
The Complex Relation between Atrial Cardiomyopathy and Thrombogenesis

 , ,
, ,
Abstract
:1. Introduction
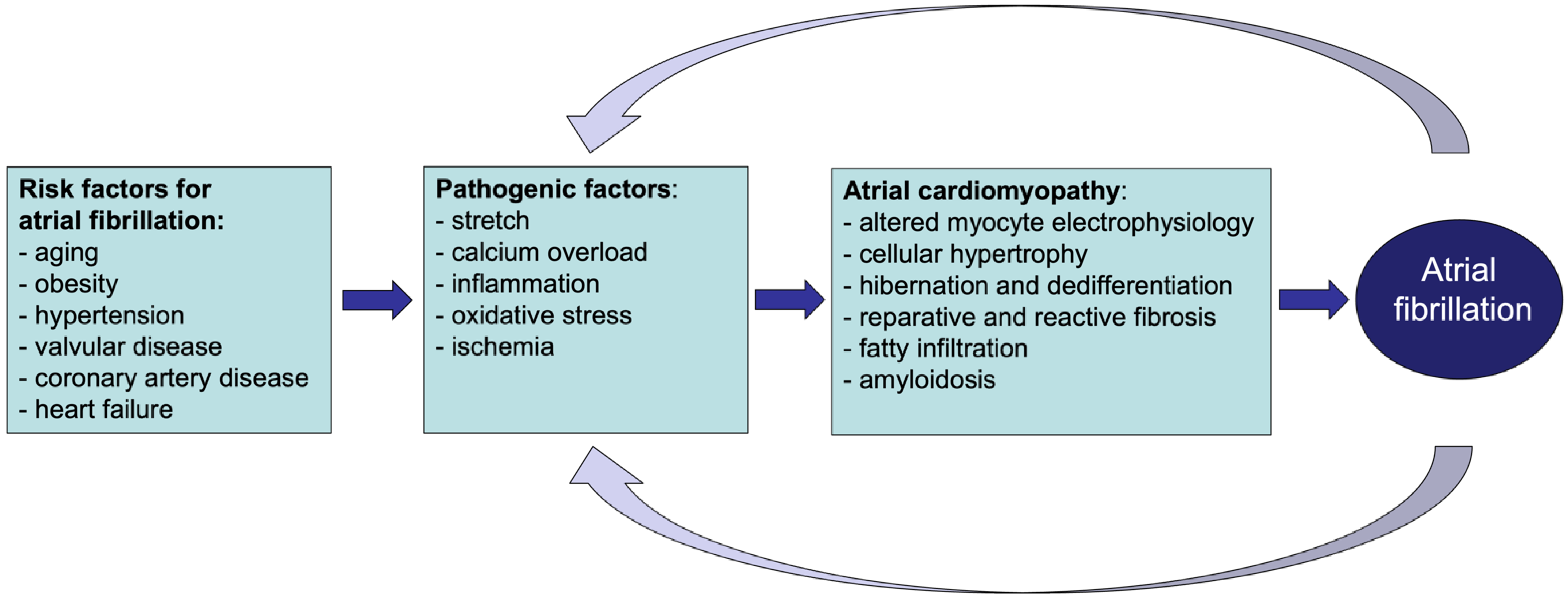
2. Atrial Cardiomyopathy and Atrial Fibrillation
3. Diversity of Atrial Cardiomyopathy in Animal Models
4. Atrial Cardiomyopathy and Thrombogenesis
4.1. Blood Stasis and Endothelial Dysfunction
4.2. Pro-Thrombotic Interstitial Changes
4.3. Hypercoagulability
5. Activation of Coagulation Supports Atrial Cardiomyopathy
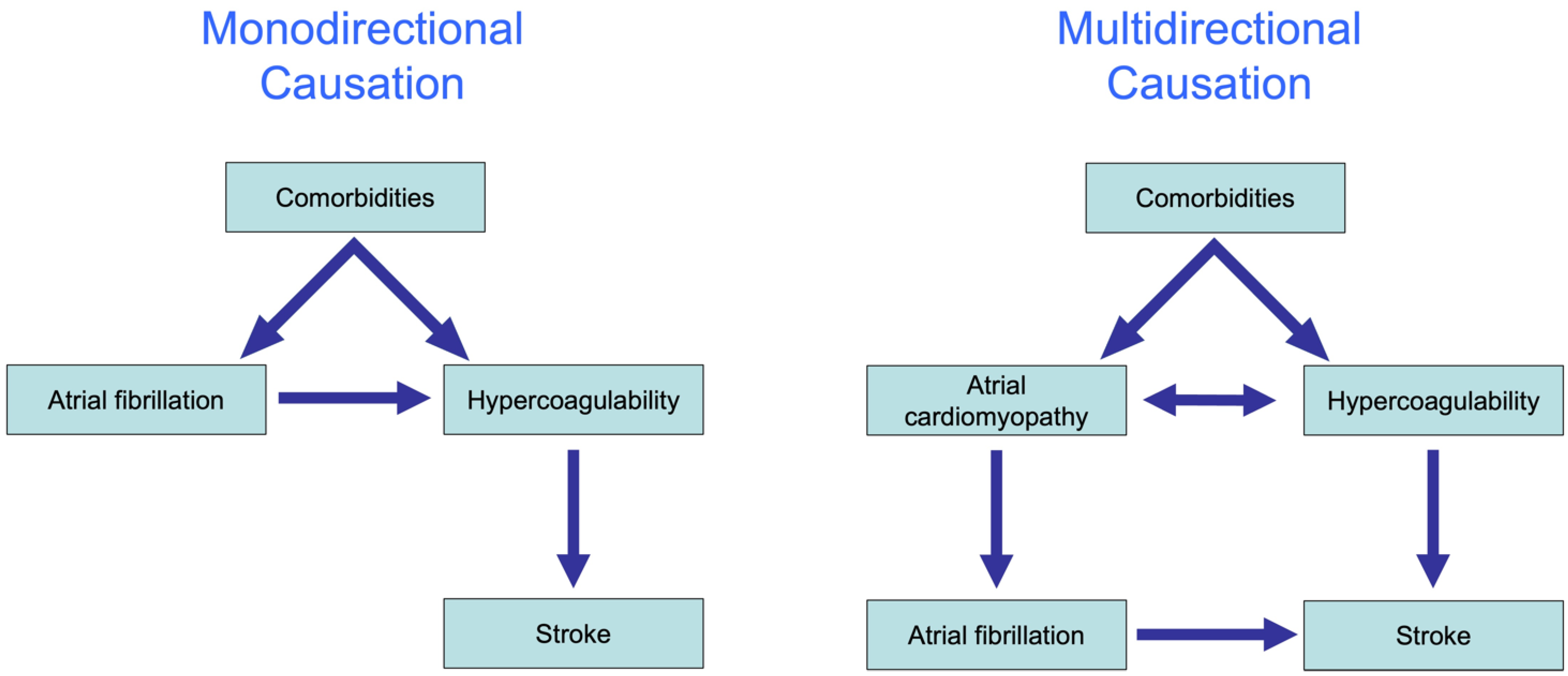
6. The Complex Association of AF and Thrombogenesis (Stroke)
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomstrom-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef] [PubMed]
- Fabritz, L.; Guasch, E.; Antoniades, C.; Bardinet, I.; Benninger, G.; Betts, T.R.; Brand, E.; Breithardt, G.; Bucklar-Suchankova, G.; Camm, A.J.; et al. Expert consensus document: Defining the major health modifiers causing atrial fibrillation: A roadmap to underpin personalized prevention and treatment. Nat. Rev. Cardiol. 2016, 13, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 120. [Google Scholar] [CrossRef]
- Rutter, M.K.; Parise, H.; Benjamin, E.J.; Levy, D.; Larson, M.G.; Meigs, J.B.; Nesto, R.W.; Wilson, P.W.; Vasan, R.S. Impact of glucose intolerance and insulin resistance on cardiac structure and function: Sex-related differences in the Framingham Heart Study. Circulation 2003, 107, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Wyse, D.G.; Van Gelder, I.C.; Ellinor, P.T.; Go, A.S.; Kalman, J.M.; Narayan, S.M.; Nattel, S.; Schotten, U.; Rienstra, M. Lone atrial fibrillation: Does it exist? J. Am. Coll. Cardiol. 2014, 63, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Goette, A.; Kalman, J.M.; Aguinaga, L.; Akar, J.; Cabrera, J.A.; Chen, S.A.; Chugh, S.S.; Corradi, D.; D’Avila, A.; Dobrev, D.; et al. EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Europace 2016, 18, 1455–1490. [Google Scholar] [CrossRef] [PubMed]
- Schotten, U.; Verheule, S.; Kirchhof, P.; Goette, A. Pathophysiological mechanisms of atrial fibrillation: A translational appraisal. Physiol. Rev. 2011, 91, 265–325. [Google Scholar] [CrossRef]
- Hatem, S.N.; Sanders, P. Epicardial adipose tissue and atrial fibrillation. Cardiovasc. Res. 2014, 102, 205–213. [Google Scholar] [CrossRef]
- Verheule, S.; Schotten, U. Electrophysiological Consequences of Cardiac Fibrosis. Cells 2021, 10, 3220. [Google Scholar] [CrossRef]
- Ausma, J.; Dispersyn, G.D.; Duimel, H.; Thone, F.; Ver Donck, L.; Allessie, M.A.; Borgers, M. Changes in ultrastructural calcium distribution in goat atria during atrial fibrillation. J. Mol. Cell Cardiol. 2000, 32, 355–364. [Google Scholar] [CrossRef]
- van Bragt, K.A.; Nasrallah, H.M.; Kuiper, M.; Luiken, J.J.; Schotten, U.; Verheule, S. Atrial supply-demand balance in healthy adult pigs: Coronary blood flow, oxygen extraction, and lactate production during acute atrial fibrillation. Cardiovasc. Res. 2014, 101, 9–19. [Google Scholar] [CrossRef]
- Eckstein, J.; Verheule, S.; de Groot, N.M.; Allessie, M.; Schotten, U. Mechanisms of perpetuation of atrial fibrillation in chronically dilated atria. Prog. Biophys. Mol. Biol. 2008, 97, 435–451. [Google Scholar] [CrossRef]
- Dudley, S.C., Jr.; Hoch, N.E.; McCann, L.A.; Honeycutt, C.; Diamandopoulos, L.; Fukai, T.; Harrison, D.G.; Dikalov, S.I.; Langberg, J. Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: Role of the NADPH and xanthine oxidases. Circulation 2005, 112, 1266–1273. [Google Scholar] [CrossRef]
- Frustaci, A.; Caldarulo, M.; Buffon, A.; Bellocci, F.; Fenici, R.; Melina, D. Cardiac biopsy in patients with “primary” atrial fibrillation. Histologic evidence of occult myocardial diseases. Chest 1991, 100, 303–306. [Google Scholar] [CrossRef]
