in Human Eyes with Glaucoma. Am. J. Ophthalmol. 1989, 107, 453–464. [Google Scholar] [CrossRef]
- Greenfield, D.S.; Weinreb, R.N. Role of Optic Nerve Imaging in Glaucoma Clinical Practice and Clinical Trials. Am. J. Ophthalmol. 2008, 145, 598–603.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Z.M.; Wollstein, G.; Schuman, J.S. Clinical Utility of Optical Coherence Tomography in Glaucoma. Investig. Opthalmol. Vis. Sci. 2016, 57, OCT556–OCT567. [Google Scholar] [CrossRef] [PubMed]
- Norman, R.E.; Flanagan, J.G.; Sigal, I.A.; Rausch, S.M.; Tertinegg, I.; Ethier, C.R. Finite element modeling of the human sclera: Influence on optic nerve head biomechanics and connections with glaucoma. Exp. Eye Res. 2011, 93, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Sigal, I.A.; Yang, H.; Roberts, M.D.; Burgoyne, C.; Downs, J.C. IOP-Induced Lamina Cribrosa Displacement and Scleral Canal Expansion: An Analysis of Factor Interactions Using Parameterized Eye-Specific Models. Investig. Opthalmol. Vis. Sci. 2011, 52, 1896–1907. [Google Scholar] [CrossRef] [PubMed]
- Grytz, R.; Fazio, M.A.; Girard, M.J.; Libertiaux, V.; Bruno, L.; Gardiner, S.; Girkin, A.C.; Downs, J.C. Material properties of the posterior human sclera. J. Mech. Behav. Biomed. Mater. 2014, 29, 602–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, C.F.; Downs, J.C.; Bellezza, A.J.; Suh, J.-K.F.; Hart, R.T. The optic nerve head as a biomechanical structure: A new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog. Retin. Eye Res. 2005, 24, 39–73. [Google Scholar] [CrossRef] [PubMed]
- Sigal, I.A.; Flanagan, J.G.; Ethier, C.R. Factors influencing optic nerve head biomechanics. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4189–4199. [Google Scholar] [CrossRef]
- Quigley, H.A.; Addicks, E.M.; Green, W.R.; Maumenee, A.E. Optic nerve damage in human glaucoma: II. The site of injury and susceptibility to damage. Arch. Ophthalmol. 1981, 99, 635–649. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef]
- Apte, R.S. Gene Therapy for Retinal Degeneration. Cell 2018, 173, 5. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Morrow, E.; Li, T.; Davis, F.C.; Cepko, C.L. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat. Genet. 1999, 23, 466–470. [Google Scholar] [CrossRef] [PubMed]
- De Roach, J.N.; McLaren, T.L.; Paterson, R.L.; O’Brien, E.C.; Hoffmann, L.; Mackey, D.A.; Hewitt, A.W.; Lamey, T.M. Establishment and evolution of the A ustralian I nherited R etinal D isease R egister and DNA B ank. Clin. Exp. Ophthalmol. 2013, 41, 476–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, A.E.; Bernstein, P.S.; Cideciyan, A.V.; Hoyng, C.; Issa, P.C.; Palczewski, K.; Rosenfeld, P.J.; Sadda, S.; Schraermeyer, U.; Sparrow, J.R.; et al. Towards Treatment of Stargardt Disease: Workshop Organized and Sponsored by the Foundation Fighting Blindness. Transl. Vis. Sci. Technol. 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Bujakowska, K.; Audo, I.; Mohand-Saïd, S.; Lancelot, M.E.; Antonio, A.; Germain, A.; Léveillard, T.; Letexier, M.; Saraiva, J.-P.; Lonjou, C.; et al. CRB1 mutations in inherited retinal dystrophies. Hum. Mutat. 2012, 33, 306–315. [Google Scholar] [CrossRef]
- Graziotto, J.J.; Farkas, M.H.; Bujakowska, K.; Deramaudt, B.M.; Zhang, Q.; Nandrot, E.; Inglehearn, C.F.; Bhattacharya, S.S.; Pierce, E.A. Three Gene-Targeted Mouse Models of RNA Splicing Factor RP Show Late-Onset RPE and Retinal Degeneration. Investig. Opthalmology Vis. Sci. 2011, 52, 190–198. [Google Scholar] [CrossRef] [Green Version]
- El-Amraoui, A.; Petit, C. The retinal phenotype of Usher syndrome: Pathophysiological insights from animal models. Comptes Rendus. Biol. 2014, 337, 167–177. [Google Scholar] [CrossRef]
