
Autologous iPSC-Derived Human Neuromuscular Junction to Model the Pathophysiology of Hereditary Spastic Paraplegia
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedures
2.1. Study Design and Ethics Statement
2.2. Generation of Integration-Free HSP hiPSCs
2.3. hiPSC Culture
2.4. Skeletal Muscle Differentiation of hiPSCs
2.5. Lower MN Differentiation of hiPSCs
2.6. NMJ Formation in Microfluidic Devices
2.7. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.8. Immunofluorescence (IF)
2.9. Western Blot (WB)
2.10. Statistical Analysis
3. Results
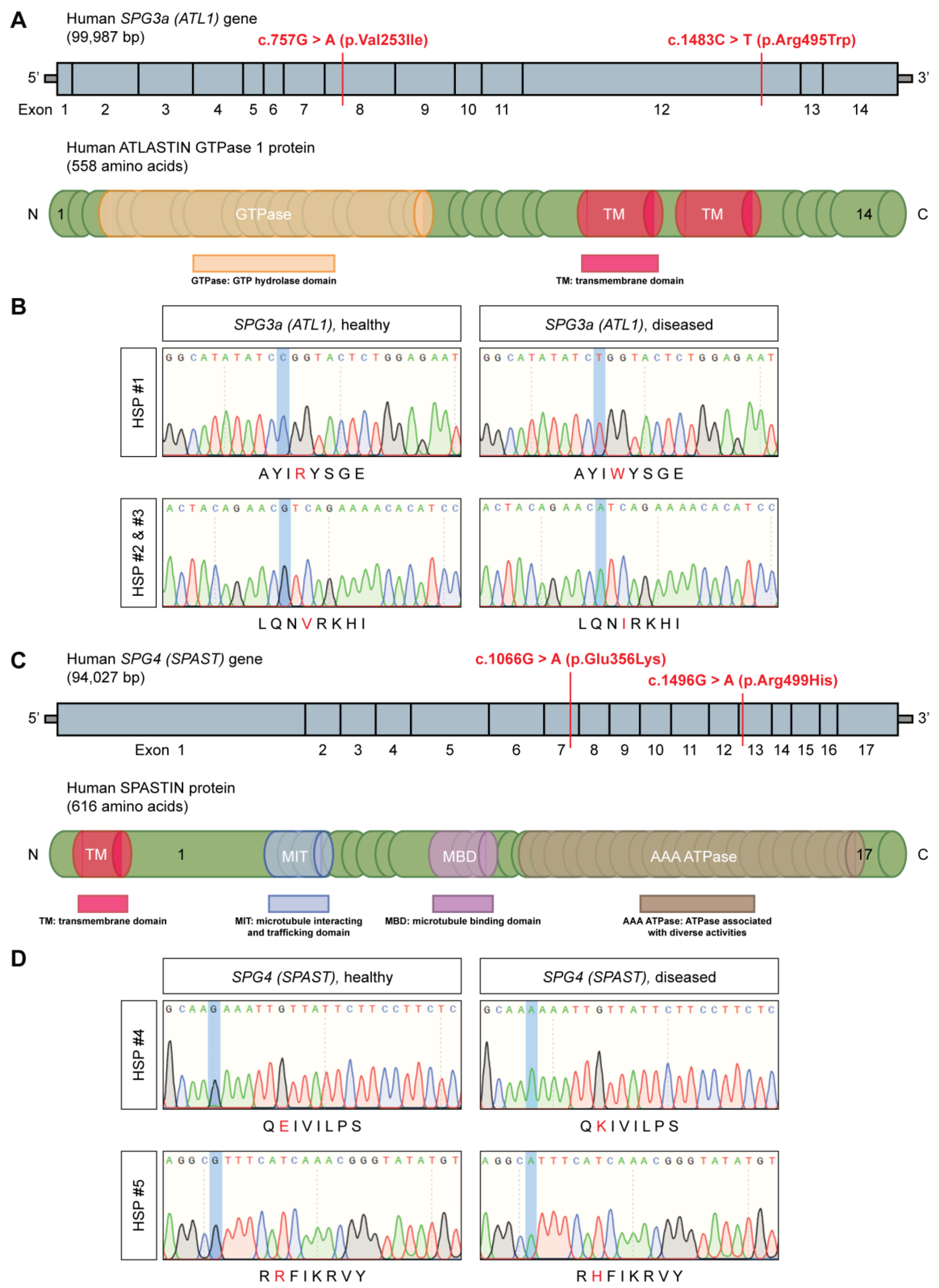
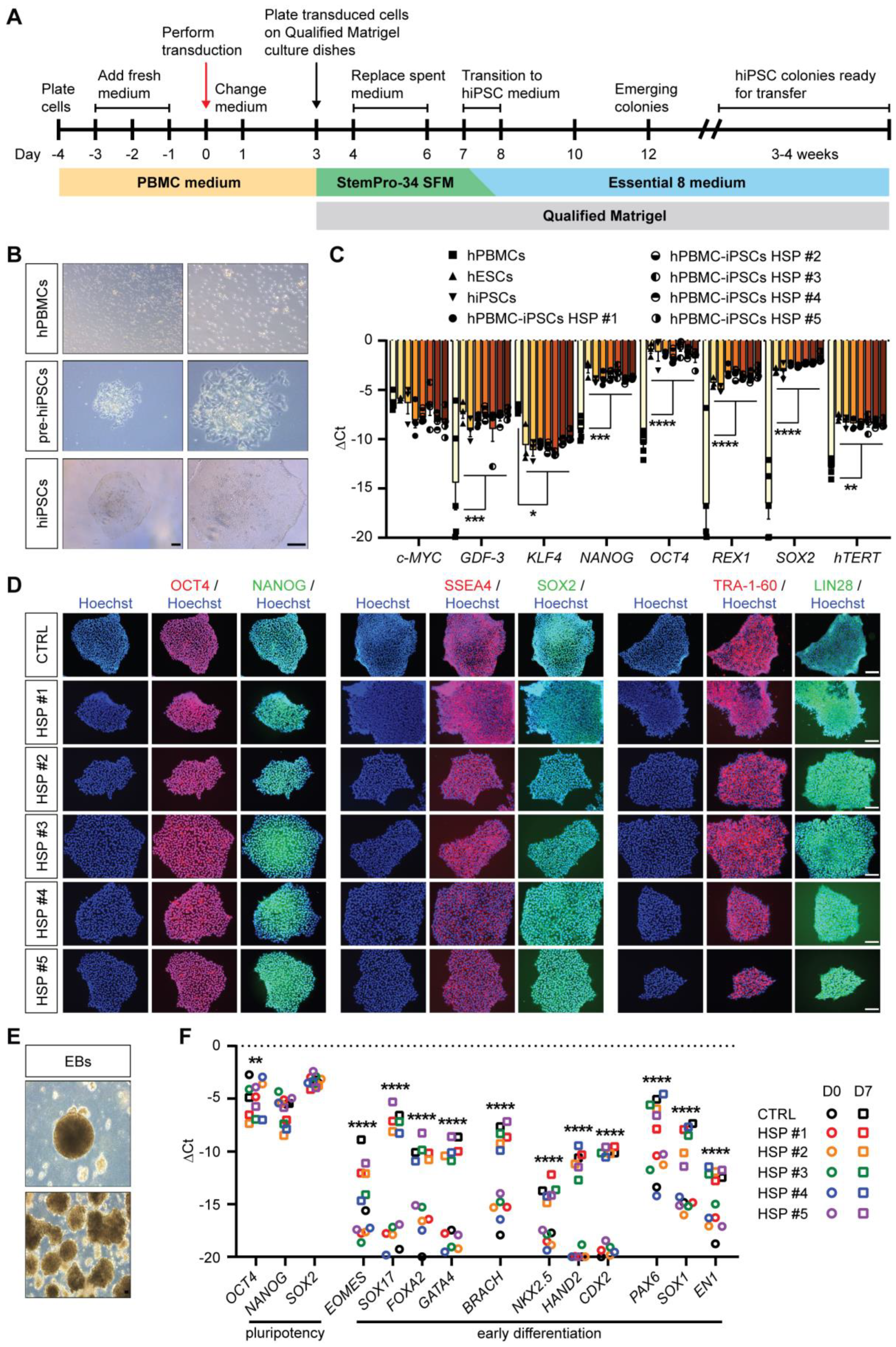
3.1. Generation of Integration-Free HSP hiPSCs
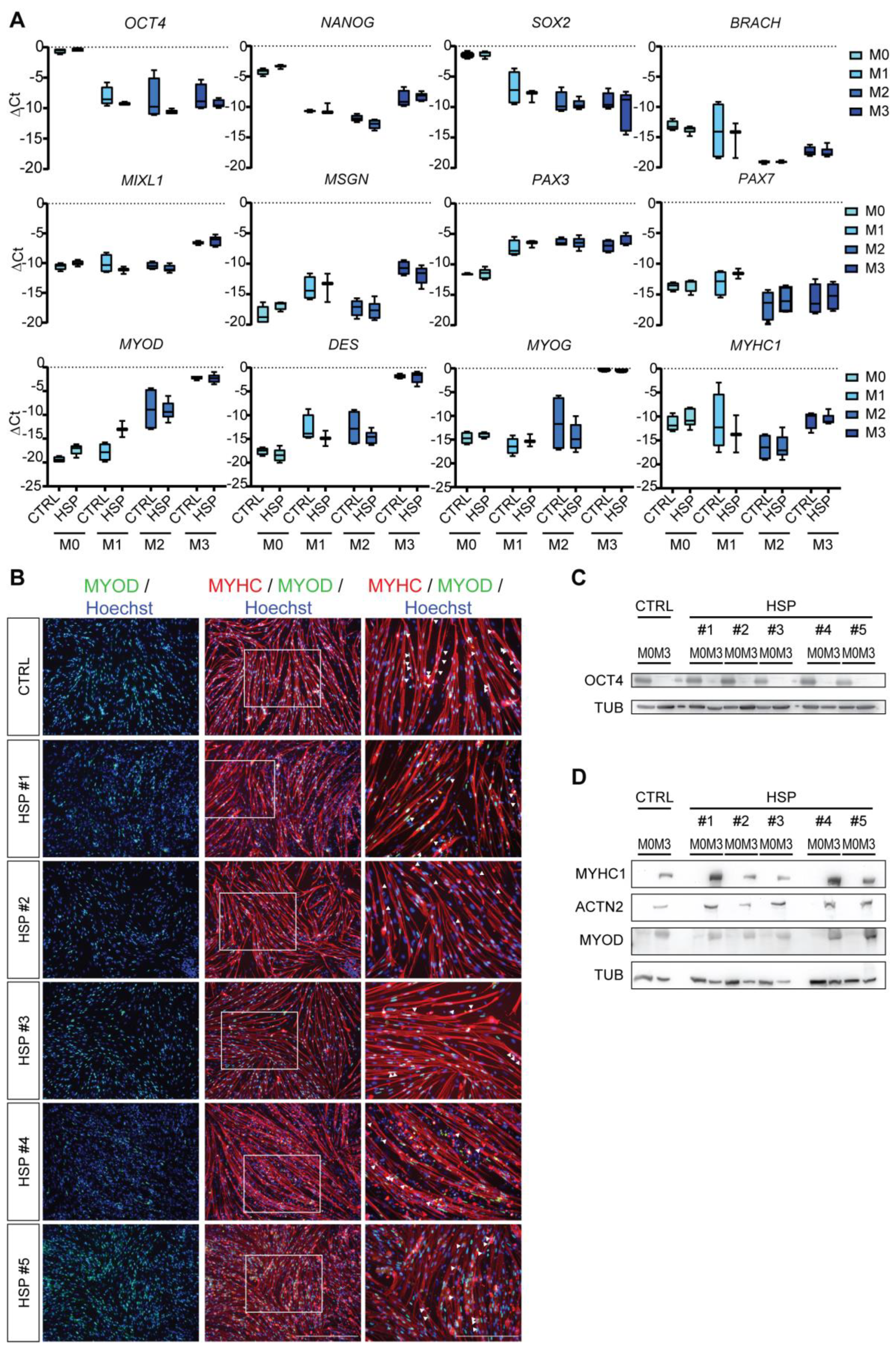
3.2. Differentiation of Mature Myotubes from HSP hiPSCs
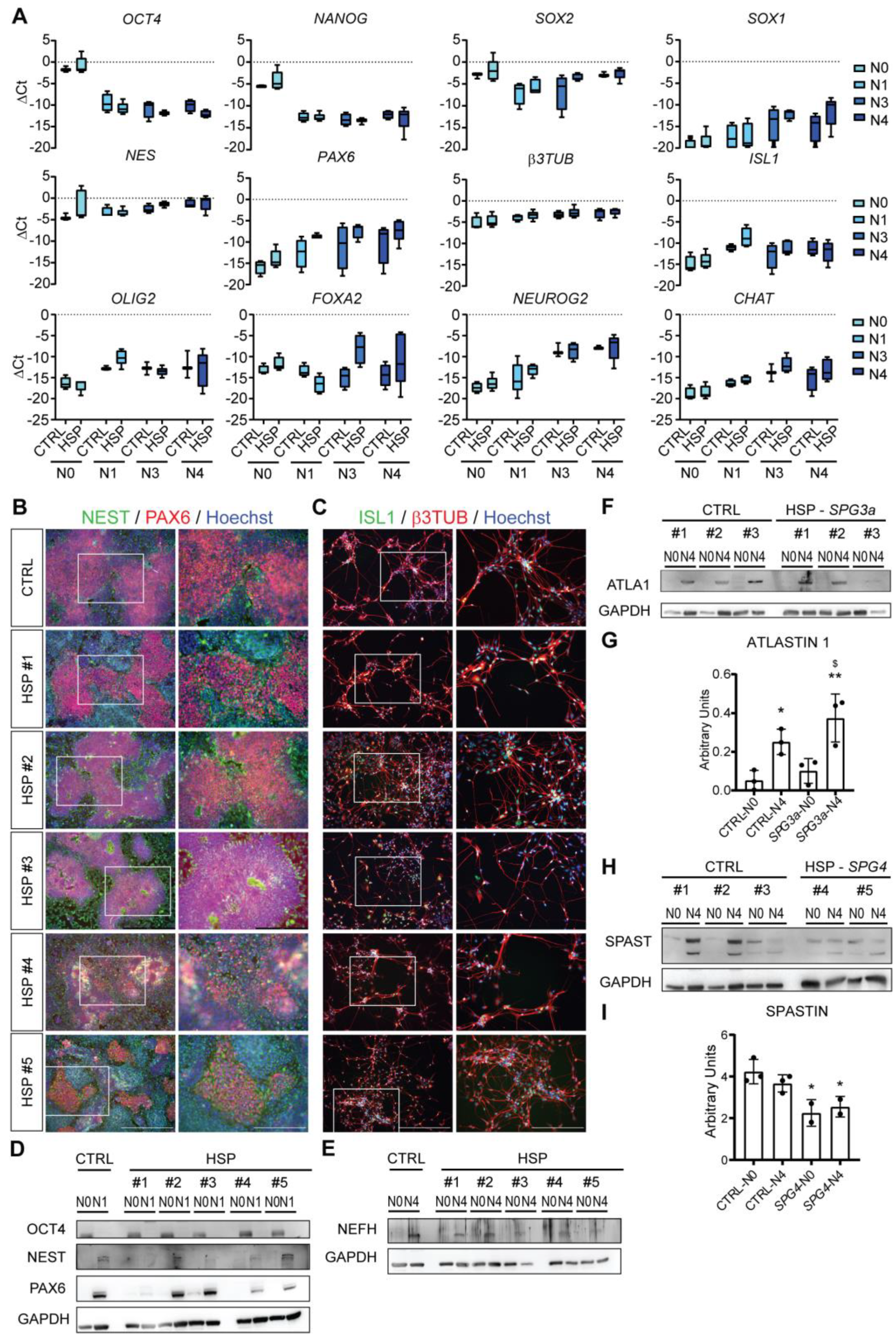
3.3. Differentiation of Lower MNs from HSP hiPSCs
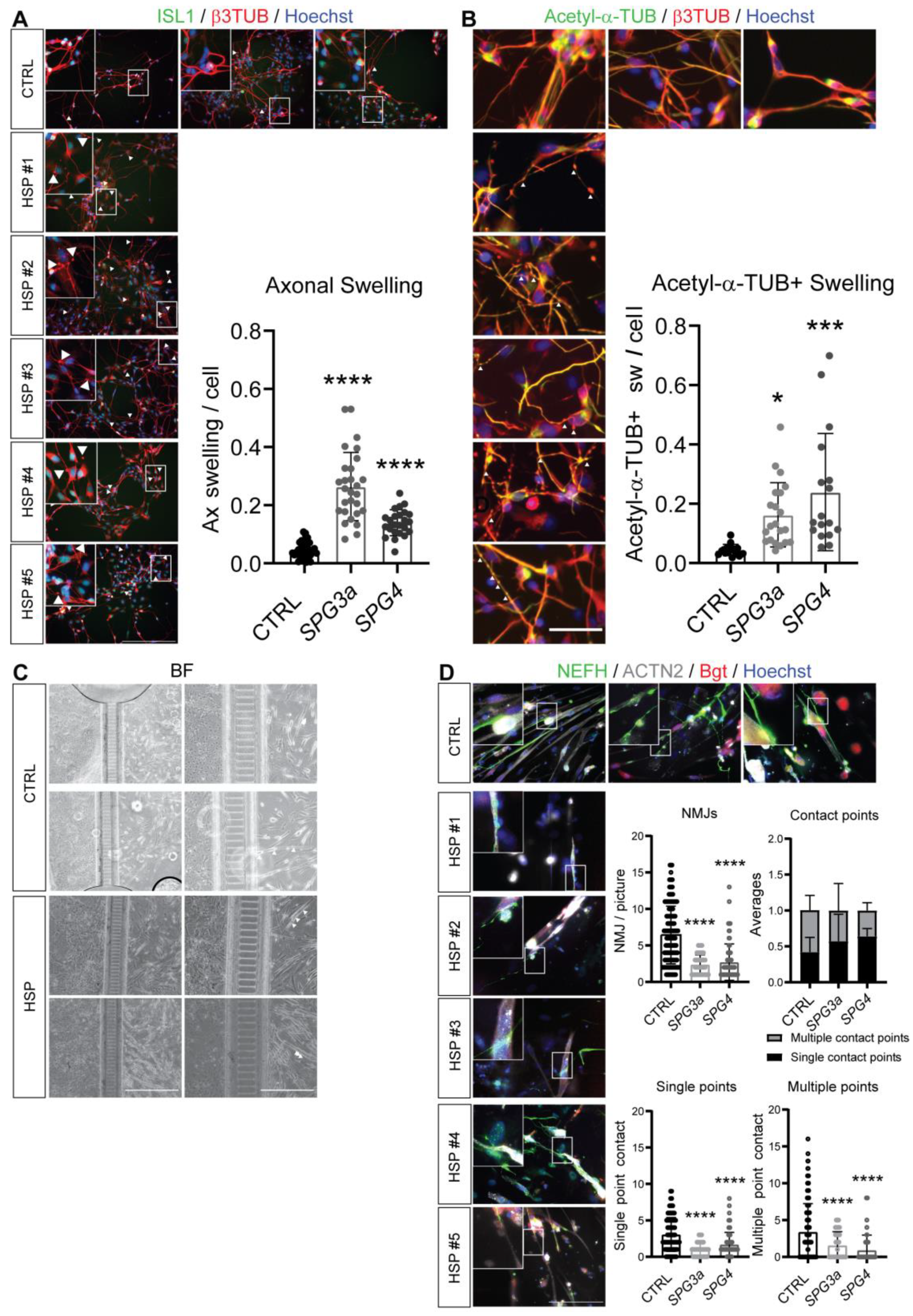
3.4. Axonal Swelling and Abnormal NMJ Formation in hiPSC-Derived MNs from HSP Patients
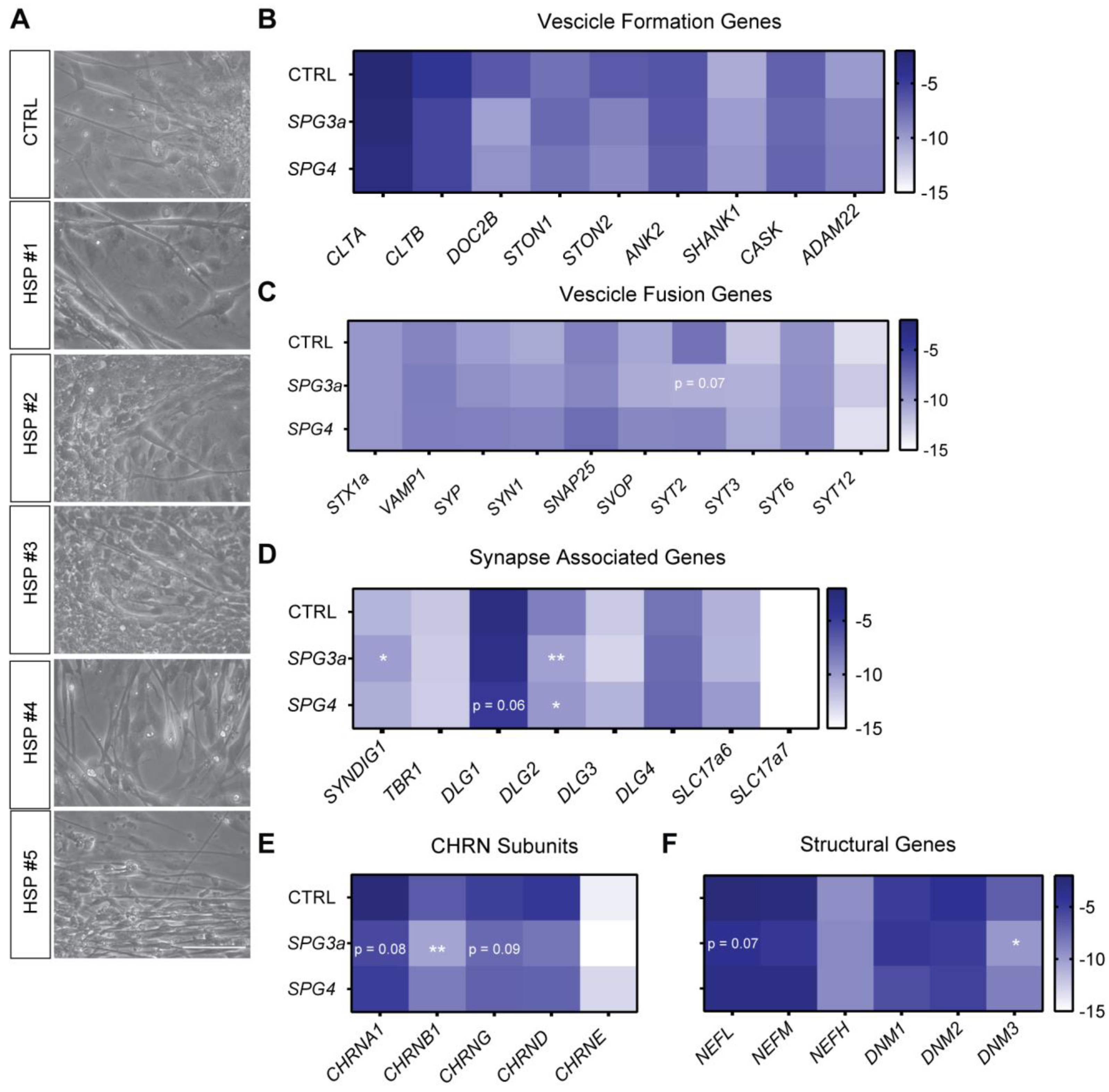
3.5. Formation of Neuromuscular Junctions from HSP hiPSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shribman, S.; Reid, E.; Crosby, A.H.; Houlden, H.; Warner, T.T. Hereditary spastic paraplegia: From diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019, 18, 1136–1146. [Google Scholar] [CrossRef]
- Erfanian Omidvar, M.; Torkamandi, S.; Rezaei, S.; Alipoor, B.; Omrani, M.D.; Darvish, H.; Ghaedi, H. Genotype-phenotype associations in hereditary spastic paraplegia: A systematic review and meta-analysis on 13,570 patients. J. Neurol. 2021, 268, 2065–2082. [Google Scholar] [CrossRef] [PubMed]
- Parodi, L.; Fenu, S.; Barbier, M.; Banneau, G.; Duyckaerts, C.; Tezenas du Montcel, S.; Monin, M.L.; Ait Said, S.; Guegan, J.; Tallaksen, C.M.E.; et al. Spastic paraplegia due to SPAST mutations is modified by the underlying mutation and sex. Brain 2018, 141, 3331–3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roll-Mecak, A.; Vale, R.D. Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 2008, 451, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X.J. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem. Cells 2014, 32, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulengin, I.; Park, J.J.; Lee, T.H. ER network formation and membrane fusion by atlastin1/SPG3A disease variants. Mol. Biol. Cell 2015, 26, 1616–1628. [Google Scholar] [CrossRef]