- Frustaci, A.; Chimenti, C.; Bellocci, F.; Morgante, E.; Russo, M.A.; Maseri, A. Histological Substrate of Atrial Biopsies in Patients With Lone Atrial Fibrillation. Circulation 1997, 96, 1180–1184. [Google Scholar] [CrossRef]
- Rocken, C.; Peters, B.; Juenemann, G.; Saeger, W.; Klein, H.; Huth, C.; Roessner, A.; Goette, A. Atrial amyloidosis: An arrhythmogenic substrate for persistent atrial fibrillation. Circulation 2002, 106, 2091–2097. [Google Scholar] [CrossRef]
- Anné, W.; Willems, R.; Roskams, T.; Sergeant, P.; Herijgers, P.; Holemans, P.; Ector, H.; Heidbüchel, H. Matrix metalloproteinases and atrial remodeling in patients with mitral valve disease and atrial fibrillation. Cardiovasc. Res. 2005, 67, 655–666. [Google Scholar] [CrossRef]
- Platonov, P.G.; Mitrofanova, L.B.; Orshanskaya, V.; Ho, S.Y. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J. Am. Coll. Cardiol. 2011, 58, 2225–2232. [Google Scholar] [CrossRef]
- Maesen, B.; Verheule, S.; Zeemering, S.; La Meir, M.; Nijs, J.; Lumeij, S.; Lau, D.H.; Granier, M.; Crijns, H.J.; Maessen, J.G.; et al. Endomysial fibrosis, rather than overall connective tissue content, is the main determinant of conduction disturbances in human atrial fibrillation. Europace 2022, 24, 1015–1024. [Google Scholar] [CrossRef]
- Verheule, S.; Tuyls, E.; Gharaviri, A.; Hulsmans, S.; van Hunnik, A.; Kuiper, M.; Serroyen, J.; Zeemering, S.; Kuijpers, N.H.; Schotten, U. Loss of continuity in the thin epicardial layer because of endomysial fibrosis increases the complexity of atrial fibrillatory conduction. Circ. Arrhythm. Electrophysiol. 2013, 6, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Schuessler, R.B.; Grayson, T.M.; Bromberg, B.I.; Cox, J.L.; Boineau, J.P. Cholinergically mediated tachyarrhythmias induced by a single extrastimulus in the isolated canine right atrium. Circ. Res. 1992, 71, 1254–1267. [Google Scholar] [CrossRef]
- Ryu, K.; Li, L.; Khrestian, C.M.; Matsumoto, N.; Sahadevan, J.; Ruehr, M.L.; Van Wagoner, D.R.; Efimov, I.R.; Waldo, A.L. Effects of sterile pericarditis on connexins 40 and 43 in the atria: Correlation with abnormal conduction and atrial arrhythmias. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1231–H1241. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Fareh, S.; Leung, T.; Nattel, S. Promotion of atrial fibrillation by heart failure in dogs: Atrial remodeling of a different sort. Circulation 1999, 100, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Wilson, E.E.; Everett, T.H.; Shanbhag, S.; Golden, C.; Olgin, J.E. Alterations in atrial electrophysiology and tissue structure in a canine model of chronic atrial dilatation due to mitral regurgitation. Circulation 2003, 107, 2615–2622. [Google Scholar] [CrossRef]
- Neuberger, H.-R.; Schotten, U.; Verheule, S.; Eijsbouts, S.; Blaauw, Y.; van Hunnik, A.; Allessie, M.A. Development of a substrate of atrial fibrillation during chronic atrioventricular block in the goat. Circulation 2005, 111, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Qi, X.Y.; Wakili, R.; Comtois, P.; Chartier, D.; Harada, M.; Iwasaki, Y.K.; Romeo, P.; Maguy, A.; Dobrev, D.; et al. Mechanisms of atrial tachyarrhythmias associated with coronary artery occlusion in a chronic canine model. Circulation 2011, 123, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Abed, H.S.; Samuel, C.S.; Lau, D.H.; Kelly, D.J.; Royce, S.G.; Alasady, M.; Mahajan, R.; Kuklik, P.; Zhang, Y.; Brooks, A.G.; et al. Obesity results in progressive atrial structural and electrical remodeling: Implications for atrial fibrillation. Heart Rhythm. 2013, 10, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; Schotten, U.; Neuberger, H.R.; Bohm, M.; Wirth, K. Negative tracheal pressure during obstructive respiratory events promotes atrial fibrillation by vagal activation. Heart Rhythm. 2011, 8, 1436–1443. [Google Scholar] [CrossRef]