- Kimberling, W.J.; Hildebrand, M.S.; Shearer, A.E.; Jensen, M.L.; Halder, J.A.; Trzupek, K.; Cohn, E.S.; Weleber, R.G.; Stone, E.M.; Smith, R.J. Frequency of Usher syndrome in two pediatric populations: Implications for genetic screening of deaf and hard of hearing children. Genet. Med. 2010, 12, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Reiners, J.; Nagel-Wolfrum, K.; Jürgens, K.; Märker, T.; Wolfrum, U. Molecular basis of human Usher syndrome: Deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp. Eye Res. 2006, 83, 97–119. [Google Scholar] [CrossRef]
- Sun, T.; Xu, K.; Ren, Y.; Xie, Y.; Zhang, X.; Tian, L.; Li, Y. Comprehensive Molecular Screening in Chinese Usher Syndrome Patients. Investig. Opthalmology Vis. Sci. 2018, 59, 1229–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Mao, Y.; Yang, J.; Li, Y.; Li, Y.; Yang, Z. Mutation screening of the USH2A gene in retinitis pigmentosa and USHER patients in a Han Chinese population. Eye 2018, 32, 1608–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Wang, P.; Ma, J.H.; Cui, Z.; Yu, Q.; Liu, S.; Xue, Y.; Zhu, D.; Cao, J.; Li, Z.; et al. Modeling Retinitis Pigmentosa: Retinal Organoids Generated from the iPSCs of a Patient with the USH2A Mutation Show Early Developmental Abnormalities. Front. Cell. Neurosci. 2019, 13, 361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutman, A.F. The Hereditary Dystrophies of the Posterior Pole of the Eye; Van Gorcum & Comp., B.V.: Assen, The Netherlands, 1971. [Google Scholar]
- Molday, L.L.; Hicks, D.; Sauer, C.G.; Weber, B.H.; Molday, R.S. Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Investig. Ophthalmol. Vis. Sci. 2001, 42, 816–825. [Google Scholar]
- Kawano, K.; Tanaka, K.; Murakami, F.; Ohba, N. Congenital hereditary retinoschisis: Evolution at the initial stage. Albrecht Graefes Arch. Klin. Exp. Ophthalmol. 1981, 217, 315–323. [Google Scholar] [CrossRef] [PubMed]
- George, N.D.L.; Yates, J.R.W.; Moore, A.T. Clinical Features in Affected Males with X-Linked Retinoschisis. Arch. Ophthalmol. 1996, 114, 274–280. [Google Scholar] [CrossRef]
- Kellner, U.; Brümmer, S.; Foerster, M.H.; Wessing, A. X-linked congenital retinoschisis. Graefe’s Arch. Clin. Exp. Ophthalmol. 1990, 228, 432–437. [Google Scholar] [CrossRef]
- Forsius, H.; Krause, U.; Helve, J.; Vuopala, V.; Mustonen, E.; Vainio-Mattila, B.; Fellman, J.; Eriksson, A.W. Visual acuity in 183 cases of X-chromosomal retinoschisis. Can. J. Ophthalmol. 1973, 8, 385–393. [Google Scholar]
- Dimaras, H.; Kimani, K.; Dimba, E.A.; Gronsdahl, P.; White, A.; Chan, H.S.; Gallie, B.L. Retinoblastoma. Nat. Rev. Dis. Primers 2015, 1, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Aerts, I.; Rouic, L.L.; Gauthier-Villars, M.; Brisse, H.; Doz, F.; Desjardins, L. Retinoblastoma. Orphanet J. Rare Dis. 2006, 1, 1–11. [Google Scholar] [CrossRef]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Sexton, C.R.; Mayhew, C.N. Role of the retinoblastoma tumor suppressor in the maintenance of genome integrity. Curr. Mol. Med. 2006, 6, 749–757. [Google Scholar]
- Indovina, P.; Pentimalli, F.; Casini, N.; Vocca, I.; Giordano, A. RB1 dual role in proliferation and apoptosis: Cell fate control and implications for cancer therapy. Oncotarget 2015, 6, 17873–17890. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Fazeli, A.; Schmitt, E.M.; Bronson, R.T.; Goodell, M.; Weinberg, R.A. Effects of an Rb mutation in the mouse. Nature 1992, 359, 295–300. [Google Scholar] [CrossRef]