- Havlicek, S.; Kohl, Z.; Mishra, H.K.; Prots, I.; Eberhardt, E.; Denguir, N.; Wend, H.; Plotz, S.; Boyer, L.; Marchetto, M.C.; et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 2014, 23, 2527–2541. [Google Scholar] [CrossRef]
- Zhu, P.P.; Denton, K.R.; Pierson, T.M.; Li, X.J.; Blackstone, C. Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum. Mol. Genet. 2014, 23, 5638–5648. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Chai, E.; Mou, Y.; Roda, R.H.; Blackstone, C.; Li, X.J. Inhibiting mitochondrial fission rescues degeneration in hereditary spastic paraplegia neurons. Brain 2022. [Google Scholar] [CrossRef]
- Husain, M. New mechanistic insights into hereditary spastic paraplegias. Brain 2022, 145, 2937–2938. [Google Scholar] [CrossRef]
- Tintignac, L.A.; Brenner, H.R.; Ruegg, M.A. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.Y.; Yoshida, M.; Li, L.T.; Ikenaka, A.; Oshima, S.; Nakagawa, K.; Sakurai, H.; Matsui, E.; Nakahata, T.; Saito, M.K. iPSC-derived functional human neuromuscular junctions model the pathophysiology of neuromuscular diseases. JCI Insight 2019, 4, e124299. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, K.; Ito, A.; Kawabe, Y.; Kamihira, M. Novel neuromuscular junction model in 2D and 3D myotubes co-cultured with induced pluripotent stem cell-derived motor neurons. J. Biosci. Bioeng. 2020, 129, 486–493. [Google Scholar] [CrossRef]
- Paranjape, S.R.; Nagendran, T.; Poole, V.; Harris, J.; Taylor, A.M. Compartmentalization of Human Stem Cell-Derived Neurons within Pre-Assembled Plastic Microfluidic Chips. J. Vis. Exp. 2019, e59250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoklund Dittlau, K.; Krasnow, E.N.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Kerstens, A.; Giacomazzi, G.; Pavie, B.; Rossaert, E.; Beckers, J.; et al. Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem. Cell Rep. 2021, 16, 2213–2227. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Duelen, R.; Costamagna, D.; Gilbert, G.; De Waele, L.; Goemans, N.; Desloovere, K.; Verfaillie, C.M.; Sipido, K.R.; Buyse, G.M.; Sampaolesi, M. Human iPSC model reveals a central role for NOX4 and oxidative stress in Duchenne cardiomyopathy. Stem. Cell Rep. 2022, 17, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdu, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem. Cell 2018, 23, 101–113.e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, C.; Feifel, E.; Kern, G.; Murphy, C.; Meier, F.; Parson, W.; Beilmann, M.; Jennings, P.; Gstraunthaler, G.; Wilmes, A. Differentiation