- Kistler, P.M.; Sanders, P.; Dodic, M.; Spence, S.J.; Samuel, C.S.; Zhao, C.; Charles, J.A.; Edwards, G.A.; Kalman, J.M. Atrial electrical and structural abnormalities in an ovine model of chronic blood pressure elevation after prenatal corticosteroid exposure: Implications for development of atrial fibrillation. Eur. Heart J. 2006, 27, 3045–3056. [Google Scholar] [CrossRef]
- Koura, T.; Hara, M.; Takeuchi, S.; Ota, K.; Okada, Y.; Miyoshi, S.; Watanabe, A.; Shiraiwa, K.; Mitamura, H.; Kodama, I.; et al. Anisotropic conduction properties in canine atria analyzed by high-resolution optical mapping: Preferential direction of conduction block changes from longitudinal to transverse with increasing age. Circulation 2002, 105, 2092–2098. [Google Scholar] [CrossRef] [Green Version]
- Spach, M.S.; Boineau, J.P. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: A major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. 1997, 20, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Duffy, H.S.; Wit, A.L. Is there a role for remodeled connexins in AF? No simple answers. J. Mol. Cell Cardiol. 2008, 44, 4–13. [Google Scholar] [CrossRef]
- Spach, M.S.; Heidlage, J.F.; Barr, R.C.; Dolber, P.C. Cell size and communication: Role in structural and electrical development and remodeling of the heart. Heart Rhythm. 2004, 1, 500–515. [Google Scholar] [CrossRef]
- Ausma, J.; Litjens, N.; Lenders, M.H.; Duimel, H.; Mast, F.; Wouters, L.; Ramaekers, F.; Allessie, M.; Borgers, M. Time course of atrial fibrillation-induced cellular structural remodeling in atria of the goat. J. Mol. Cell Cardiol. 2001, 33, 2083–2094. [Google Scholar] [CrossRef]
- Ausma, J.; van der Velden, H.; Lenders, M.; van Ankeren, E.; Jongsma, H.; Ramaekers, F.; Borgers, M.; Allessie, M. Reverse structural and gap-junctional remodeling after prolonged atrial fibrillation in the goat. Circulation 2003, 107, 2051–2058. [Google Scholar] [CrossRef]
- Eijsbouts, S.; Ausma, J.; Blaauw, Y.; Schotten, U.; Duytschaever, M.; Allessie, M. Serial cardioversion by class IC Drugs during 4 months of persistent atrial fibrillation in the goat. J. Cardiovasc. Electrophysiol. 2006, 17, 648–654. [Google Scholar] [CrossRef]
- Verheule, S.; Tuyls, E.; van Hunnik, A.; Kuiper, M.; Schotten, U.; Allessie, M. Fibrillatory conduction in the atrial free walls of goats in persistent and permanent atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2010, 3, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Schotten, U.; Smaill, B.; Verheule, S. Loss of Side-to-Side Connections Affects the Relative Contributions of the Sodium and Calcium Current to Transverse Propagation between Strands of Atrial Myocytes. Front. Physiol. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Verheule, S.; Eckstein, J.; Linz, D.; Maesen, B.; Bidar, E.; Gharaviri, A.; Schotten, U. Role of endo-epicardial dissociation of electrical activity and transmural conduction in the development of persistent atrial fibrillation. Prog. Biophys. Mol. Biol. 2014, 115, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Dispersyn, G.D.; Ausma, J.; Thone, F.; Flameng, W.; Vanoverschelde, J.L.; Allessie, M.A.; Ramaekers, F.C.; Borgers, M. Cardiomyocyte remodelling during myocardial hibernation and atrial fibrillation: Prelude to apoptosis. Cardiovasc. Res. 1999, 43, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Cardin, S.; Li, D.; Thorin-Trescases, N.; Leung, T.-K.; Thorin, E.; Nattel, S. Evolution of the atrial fibrillation substrate in experimental congestive heart failure: Angiotensin-dependent and -independent pathways. Cardiovasc. Res. 2003, 60, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Cardin, S.; Libby, E.; Pelletier, P.; Le Bouter, S.; Shiroshita-Takeshita, A.; Le Meur, N.; Léger, J.; Demolombe, S.; Ponton, A.; Glass, L.; et al. Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ. Res. 2007, 100, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Olsen, F.J.; Mogelvang, R.; Modin, D.; Schnohr, P.; Jensen, G.B.; Biering-Sorensen, T. Association between Isometric and Allometric Height-Indexed Left Atrial Size and Atrial Fibrillation. J. Am. Soc. Echocardiogr. 2022, 35, 141–150.e144. [Google Scholar] [CrossRef]