- Clarke, A.R.; Maandag, E.R.; van Roon, M.; van der Lugt, N.M.; van der Valk, M.; Hooper, M.L.; Berns, A.; Te Rielef, H. Requirement for a functional Rb-1 gene in murine development. Nature 1992, 359, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.-H.P.; Chang, C.-Y.; Hu, N.; Wang, Y.-C.J.; Lai, C.-C.; Herrup, K.; Lee, W.-H.; Bradley, A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 1992, 359, 288–294. [Google Scholar] [CrossRef]
- Zhang, J.; Gray, J.; Wu, L.; Leone, G.; Rowan, S.; Cepko, C.L.; Zhu, X.; Craft, C.M.; Dyer, M.A. Rb regulates proliferation and rod photoreceptor development in the mouse retina. Nat. Genet. 2004, 36, 351–360. [Google Scholar] [CrossRef]
- Zhang, J.; Schweers, B.; Dyer, M.A. The First Knockout Mouse Model of Retinoblastoma. Cell Cycle 2004, 3, 950–957. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Livne-Bar, I.; Vanderluit, J.L.; Slack, R.; Agochiya, M.; Bremner, R. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell 2004, 5, 539–551. [Google Scholar] [CrossRef]
- Norrie, J.L.; Nityanandam, A.; Lai, K.; Chen, X.; Wilson, M.; Stewart, E.; Griffiths, L.; Jin, H.; Wu, G.; Orr, B.; et al. Retinoblastoma from human stem cell-derived retinal organoids. Nat. Commun. 2021, 12, 4535. [Google Scholar] [CrossRef]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.S.; FitzPatrick, D.R. Anophthalmia and microphthalmia. Orphanet J. Rare Dis. 2007, 2, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, R.; Sowden, J.; Gerth-Kahlert, C.; Moore, A.T.; Moosajee, M. Clinical utility gene card for: Non-Syndromic Microphthalmia Including Next-Generation Sequencing-Based Approaches. Eur. J. Hum. Genet. 2017, 25, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragge, N.K.; Subak-Sharpe, I.D.; Collin, J.R.O. A practical guide to the management of anophthalmia and microphthalmia. Eye 2007, 21, 1290–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Taylor, A.E.; Sowden, J.; Ragge, N.K.; Russell-Eggitt, I.; Rahi, J.; Gilbert, C.E. Anophthalmos, Microphthalmos, and Typical Coloboma in the United Kingdom: A Prospective Study of Incidence and Risk. Investig. Opthalmol. Vis. Sci. 2011, 52, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Horsford, D.J.; Nguyen, M.-T.T.; Sellar, G.C.; Kothary, R.; Arnheiter, H.; McInnes, R.R. Chx10 repression of Mitf is required for the maintenance of mammalian neuroretinal identity. Development 2005, 132, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Green, E.S.; Stubbs, J.L.; Levine, E. Genetic rescue of cell number in a mouse model of microphthalmia:interactions between Chx10 and G1-phase cell cycle regulators. Development 2003, 130, 539–552. [Google Scholar] [CrossRef] [Green Version]
- Rowan, S.; Chen, C.-M.A.; Young, T.L.; Fisher, D.E.; Cepko, C.L. Transdifferentiation of the retina into pigmented cells in ocular retardation mice defines a new function of the homeodomain gene Chx10. Development 2004, 131, 5139–5152. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.J.; Perez, E.T.; Martin, J.M.; Reshel, S.T.; Wallace, K.A.; Capowski, E.E.; Singh, R.; Wright, L.S.; Clark, E.M.; Barney, P.M.; et al. Modeling Human Retinal Development with Patient-Specific Induced Pluripotent Stem Cells Reveals Multiple Roles for Visual System Homeobox 2. Stem Cells 2014, 32, 1480–1492. [Google Scholar] [CrossRef]
- Gonzalez-Cordero, A.; Kruczek, K.; Naeem, A.; Fernando, M.; Kloc, M.; Ribeiro, J.; Goh, D.; Duran, Y.; Blackford, S.J.; Abelleira-Hervas, L.; et al. Recapitulation of Human Retinal Development from Human Pluripotent Stem Cells Generates Transplantable Populations of Cone Photoreceptors. Stem Cell Rep. 2017, 9, 820–837. [Google Scholar] [CrossRef] [Green Version]