of human iPSCs into functional podocytes. PLoS ONE 2018, 13, e0203869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, L.; Kher, D.; Lee, K.L.; McKernan, R.; Dumevska, B.; Hidalgo, A.; Li, J.; Yang, H.; Main, H.; Ferri, G.; et al. A Human Pluripotent Stem Cell Model of Facioscapulohumeral Muscular Dystrophy-Affected Skeletal Muscles. Stem. Cells Transl. Med. 2016, 5, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.; Malboubi, M.; Li, Y.; George, J.H.; Jerusalem, A.; Szele, F.; Thompson, M.S.; Ye, H. Rapid and efficient differentiation of functional motor neurons from human iPSC for neural injury modelling. Stem. Cell Res. 2018, 32, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, J.A.; Jaiswal, M.K.; Calder, E.L.; Kishinevsky, S.; Weishaupt, A.; Toyka, K.V.; Goldstein, P.A.; Studer, L. Functional Connectivity under Optogenetic Control Allows Modeling of Human Neuromuscular Disease. Cell Stem. Cell 2016, 18, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Parodi, L.; Fenu, S.; Stevanin, G.; Durr, A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev. Neurol. 2017, 173, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Mackay-Sim, A. Hereditary Spastic Paraplegia: From Genes, Cells and Networks to Novel Pathways for Drug Discovery. Brain Sci. 2021, 11, 403. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, L.E.O.; Eltazi, I.Z.M.; Ahmed, A.E.M.; Stevanin, G. Hereditary spastic paraplegias: Time for an objective case definition and a new nosology for neurogenetic disorders to facilitate biomarker/therapeutic studies. Expert Rev. Neurother. 2019, 19, 409–415. [Google Scholar] [CrossRef]
- Vila, O.F.; Chavez, M.; Ma, S.P.; Yeager, K.; Zholudeva, L.V.; Colon-Mercado, J.M.; Qu, Y.; Nash, T.R.; Lai, C.; Feliciano, C.M.; et al. Bioengineered optogenetic model of human neuromuscular junction. Biomaterials 2021, 276, 121033. [Google Scholar] [CrossRef]
- Al Tanoury, Z.; Zimmerman, J.F.; Rao, J.; Sieiro, D.; McNamara, H.M.; Cherrier, T.; Rodriguez-delaRosa, A.; Hick-Colin, A.; Bousson, F.; Fugier-Schmucker, C.; et al. Prednisolone rescues Duchenne muscular dystrophy phenotypes in human pluripotent stem cell-derived skeletal muscle in vitro. Proc. Natl. Acad. Sci. USA 2021, 118, e2022960118. [Google Scholar] [CrossRef]