- Boyden, P.; Hoffman, B. The effects on atrial electrophysiology and structure of surgically induced right atrial enlargement in dogs. Circ. Res. 1981, 49, 1319–1331. [Google Scholar] [CrossRef]
- Zhang, Y.; Dedkov, E.I.; Teplitsky, D.; Weltman, N.Y.; Pol, C.J.; Rajagopalan, V.; Lee, B.; Gerdes, A.M. Both hypothyroidism and hyperthyroidism increase atrial fibrillation inducibility in rats. Circ. Arrhythm. Electrophysiol. 2013, 6, 952–959. [Google Scholar] [CrossRef]
- Liu, L.; Yun, F.; Zhao, H.; Zhang, S.; Liu, Z.; Wang, X.; Wang, D.; Peng, W.; Li, S.; Xiu, C.; et al. Atrial sympathetic remodeling in experimental hyperthyroidism and hypothyroidism rats. Int. J. Cardiol. 2015, 187, 148–150. [Google Scholar] [CrossRef]
- Spach, M.S.; Dolber, P.C. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle. Evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ. Res. 1986, 58, 356–371. [Google Scholar] [CrossRef]
- Yue, L.; Melnyk, P.; Gaspo, R.; Wang, Z.; Nattel, S. Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circ. Res. 1999, 84, 776–784. [Google Scholar] [CrossRef]
- Cha, T.J.; Ehrlich, J.R.; Zhang, L.; Nattel, S. Atrial ionic remodeling induced by atrial tachycardia in the presence of congestive heart failure. Circulation 2004, 110, 1520–1526. [Google Scholar] [CrossRef]
- Molina, C.E.; Abu-Taha, I.H.; Wang, Q.; Rosello-Diez, E.; Kamler, M.; Nattel, S.; Ravens, U.; Wehrens, X.H.T.; Hove-Madsen, L.; Heijman, J.; et al. Profibrotic, Electrical, and Calcium-Handling Remodeling of the Atria in Heart Failure Patients with and without Atrial Fibrillation. Front. Physiol. 2018, 9, 1383. [Google Scholar] [CrossRef] [Green Version]
- Geng, M.; Lin, A.; Nguyen, T.P. Revisiting Antiarrhythmic Drug Therapy for Atrial Fibrillation: Reviewing Lessons Learned and Redefining Therapeutic Paradigms. Front. Pharm. 2020, 11, 581837. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.C.; Singer, D.E.; Chang, Y.; Hylek, E.M.; Henault, L.E.; Jensvold, N.G.; Go, A.S. Gender differences in the risk of ischemic stroke and peripheral embolism in atrial fibrillation: The AnTicoagulation and Risk factors in Atrial fibrillation (ATRIA) study. Circulation 2005, 112, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.; Rahman, F.; Schnabel, R.B.; Yin, X.; Benjamin, E.J.; Christophersen, I.E. Atrial fibrillation in women: Epidemiology, pathophysiology, presentation, and prognosis. Nat. Rev. Cardiol. 2016, 13, 321–332. [Google Scholar] [CrossRef]
- Wong, G.R.; Nalliah, C.J.; Lee, G.; Voskoboinik, A.; Chieng, D.; Prabhu, S.; Parameswaran, R.; Sugumar, H.; Al-Kaisey, A.; McLellan, A.; et al. Sex-Related Differences in Atrial Remodeling in Patients with Atrial Fibrillation: Relationship to Ablation Outcomes. Circ. Arrhythm. Electrophysiol. 2022, 15, e009925. [Google Scholar] [CrossRef]
- Patel, D.; Mohanty, P.; Di Biase, L.; Sanchez, J.E.; Shaheen, M.H.; Burkhardt, J.D.; Bassouni, M.; Cummings, J.; Wang, Y.; Lewis, W.R.; et al. Outcomes and complications of catheter ablation for atrial fibrillation in females. Heart Rhythm. 2010, 7, 167–172. [Google Scholar] [CrossRef]
- Kloosterman, M.; Crijns, H.; Mulder, B.A.; Groenveld, H.F.; Van Veldhuisen, D.J.; Rienstra, M.; Van Gelder, I.C. Sex-related differences in risk factors, outcome, and quality of life in patients with permanent atrial fibrillation: Results from the RACE II study. Europace 2020, 22, 1619–1627. [Google Scholar] [CrossRef]