- Lamba, D.A.; Gust, J.; Reh, T.A. Transplantation of Human Embryonic Stem Cell-Derived Photoreceptors Restores Some Visual Function in Crx-Deficient Mice. Cell Stem Cell 2009, 4, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of Human Induced Pluripotent Stem Cell-Derived Retinal Pigment Epithelium Cell Sheets Aiming for Clinical Application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, H.; Mandai, M.; Matsushita, K.; Kuwahara, A.; Yonemura, S.; Nakano, T.; Assawachananont, J.; Kimura, T.; Saito, K.; Terasaki, H.; et al. Transplantation of human embryonic stem cell-derived retinal tissue in two primate models of retinal degeneration. Proc. Natl. Acad. Sci. USA 2015, 113, E81–E90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assawachananont, J.; Mandai, M.; Okamoto, S.; Yamada, C.; Eiraku, M.; Yonemura, S.; Sasai, Y.; Takahashi, M. Transplantation of Embryonic and Induced Pluripotent Stem Cell-Derived 3D Retinal Sheets into Retinal Degenerative Mice. Stem Cell Rep. 2014, 2, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, U.; Oriyakhel, W.; Kenna, P.F.; Linke, S.; Richard, G.; Petrowitz, B.; Humphries, P.; Farrar, G.J.; Ader, M. Retinal cells integrate into the outer nuclear layer and differentiate into mature photoreceptors after subretinal transplantation into adult mice. Exp. Eye Res. 2008, 86, 691–700. [Google Scholar] [CrossRef]
- MacLaren, R.; Pearson, R.; MacNeil, A.; Douglas, R.H.; Salt, T.E.; Akimoto, M.; Swaroop, A.; Sowden, J.; Ali, R. Retinal repair by transplantation of photoreceptor precursors. Nature 2006, 444, 203–207. [Google Scholar] [CrossRef]
- Eastlake, K.; Wang, W.; Jayaram, H.; Murray-Dunning, C.; Carr, A.J.F.; Ramsden, C.M.; Vugler, A.; Gore, K.; Clemo, N.; Stewart, M.; et al. Phenotypic and Functional Characterization of Müller Glia Isolated from Induced Pluripotent Stem Cell-Derived Retinal Organoids: Improvement of Retinal Ganglion Cell Function upon Transplantation. STEM CELLS Transl. Med. 2019, 8, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Miltner, A.M.; La Torre, A. Retinal Ganglion Cell Replacement: Current Status and Challenges Ahead. Dev. Dyn. 2018, 248, 118–128. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, G.; M’Barek, K.B.; Chaffiol, A.; Slembrouck-Brec, A.; Conart, J.B.; Nanteau, C.; Rabesandratana, O.; Sahel, J.-A.; Duebel, A.; Orieux, G.; et al. Characterization and Transplantation of CD73-Positive Photoreceptors Isolated from Human iPSC-Derived Retinal Organoids. Stem Cell Rep. 2018, 11, 665–680. [Google Scholar] [CrossRef]
- McLelland, B.T.; Lin, B.; Mathur, A.; Aramant, R.B.; Thomas, B.B.; Nistor, G.; Keirstead, H.S.; Seiler, M.J. Transplanted hESC-derived retina organoid sheets differentiate, integrate, and improve visual function in retinal degenerate rats. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2586–2603. [Google Scholar] [CrossRef] [Green Version]
- Hallam, D.; Hilgen, G.; Dorgau, B.; Zhu, L.; Yu, M.; Bojic, S.; Hewitt, P.; Schmitt, M.; Uteng, M.; Kustermann, S.; et al. Human-Induced Pluripotent Stem Cells Generate Light Responsive Retinal Organoids with Variable and Nutrient-Dependent Efficiency. Stem Cells 2018, 36, 1535–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, K.C.; Aotaki-Keen, A.E.; Putkey, F.R.; Hjelmeland, L.M. ARPE-19, A Human Retinal Pigment Epithelial Cell Line with Differentiated Properties. Exp. Eye Res. 1996, 62, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Seigel, G.M. The golden age of retinal cell culture. Mol. Vis. 1999, 5, 4. [Google Scholar]
- Limb, G.A.; Salt, T.E.; Munro, P.M.G.; Moss, S.E.; Khaw, P.T. In vitro characterization of a spontaneously immortalized human Müller cell line (MIO-M1). Investig. Ophthalmol. Vis. Sci. 2002, 43, 864–869. [Google Scholar]