- Ryan, T.; Liu, J.; Chu, A.; Wang, L.; Blais, A.; Skerjanc, I.S. Retinoic acid enhances skeletal myogenesis in human embryonic stem cells by expanding the premyogenic progenitor population. Stem. Cell Rev. Rep. 2012, 8, 482–493. [Google Scholar] [CrossRef]
- Borchin, B.; Chen, J.; Barberi, T. Derivation and FACS-mediated purification of PAX3+/PAX7+ skeletal muscle precursors from human pluripotent stem cells. Stem. Cell Rep. 2013, 1, 620–631. [Google Scholar] [CrossRef] [Green Version]
- Maury, Y.; Come, J.; Piskorowski, R.A.; Salah-Mohellibi, N.; Chevaleyre, V.; Peschanski, M.; Martinat, C.; Nedelec, S. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat. Biotechnol. 2015, 33, 89–96. [Google Scholar] [CrossRef]
- Qu, Q.; Li, D.; Louis, K.R.; Li, X.; Yang, H.; Sun, Q.; Crandall, S.R.; Tsang, S.; Zhou, J.; Cox, C.L.; et al. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat. Commun. 2014, 5, 3449. [Google Scholar] [CrossRef] [Green Version]
- Salimi, A.; Nadri, S.; Ghollasi, M.; Khajeh, K.; Soleimani, M. Comparison of different protocols for neural differentiation of human induced pluripotent stem cells. Mol. Biol. Rep. 2014, 41, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, M.W.; Croft, G.F.; Williams, D.J.; O’Keeffe, S.; Carrasco, M.A.; Davis, A.R.; Roybon, L.; Oakley, D.H.; Maniatis, T.; Henderson, C.E.; et al. Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J. Neurosci. 2013, 33, 574–586. [Google Scholar] [CrossRef] [Green Version]
- Frati, A.; Cerretani, D.; Fiaschi, A.I.; Frati, P.; Gatto, V.; La Russa, R.; Pesce, A.; Pinchi, E.; Santurro, A.; Fraschetti, F.; et al. Diffuse Axonal Injury and Oxidative Stress: A Comprehensive Review. Int. J. Mol. Sci. 2017, 18, 2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, K.R.; Xu, C.C.; Li, X.J. Modeling Axonal Phenotypes with Human Pluripotent Stem Cells. Methods Mol. Biol. 2016, 1353, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon degeneration: Molecular mechanisms of a self-destruction pathway. J. Cell Biol. 2012, 196, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Akiyama, T.; Warita, H.; Aoki, M. Omics Approach to Axonal Dysfunction of Motor Neurons in Amyotrophic Lateral Sclerosis (ALS). Front. Neurosci. 2020, 14, 194. [Google Scholar] [CrossRef] [Green Version]
- Claudiani, P.; Riano, E.; Errico, A.; Andolfi, G.; Rugarli, E.I. Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp. Cell Res. 2005, 309, 358–369. [Google Scholar] [CrossRef]