- Cochet, H.; Mouries, A.; Nivet, H.; Sacher, F.; Derval, N.; Denis, A.; Merle, M.; Relan, J.; Hocini, M.; Haissaguerre, M.; et al. Age, atrial fibrillation, and structural heart disease are the main determinants of left atrial fibrosis detected by delayed-enhanced magnetic resonance imaging in a general cardiology population. J. Cardiovasc. Electrophysiol. 2015, 26, 484–492. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Qiao, S.B.; Hu, F.H.; Yuan, J.S.; Yang, W.X.; Cui, J.G.; Zhang, Y.; Zhang, C.L. Left ventricular remodeling and fibrosis: Sex differences and relationship with diastolic function in hypertrophic cardiomyopathy. Eur. J. Radiol. 2015, 84, 1487–1492. [Google Scholar] [CrossRef]
- Akoum, N.; Mahnkopf, C.; Kholmovski, E.G.; Brachmann, J.; Marrouche, N.F. Age and sex differences in atrial fibrosis among patients with atrial fibrillation. Europace 2018, 20, 1086–1092. [Google Scholar] [CrossRef]
- Lip, G.Y.; Nieuwlaat, R.; Pisters, R.; Lane, D.A.; Crijns, H.J. Refining clinical risk stratification for predicting stroke and thromboembolism in atrial fibrillation using a novel risk factor-based approach: The euro heart survey on atrial fibrillation. Chest 2010, 137, 263–272. [Google Scholar] [CrossRef]
- Watson, T.; Shantsila, E.; Lip, G.Y. Mechanisms of thrombogenesis in atrial fibrillation: Virchow’s triad revisited. Lancet 2009, 373, 155–166. [Google Scholar] [CrossRef]
- Lip, G.Y. Does atrial fibrillation confer a hypercoagulable state? Lancet 1995, 346, 1313–1314. [Google Scholar] [CrossRef]
- Schotten, U.; Duytschaever, M.; Ausma, J.; Eijsbouts, S.; Neuberger, H.R.; Allessie, M. Electrical and contractile remodeling during the first days of atrial fibrillation go hand in hand. Circulation 2003, 107, 1433–1439. [Google Scholar] [CrossRef]
- Manning, W.J.; Silverman, D.I.; Katz, S.E.; Riley, M.F.; Come, P.C.; Doherty, R.M.; Munson, J.T.; Douglas, P.S. Impaired left atrial mechanical function after cardioversion: Relation to the duration of atrial fibrillation. J. Am. Coll. Cardiol. 1994, 23, 1535–1540. [Google Scholar] [CrossRef]
- Vincenti, A.; Genovesi, S.; Sonaglioni, A.; Binda, G.; Rigamonti, E.; Lombardo, M.; Anza, C. Mechanical atrial recovery after cardioversion in persistent atrial fibrillation evaluated by bidimensional speckle tracking echocardiography. J. Cardiovasc. Med. 2019, 20, 745–751. [Google Scholar] [CrossRef]
- Fatkin, D.; Kuchar, D.L.; Thorburn, C.W.; Feneley, M.P. Transesophageal echocardiography before and during direct current cardioversion of atrial fibrillation: Evidence for “atrial stunning” as a mechanism of thromboembolic complications. J. Am. Coll. Cardiol. 1994, 23, 307–316. [Google Scholar] [CrossRef]
- Airaksinen, K.E.; Gronberg, T.; Nuotio, I.; Nikkinen, M.; Ylitalo, A.; Biancari, F.; Hartikainen, J.E. Thromboembolic complications after cardioversion of acute atrial fibrillation: The FinCV (Finnish CardioVersion) study. J. Am. Coll. Cardiol. 2013, 62, 1187–1192. [Google Scholar] [CrossRef]
- Spartera, M.; Stracquadanio, A.; Pessoa-Amorim, G.; Von Ende, A.; Fletcher, A.; Manley, P.; Ferreira, V.M.; Hess, A.T.; Hopewell, J.C.; Neubauer, S.; et al. The impact of atrial fibrillation and stroke risk factors on left atrial blood flow characteristics. Eur. Heart J. Cardiovasc. Imaging 2021, 23, 115–123. [Google Scholar] [CrossRef]
- Uematsu, M.; Ohara, Y.; Navas, J.P.; Nishida, K.; Murphy, T.J.; Alexander, R.W.; Nerem, R.M.; Harrison, D.G. Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. Am. J. Physiol. 1995, 269, C1371–C1378. [Google Scholar] [CrossRef]