- Eade, K.; Giles, S.; Harkins-Perry, S.; Friedlander, M. Toxicity Screens in Human Retinal Organoids for Pharmaceutical Discovery. J. Vis. Exp. 2021, 169, e62269. [Google Scholar] [CrossRef]
- Klingeborn, M.; Dismuke, W.M.; Skiba, N.P.; Kelly, U.; Stamer, W.D.; Rickman, C.B. Directional Exosome Proteomes Reflect Polarity-Specific Functions in Retinal Pigmented Epithelium Monolayers. Sci. Rep. 2017, 7, 4901. [Google Scholar] [CrossRef] [Green Version]
- Lakkaraju, A.; Umapathy, A.; Tan, L.X.; Daniele, L.; Philp, N.J.; Boesze-Battaglia, K.; Williams, D.S. The cell biology of the retinal pigment epithelium. Prog. Retin. Eye Res. 2020, 78, 100846. [Google Scholar] [CrossRef]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and Autophagy: Coordinated Mechanisms for the Maintenance of Cellular Fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Camfield, R.; Gorski, S.M. The interplay between exosomes and autophagy—Partners in crime. J. Cell Sci. 2018, 131, jcs215210. [Google Scholar] [CrossRef] [Green Version]
- Schiffelers, R.; Kooijmans, S.; Vader, P.; Dommelen, V.; Van Solinge, W. Exosome mimetics: A novel class of drug delivery systems. Int. J. Nanomed. 2012, 7, 1525–1541. [Google Scholar] [CrossRef] [Green Version]
- Schiller, M.; Bekeredjian-Ding, I.; Heyder, P.; Blank, N.; Ho, A.D.; Lorenz, H.-M. Autoantigens are translocated into small apoptotic bodies during early stages of apoptosis. Cell Death Differ. 2007, 15, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Gregory, C.D.; Pound, J.D. Microenvironmental influences of apoptosis in vivo and in vitro. Apoptosis 2010, 15, 1029–1049. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, D.; Ho, E.A. Challenges in the development and establishment of exosome-based drug delivery systems. J. Control. Release 2020, 329, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, Y.; Zhao, B.; Niu, X.; Hu, B.; Li, Q.; Zhang, J.; Ding, J.; Chen, Y.; Wang, Y. Comparison of exosomes secreted by induced pluripotent stem cell-derived mesenchymal stem cells and synovial membrane-derived mesenchymal stem cells for the treatment of osteoarthritis. Stem Cell Res. Ther. 2017, 8, 64. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Sun, X.; Liu, L.; Jiang, H.; Shen, Y.; Xu, X.; Li, J.; Zhang, G.; Huang, J.; Lin, Z.; et al. Exosomes and Their Therapeutic Potentials of Stem Cells. Stem Cells Int. 2016, 2016, 7653489. [Google Scholar] [CrossRef] [Green Version]
- Fuster-Matanzo, A.; Gessler, F.; Leonardi, T.; Iraci, N.; Pluchino, S. Acellular approaches for regenerative medicine: On the verge of clinical trials with extracellular membrane vesicles? Stem Cell Res. Ther. 2015, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Jiang, F.; Jiang, Y.; Wang, Y.; Li, Z.; Shi, X.; Zhu, Y.; Wang, H.; Zhang, Z. Roles of exosomes in ocular diseases. Int. J. Nanomed. 2020, 15, 10519. [Google Scholar] [CrossRef]
- Li, S.-F.; Han, Y.; Wang, F.; Su, Y. Progress in exosomes and their potential use in ocular diseases. Int. J. Ophthalmol. 2020, 13, 1493–1498. [Google Scholar] [CrossRef]
- Rajool Dezfuly, A.; Safaee, A.; Salehi, H. Therapeutic effects of mesenchymal stem cells-derived extracellular vesicles’ miRNAs on retinal regeneration: A review. Stem Cell Res. Ther. 2021, 12, 530. [Google Scholar] [CrossRef]
- Elliott, R.; He, M. Unlocking the Power of Exosomes for Crossing Biological Barriers in Drug Delivery. Pharmaceutics 2021, 13, 122. [Google Scholar] [CrossRef]