- Mancuso, G.; Rugarli, E.I. A cryptic promoter in the first exon of the SPG4 gene directs the synthesis of the 60-kDa spastin isoform. BMC Biol. 2008, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, M.; Brown, R.H., Jr. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect Med. 2018, 8, a024125. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.T.; Hausrat, T.J.; Heisler, F.F.; Gromova, K.V.; Lombino, F.L.; Fischer, T.; Ruschkies, L.; Breiden, P.; Thies, E.; Hermans-Borgmeyer, I.; et al. Spastin depletion increases tubulin polyglutamylation and impairs kinesin-mediated neuronal transport, leading to working and associative memory deficits. PLoS Biol. 2020, 18, e3000820. [Google Scholar] [CrossRef] [PubMed]
- Wali, G.; Liyanage, E.; Blair, N.F.; Sutharsan, R.; Park, J.S.; Mackay-Sim, A.; Sue, C.M. Oxidative Stress-Induced Axon Fragmentation Is a Consequence of Reduced Axonal Transport in Hereditary Spastic Paraplegia SPAST Patient Neurons. Front. Neurosci. 2020, 14, 401. [Google Scholar] [CrossRef]
- Zhu, P.P.; Soderblom, C.; Tao-Cheng, J.H.; Stadler, J.; Blackstone, C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet. 2006, 15, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Namekawa, M.; Muriel, M.P.; Janer, A.; Latouche, M.; Dauphin, A.; Debeir, T.; Martin, E.; Duyckaerts, C.; Prigent, A.; Depienne, C.; et al. Mutations in the SPG3A gene encoding the GTPase atlastin interfere with vesicle trafficking in the ER/Golgi interface and Golgi morphogenesis. Mol. Cell Neurosci. 2007, 35, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.; Lumb, J.H.; Fassier, C.; Connell, J.W.; Ten Martin, D.; Seaman, M.N.; Hazan, J.; Reid, E. An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome. J. Cell Biol. 2013, 202, 527–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connell, J.W.; Allison, R.J.; Rodger, C.E.; Pearson, G.; Zlamalova, E.; Reid, E. ESCRT-III-associated proteins and spastin inhibit protrudin-dependent polarised membrane traffic. Cell Mol. Life Sci. 2020, 77, 2641–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Hedera, P. Hereditary spastic paraplegia-causing mutations in atlastin-1 interfere with BMPRII trafficking. Mol. Cell Neurosci. 2013, 52, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Fassier, C.; Hutt, J.A.; Scholpp, S.; Lumsden, A.; Giros, B.; Nothias, F.; Schneider-Maunoury, S.; Houart, C.; Hazan, J. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat. Neurosci. 2010, 13, 1380–1387. [Google Scholar] [CrossRef]