- Greiser, M.; Neuberger, H.R.; Harks, E.; El-Armouche, A.; Boknik, P.; de Haan, S.; Verheyen, F.; Verheule, S.; Schmitz, W.; Ravens, U.; et al. Distinct contractile and molecular differences between two goat models of atrial dysfunction: AV block-induced atrial dilatation and atrial fibrillation. J. Mol. Cell Cardiol. 2009, 46, 385–394. [Google Scholar] [CrossRef]
- Benjamin, E.J.; D’Agostino, R.B.; Belanger, A.J.; Wolf, P.A.; Levy, D. Left atrial size and the risk of stroke and death. The Framingham Heart Study. Circulation 1995, 92, 835–841. [Google Scholar] [CrossRef]
- Ammash, N.; Konik, E.A.; McBane, R.D.; Chen, D.; Tange, J.I.; Grill, D.E.; Herges, R.M.; McLeod, T.G.; Friedman, P.A.; Wysokinski, W.E. Left atrial blood stasis and Von Willebrand factor-ADAMTS13 homeostasis in atrial fibrillation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2760–2766. [Google Scholar] [CrossRef]
- Fukuchi, M.; Watanabe, J.; Kumagai, K.; Katori, Y.; Baba, S.; Fukuda, K.; Yagi, T.; Iguchi, A.; Yokoyama, H.; Miura, M.; et al. Increased von Willebrand factor in the endocardium as a local predisposing factor for thrombogenesis in overloaded human atrial appendage. J. Am. Coll. Cardiol. 2001, 37, 1436–1442. [Google Scholar] [CrossRef]
- Lip, G.Y.; Blann, A. von Willebrand factor: A marker of endothelial dysfunction in vascular disorders? Cardiovasc. Res. 1997, 34, 255–265. [Google Scholar] [CrossRef]
- Wysokinski, W.E.; Melduni, R.M.; Ammash, N.M.; Vlazny, D.T.; Konik, E.; Saadiq, R.A.; Gosk-Bierska, I.; Slusser, J.; Grill, D.; McBane, R.D. Von Willebrand Factor and ADAMTS13 as Predictors of Adverse Outcomes in Patients with Nonvalvular Atrial Fibrillation. CJC Open 2021, 3, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Nso, N.; Bookani, K.R.; Metzl, M.; Radparvar, F. Role of inflammation in atrial fibrillation: A comprehensive review of current knowledge. J. Arrhythm. 2021, 37, 1–10. [Google Scholar] [CrossRef]
- Korantzopoulos, P.; Letsas, K.P.; Tse, G.; Fragakis, N.; Goudis, C.A.; Liu, T. Inflammation and atrial fibrillation: A comprehensive review. J. Arrhythm. 2018, 34, 394–401. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef]
- Nakamura, Y.; Nakamura, K.; Fukushima-Kusano, K.; Ohta, K.; Matsubara, H.; Hamuro, T.; Yutani, C.; Ohe, T. Tissue factor expression in atrial endothelia associated with nonvalvular atrial fibrillation: Possible involvement in intracardiac thrombogenesis. Thromb. Res. 2003, 111, 137–142. [Google Scholar] [CrossRef]
- Nightingale, T.; Cutler, D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J. Thromb. Haemost. 2013, 11 (Suppl. 1), 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessandro, E.; Posma, J.J.N.; Spronk, H.M.H.; Ten Cate, H. Tissue factor (:Factor VIIa) in the heart and vasculature: More than an envelope. Thromb. Res. 2018, 168, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhang, H.; Guo, L.; Hong, K. Relationship between epicardial adipose tissue volume and atrial fibrillation: A systematic review and meta-analysis. Herz 2016, 41, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Kocyigit, D.; Gurses, K.M.; Yalcin, M.U.; Turk, G.; Evranos, B.; Yorgun, H.; Sahiner, M.L.; Kaya, E.B.; Hazirolan, T.; Tokgozoglu, L.; et al. Periatrial epicardial adipose tissue thickness is an independent predictor of atrial fibrillation recurrence after cryoballoon-based pulmonary vein isolation. J. Cardiovasc. Comput. Tomogr. 2015, 9, 295–302. [Google Scholar] [CrossRef]
- Antonopoulos, A.S.; Margaritis, M.; Verheule, S.; Recalde, A.; Sanna, F.; Herdman, L.; Psarros, C.; Nasrallah, H.; Coutinho, P.; Akoumianakis, I.; et al. Mutual Regulation of Epicardial Adipose Tissue and Myocardial Redox State by PPAR-gamma/Adiponectin Signalling. Circ. Res. 2016, 118, 842–855. [Google Scholar] [CrossRef]