- Yu, Y.; Li, L.; Lin, S.; Hu, J. Update of application of olfactory ensheathing cells and stem cells/exosomes in the treatment of retinal disorders. Stem Cell Res. Ther. 2022, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Flores-Bellver, M.; Pan, J.; Benito-Martin, A.; Shi, C.; Onwumere, O.; Mighty, J.; Qian, J.; Zhong, X.; Hogue, T.; et al. Human retinal organoids release extracellular vesicles that regulate gene expression in target human retinal progenitor cells. Sci. Rep. 2021, 11, 21128. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Kommineni, N.; Surapaneni, S.K.; Kalvala, A.; Yaun, X.; Gebeyehu, A.; Arthur, P.; Duke, L.C.; York, S.B.; Bagde, A.; et al. Cannabidiol loaded extracellular vesicles sensitize triple-negative breast cancer to doxorubicin in both in-vitro and in vivo models. Int. J. Pharm. 2021, 607, 120943. [Google Scholar] [CrossRef]
- Arthur, P.; Patel, N.; Surapaneni, S.K.; Mondal, A.; Gebeyehu, A.; Bagde, A.; Kutlehria, S.; Nottingham, E.; Singh, M. Targeting lung cancer stem cells using combination of Tel and Docetaxel liposomes in 3D cultures and tumor xenografts. Toxicol. Appl. Pharmacol. 2020, 401, 115112. [Google Scholar] [CrossRef]
- Milman, N.; Ginini, L.; Gil, Z. Exosomes and their role in tumorigenesis and anticancer drug resistance. Drug Resist. Updat. 2019, 45, 1–12. [Google Scholar] [CrossRef]
- Tan, A.; Rajadas, J.; Seifalian, A. Exosomes as nano-theranostic delivery platforms for gene therapy. Adv. Drug Deliv. Rev. 2013, 65, 357–367. [Google Scholar] [CrossRef]
- Jin, N.; Sha, W.; Gao, L. Shaping the Microglia in Retinal Degenerative Diseases Using Stem Cell Therapy: Practice and Prospects. Front. Cell Dev. Biol. 2021, 9, 3587. [Google Scholar] [CrossRef]
- Flores-Bellver, M.; Mighty, J.; Aparicio-Domingo, S.; Li, K.V.; Shi, C.; Zhou, J.; Cobb, H.; McGrath, P.; Michelis, G.; Lenhart, P.; et al. Extracellular vesicles released by human retinal pigment epithelium mediate increased polarised secretion of drusen proteins in response to AMD stressors. J. Extracell. Vesicles 2021, 10, e12165. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, Z.; Mugisha, A.; Fransisca, S.; Liu, Q.; Xie, P. Emerging role of exosomes in retinal diseases. Front. Cell Dev. Biol. 2021, 9, 679. [Google Scholar]
- Marzano, M.; Bejoy, J.; Cheerathodi, M.R.; Sun, L.; York, S.B.; Zhao, J.; Kanekiyo, T.; Bu, G.; Meckes, D.; Li, Y. Differential effects of extracellular vesicles of lineage-specific human pluripotent stem cells on cellular behaviours of isogenic cortical spheroids. Cells 2019, 8, 993. [Google Scholar] [CrossRef] [PubMed]
- Jeske, R.; Bejoy, J.; Marzano, M.; Li, Y. Human induced pluripotent stem cell-derived extracellular vesicles: Characteristics and applications. Tissue Eng. Part B Rev. 2020, 26, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Marzano, M.; Bou-Dargham, M.J.; Cone, A.S.; York, S.; Helsper, S.; Grant, S.C.; Meckes, D.G., Jr.; Sang, Q.-X.A.; Li, Y. Biogenesis of Extracellular Vesicles Produced from Human-Stem-Cell-Derived Cortical Spheroids Exposed to Iron Oxides. ACS Biomater. Sci. Eng. 2021, 7, 1111–1122. [Google Scholar] [CrossRef]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.-J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.-S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.-È.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef]
- Li, Z.; Guo, X.; Guan, J. An oxygen release system to augment cardiac progenitor cell survival and differentiation under hypoxic condition. Biomaterials 2012, 33, 5914–5923. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia Regulate the Number of Neural Precursor Cells in the Developing Cerebral Cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, M.; Lancaster, M.A.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; Knoblich, J.A. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Hölscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.W.H.; Koizumi, K.S.S.; Moorhouse, A.; Yoshimura, A.W.I.Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef] [Green Version]