- Toupenet Marchesi, L.; Leblanc, M.; Stevanin, G. Current Knowledge of Endolysosomal and Autophagy Defects in Hereditary Spastic Paraplegia. Cells 2021, 10, 1678. [Google Scholar] [CrossRef]
- Nijssen, J.; Aguila, J.; Hoogstraaten, R.; Kee, N.; Hedlund, E. Axon-Seq Decodes the Motor Axon Transcriptome and Its Modulation in Response to ALS. Stem. Cell Rep. 2018, 11, 1565–1578. [Google Scholar] [CrossRef]
- Greek, R.; Hansen, L.A. Questions regarding the predictive value of one evolved complex adaptive system for a second: Exemplified by the SOD1 mouse. Prog. Biophys. Mol. Biol. 2013, 113, 231–253. [Google Scholar] [CrossRef] [Green Version]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen. Res. 2018, 13, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.; Jarvis, P. Review of the potential use of blood neuro-biomarkers in the diagnosis of mild traumatic brain injury. Clin. Exp. Emerg. Med. 2017, 4, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Natali Sora, M.G.; Scarlato, M.; et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol. Med. 2015, 7, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, S.M.; Smith, A.S.T.; Mack, D.L. Creating stem cell-derived neuromuscular junctions in vitro. Muscle Nerve 2021, 64, 388–403. [Google Scholar] [CrossRef]
- Trotta, N.; Orso, G.; Rossetto, M.G.; Daga, A.; Broadie, K. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr. Biol. 2004, 14, 1135–1147. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, N.T.; Sun, Q.; Xue, M.; Zhang, B.; Zinn, K. Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2004, 2, e429. [Google Scholar] [CrossRef] [Green Version]
- Orso, G.; Martinuzzi, A.; Rossetto, M.G.; Sartori, E.; Feany, M.; Daga, A. Disease-related phenotypes in a Drosophila model of hereditary spastic paraplegia are ameliorated by treatment with vinblastine. J. Clin. Investig. 2005, 115, 3026–3034. [Google Scholar] [CrossRef] [Green Version]
- Slater, C.R. ‘Fragmentation’ of NMJs: A sign of degeneration or regeneration? A long journey with many junctions. Neuroscience 2020, 439, 28–40. [Google Scholar] [CrossRef]
- Diniz de Lima, F.; Faber, I.; Servelhere, K.R.; Bittar, M.F.R.; Martinez, A.R.M.; Piovesana, L.G.; Martins, M.P.; Martins, C.R., Jr.; Benaglia, T.; de Sa Carvalho, B.; et al. Randomized Trial of Botulinum Toxin Type A in Hereditary Spastic Paraplegia-The SPASTOX Trial. Mov. Disord. 2021, 36, 1654–1663. [Google Scholar] [CrossRef]