- Mahajan, R.; Nelson, A.; Pathak, R.K.; Middeldorp, M.E.; Wong, C.X.; Twomey, D.J.; Carbone, A.; Teo, K.; Agbaedeng, T.; Linz, D.; et al. Electroanatomical Remodeling of the Atria in Obesity: Impact of Adjacent Epicardial Fat. JACC Clin. Electrophysiol. 2018, 4, 1529–1540. [Google Scholar] [CrossRef]
- Yamashita, T.; Sekiguchi, A.; Suzuki, S.; Ohtsuka, T.; Sagara, K.; Tanabe, H.; Kunihara, T.; Sawada, H.; Aizawa, T. Enlargement of the left atrium is associated with increased infiltration of immune cells in patients with atrial fibrillation who had undergone surgery. J. Arrhythm. 2015, 31, 78–82. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, D.; Xie, X.; Wang, S.; Wang, Z.; Zhao, W.; Xu, H.; Zheng, L. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res. Cardiol. 2016, 111, 63. [Google Scholar] [CrossRef]
- Xia, R.; Tomsits, P.; Loy, S.; Zhang, Z.; Pauly, V.; Schuttler, D.; Clauss, S. Cardiac Macrophages and Their Effects on Arrhythmogenesis. Front. Physiol. 2022, 13, 900094. [Google Scholar] [CrossRef]
- Yue, Y.; Huang, S.; Li, H.; Li, W.; Hou, J.; Luo, L.; Liu, Q.; Wang, C.; Yang, S.; Lv, L.; et al. M2b macrophages protect against myocardial remodeling after ischemia/reperfusion injury by regulating kinase activation of platelet-derived growth factor receptor of cardiac fibroblast. Ann. Transl. Med. 2020, 8, 1409. [Google Scholar] [CrossRef]
- van Nieuwenhoven, F.A.; Turner, N.A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharm. 2013, 58, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Erlich, J.H.; Boyle, E.M.; Labriola, J.; Kovacich, J.C.; Santucci, R.A.; Fearns, C.; Morgan, E.N.; Yun, W.; Luther, T.; Kojikawa, O.; et al. Inhibition of the tissue factor-thrombin pathway limits infarct size after myocardial ischemia-reperfusion injury by reducing inflammation. Am. J. Pathol. 2000, 157, 1849–1862. [Google Scholar] [CrossRef]
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms regulating endothelial permeability. Pulm. Circ. 2014, 4, 535–551. [Google Scholar] [CrossRef]
- Choudhury, A.; Lip, G.Y. Atrial fibrillation and the hypercoagulable state: From basic science to clinical practice. Pathophysiol. Haemost. Thromb. 2003, 33, 282–289. [Google Scholar] [CrossRef]
- Lim, H.S.; Willoughby, S.R.; Schultz, C.; Gan, C.; Alasady, M.; Lau, D.H.; Leong, D.P.; Brooks, A.G.; Young, G.D.; Kistler, P.M.; et al. Effect of atrial fibrillation on atrial thrombogenesis in humans: Impact of rate and rhythm. J. Am. Coll. Cardiol. 2013, 61, 852–860. [Google Scholar] [CrossRef]
- Bartus, K.; Litwinowicz, R.; Natorska, J.; Zabczyk, M.; Undas, A.; Kapelak, B.; Lakkireddy, D.; Lee, R.J. Coagulation factors and fibrinolytic activity in the left atrial appendage and other heart chambers in patients with atrial fibrillation: Is there a local intracardiac prothrombotic state? (HEART-CLOT study). Int. J. Cardiol. 2020, 301, 103–107. [Google Scholar] [CrossRef]
- Hobbelt, A.H.; Spronk, H.M.; Crijns, H.; Ten Cate, H.; Rienstra, M.; Van Gelder, I.C. Prethrombotic State in Young Very Low-Risk Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 69, 1990–1992. [Google Scholar] [CrossRef]
- Kopecky, S.L.; Gersh, B.J.; McGoon, M.D.; Chu, C.P.; Ilstrup, D.M.; Chesebro, J.H.; Whisnant, J.P. Lone atrial fibrillation in elderly persons: A marker for cardiovascular risk. Arch. Intern. Med. 1999, 159, 1118–1122. [Google Scholar] [CrossRef]
- Jahangir, A.; Lee, V.; Friedman, P.A.; Trusty, J.M.; Hodge, D.O.; Kopecky, S.L.; Packer, D.L.; Hammill, S.C.; Shen, W.K.; Gersh, B.J. Long-term progression and outcomes with aging in patients with lone atrial fibrillation: A 30-year follow-up study. Circulation 2007, 115, 3050–3056. [Google Scholar] [CrossRef]
- Overvad, T.F.; Skjoth, F.; Lip, G.Y.; Lane, D.A.; Albertsen, I.E.; Rasmussen, L.H.; Larsen, T.B. Duration of Diabetes Mellitus and Risk of Thromboembolism and Bleeding in Atrial Fibrillation: Nationwide Cohort Study. Stroke 2015, 46, 2168–2174. [Google Scholar] [CrossRef]