- Fagerlund, I.; Dougalis, A.; Shakirzyanova, A.; Gómez-Budia, M.; Pelkonen, A.; Konttinen, H.; Ohtonen, S.; Fazaludeen, M.F.; Koskuvi, M.; Kuusisto, J.; et al. Microglia-like Cells Promote Neuronal Functions in Cerebral Organoids. Cells 2021, 11, 124. [Google Scholar] [CrossRef]
- German, O.L.; Buzzi, E.; Rotstein, N.P.; Rodríguez-Boulan, E.; Politi, L.E. Retinal pigment epithelial cells promote spatial reorganization and differentiation of retina photoreceptors. J. Neurosci. Res. 2008, 86, 3503–3514. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, T.; Xie, H.; Khan, M.I.; Zhao, H.; Bao, J.; Zhang, M.; Xue, T. Accelerated photoreceptor differentiation of hiPSC-derived retinal organoids by contact co-culture with retinal pigment epithelium. Stem Cell Res. 2019, 39, 101491. [Google Scholar] [CrossRef]
- Wahlin, K.J.; Maruotti, J.A.; Sripathi, S.R.; Ball, J.; Angueyra, J.M.; Kim, C.; Grebe, R.; Li, W.; Jones, B.W.; Zack, D.J. Photoreceptor outer segment-like structures in long-term 3D retinas from human pluripotent stem cells. Sci. Rep. 2017, 7, 766. [Google Scholar] [CrossRef] [Green Version]
- Eldred, K.C.; Hadyniak, S.E.; Hussey, K.A.; Brenerman, B.; Zhang, P.W.; Chamling, X.; Sluch, V.M.; Welsbie, D.S.; Hattar, S.; Taylor, J.; et al. Thyroid hormone signaling specifies cone subtypes in human retinal organoids. Science 2018, 362, eaau6348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crair, M.C.; Mason, C.A. Reconnecting Eye to Brain. J. Neurosci. 2016, 36, 10707–10722. [Google Scholar] [CrossRef] [PubMed]
- Fligor, C.M.; Lavekar, S.S.; Harkin, J.; Shields, P.K.; VanderWall, K.B.; Huang, K.-C.; Gomes, C.; Meyer, J.S. Extension of retinofugal projections in an assembled model of human pluripotent stem cell-derived organoids. Stem Cell Rep. 2021, 16, 2228–2241. [Google Scholar] [CrossRef] [PubMed]
- Berber, P.; Milenkovic, A.; Michaelis, L.; Weber, B.H.F. Retinal organoid differentiation methods determine organoid cellular composition. J. Transl. Genet. Genom. 2021, 5, 292–303. [Google Scholar] [CrossRef]
- Hiler, D.; Chen, X.; Hazen, J.; Kupriyanov, S.; Carroll, P.A.; Qu, C.; Xu, B.; Johnson, D.; Griffiths, L.; Frase, S.; et al. Quantification of Retinogenesis in 3D Cultures Reveals Epigenetic Memory and Higher Efficiency in iPSCs Derived from Rod Photoreceptors. Cell Stem Cell 2015, 17, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Mellough, C.B.; Collin, J.; Queen, R.; Hilgen, G.; Dorgau, B.; Zerti, D.; Felemban, M.; White, K.; Sernagor, E.; Lako, M. Systematic Comparison of Retinal Organoid Differentiation from Human Pluripotent Stem Cells Reveals Stage Specific, Cell Line, and Methodological Differences. STEM CELLS Transl. Med. 2019, 8, 694–706. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arthur, P.; Muok, L.; Nathani, A.; Zeng, E.Z.; Sun, L.; Li, Y.; Singh, M. Bioengineering Human Pluripotent Stem Cell-Derived Retinal Organoids and Optic Vesicle-Containing Brain Organoids for Ocular Diseases. Cells 2022, 11, 3429. https://doi.org/10.3390/cells11213429
Arthur P, Muok L, Nathani A, Zeng EZ, Sun L, Li Y, Singh M. Bioengineering Human Pluripotent Stem Cell-Derived Retinal Organoids and Optic Vesicle-Containing Brain Organoids for Ocular Diseases. Cells. 2022; 11(21):3429. https://doi.org/10.3390/cells11213429
Chicago/Turabian StyleArthur, Peggy, Laureana Muok, Aakash Nathani, Eric Z. Zeng, Li Sun, Yan Li, and Mandip Singh. 2022. "Bioengineering Human Pluripotent Stem Cell-Derived Retinal Organoids and Optic Vesicle-Containing Brain Organoids for Ocular Diseases" Cells 11, no. 21: 3429. https://doi.org/10.3390/cells11213429