- Servelhere, K.R.; Faber, I.; Martinez, A.; Nickel, R.; Moro, A.; Germiniani, F.M.B.; Moscovich, M.; Blume, T.R.; Munhoz, R.P.; Teive, H.A.G.; et al. Botulinum toxin for hereditary spastic paraplegia: Effects on motor and non-motor manifestations. Arq. Neuropsiquiatr. 2018, 76, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Servelhere, K.R.; Faber, I.; Saute, J.A.; Moscovich, M.; D’Abreu, A.; Jardim, L.B.; Teive, H.A.; Lopes-Cendes, I.; Franca, M.C., Jr. Non-motor symptoms in patients with hereditary spastic paraplegia caused by SPG4 mutations. Eur. J. Neurol. 2016, 23, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Cvetkovic, C.; Rich, M.H.; Raman, R.; Kong, H.; Bashir, R. A 3D-printed platform for modular neuromuscular motor units. Microsyst. Nanoeng. 2017, 3, 17015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, T.A.; Cohen, E.; Cairns, D.M.; Rodriguez, M.; Mathews, J.; Jose, R.R.; Kaplan, D.L. Bioinspired Three-Dimensional Human Neuromuscular Junction Development in Suspended Hydrogel Arrays. Tissue Eng. Part C Methods 2018, 24, 346–359. [Google Scholar] [CrossRef] [Green Version]
- Osaki, T.; Sivathanu, V.; Kamm, R.D. Engineered 3D vascular and neuronal networks in a microfluidic platform. Sci. Rep. 2018, 8, 5168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutry, M.; Morais, S.; Stevanin, G. Update on the Genetics of Spastic Paraplegias. Curr. Neurol. Neurosci. Rep. 2019, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- Tesson, C.; Koht, J.; Stevanin, G. Delving into the complexity of hereditary spastic paraplegias: How unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015, 134, 511–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.H.; Liu, X.M. Clinical features and genotype-phenotype correlation analysis in patients with ATL1 mutations: A literature reanalysis. Transl. Neurodegener. 2017, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyyazhagan, A.; Orlacchio, A. Hereditary Spastic Paraplegia: An Update. Int. J. Mol. Sci. 2022, 23, 1697. [Google Scholar] [CrossRef] [PubMed]
- Chrestian, N.; Dupre, N.; Gan-Or, Z.; Szuto, A.; Chen, S.; Venkitachalam, A.; Brisson, J.D.; Warman-Chardon, J.; Ahmed, S.; Ashtiani, S.; et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol. Genet. 2017, 3, e122. [Google Scholar] [CrossRef]
- Martinuzzi, A.; Montanaro, D.; Vavla, M.; Paparella, G.; Bonanni, P.; Musumeci, O.; Brighina, E.; Hlavata, H.; Rossi, G.; Aghakhanyan, G.; et al. Clinical and Paraclinical Indicators of Motor System Impairment in Hereditary Spastic Paraplegia: A Pilot Study. PLoS ONE 2016, 11, e0153283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, C.J.; Burness, C.E.; Kirby, J.; Cox, L.E.; Rao, D.G.; Hewamadduma, C.; Sharrack, B.; Hadjivassiliou, M.; Chinnery, P.F.; Dalton, A.; et al. Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology 2006, 67, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Lo Giudice, T.; Lombardi, F.; Santorelli, F.M.; Kawarai, T.; Orlacchio, A. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Exp. Neurol. 2014, 261, 518–539. [Google Scholar] [CrossRef] [PubMed]
- Solowska, J.M.; Baas, P.W. Hereditary spastic paraplegia SPG4: What is known and not known about the disease. Brain 2015, 138, 2471–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Beukelaer, N.; Weide, G.; Huyghe, E.; Vandekerckhove, I.; Hanssen, B.; Peeters, N.; Uytterhoeven, J.; Deschrevel, J.; Maes, K.; Corvelyn, M.; et al. Reduced Cross-Sectional Muscle Growth Six Months after Botulinum Toxin Type-A Injection in Children with Spastic Cerebral Palsy. Toxins 2022, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- Walhain, F.; Desloovere, K.; Declerck, M.; Van Campenhout, A.; Bar-On, L. Interventions and lower-limb macroscopic muscle morphology in children with spastic cerebral palsy: A scoping review. Dev. Med. Child. Neurol. 2021, 63, 274–286. [Google Scholar] [CrossRef]
- Howard, J.J.; Herzog, W. Skeletal Muscle in Cerebral Palsy: From Belly to Myofibril. Front. Neurol. 2021, 12, 620852. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costamagna, D.; Casters, V.; Beltrà, M.; Sampaolesi, M.; Van Campenhout, A.; Ortibus, E.; Desloovere, K.; Duelen, R. Autologous iPSC-Derived Human Neuromuscular Junction to Model the Pathophysiology of Hereditary Spastic Paraplegia. Cells 2022, 11, 3351. https://doi.org/10.3390/cells11213351
Costamagna D, Casters V, Beltrà M, Sampaolesi M, Van Campenhout A, Ortibus E, Desloovere K, Duelen R. Autologous iPSC-Derived Human Neuromuscular Junction to Model the Pathophysiology of Hereditary Spastic Paraplegia. Cells. 2022; 11(21):3351. https://doi.org/10.3390/cells11213351
Chicago/Turabian StyleCostamagna, Domiziana, Valérie Casters, Marc Beltrà, Maurilio Sampaolesi, Anja Van Campenhout, Els Ortibus, Kaat Desloovere, and Robin Duelen. 2022. "Autologous iPSC-Derived Human Neuromuscular Junction to Model the Pathophysiology of Hereditary Spastic Paraplegia" Cells 11, no. 21: 3351. https://doi.org/10.3390/cells11213351