- Li, X.; Weber, N.C.; Cohn, D.M.; Hollmann, M.W.; DeVries, J.H.; Hermanides, J.; Preckel, B. Effects of Hyperglycemia and Diabetes Mellitus on Coagulation and Hemostasis. J. Clin. Med. 2021, 10, 2419. [Google Scholar] [CrossRef]
- Javorschi, S.; Richard-Harston, S.; Labrouche, S.; Manciet, G.; Freyburger, G. Relative influence of age and thrombotic history on hemostatic parameters. Thromb. Res. 1998, 91, 241–248. [Google Scholar] [CrossRef]
- Spronk, H.M.; de Jong, A.M.; Crijns, H.J.; Schotten, U.; Van Gelder, I.C.; Ten Cate, H. Pleiotropic effects of factor Xa and thrombin: What to expect from novel anticoagulants. Cardiovasc. Res. 2014, 101, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Ten Cate, H.; Guzik, T.J.; Eikelboom, J.; Spronk, H.M.H. Pleiotropic actions of factor Xa inhibition in cardiovascular prevention: Mechanistic insights and implications for anti-thrombotic treatment. Cardiovasc. Res. 2021, 117, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Rothmeier, A.S.; Ruf, W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal. 2013, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Spronk, H.M.; De Jong, A.M.; Verheule, S.; De Boer, H.C.; Maass, A.H.; Lau, D.H.; Rienstra, M.; van Hunnik, A.; Kuiper, M.; Lumeij, S.; et al. Hypercoagulability causes atrial fibrosis and promotes atrial fibrillation. Eur. Heart J. 2017, 38, 38–50. [Google Scholar] [CrossRef]
- Guo, X.; Kolpakov, M.A.; Hooshdaran, B.; Schappell, W.; Wang, T.; Eguchi, S.; Elliott, K.J.; Tilley, D.G.; Rao, A.K.; Andrade-Gordon, P.; et al. Cardiac Expression of Factor X Mediates Cardiac Hypertrophy and Fibrosis in Pressure Overload. JACC Basic Transl. Sci. 2020, 5, 69–83. [Google Scholar] [CrossRef]
- Matsuura, T.; Soeki, T.; Fukuda, D.; Uematsu, E.; Tobiume, T.; Hara, T.; Kusunose, K.; Ise, T.; Yamaguchi, K.; Yagi, S.; et al. Activated Factor X Signaling Pathway via Protease-Activated Receptor 2 Is a Novel Therapeutic Target for Preventing Atrial Fibrillation. Circ. J. 2021, 85, 1383–1391. [Google Scholar] [CrossRef]
- Kondo, H.; Abe, I.; Fukui, A.; Saito, S.; Miyoshi, M.; Aoki, K.; Shinohara, T.; Teshima, Y.; Yufu, K.; Takahashi, N. Possible role of rivaroxaban in attenuating pressure-overload-induced atrial fibrosis and fibrillation. J. Cardiol. 2018, 71, 310–319. [Google Scholar] [CrossRef]
- D’Alessandro, E.; Scaf, B.; Munts, C.; van Hunnik, A.; Trevelyan, C.J.; Verheule, S.; Spronk, H.M.H.; Turner, N.A.; Ten Cate, H.; Schotten, U.; et al. Coagulation Factor Xa Induces Proinflammatory Responses in Cardiac Fibroblasts via Activation of Protease-Activated Receptor-1. Cells 2021, 10, 2958. [Google Scholar] [CrossRef]
- Brambatti, M.; Connolly, S.J.; Gold, M.R.; Morillo, C.A.; Capucci, A.; Muto, C.; Lau, C.P.; Van Gelder, I.C.; Hohnloser, S.H.; Carlson, M.; et al. Temporal relationship between subclinical atrial fibrillation and embolic events. Circulation 2014, 129, 2094–2099. [Google Scholar] [CrossRef]
- Shi, Y.; Ducharme, A.; Li, D.; Gaspo, R.; Nattel, S.; Tardif, J.C. Remodeling of atrial dimensions and emptying function in canine models of atrial fibrillation. Cardiovasc. Res. 2001, 52, 217–225. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease in Humans | Intervention in Animal Model | Species | Max. Duration | Main Feature of Structural Remodeling | Reference |
|---|---|---|---|---|---|
| Lone AF’ | Rapid atrial pacing | Goat/dog/sheep | 6 months | Myocyte hypertrophy and endomysial fibrosis | Verheule, Circ AE, 2013 [20] |
| Vagal AF | Acetylcholine administration | Dog/sheep perfused atria | Seconds | None; acute model | Schuessler, Circ Res 1992 [21] |
| Post-operative AF | Sterile pericarditis | Dog | 3–4 days | Gap junction redistribution | Ryu, Am J Physiol Heart Circ Physiol, 2007 [22] |
| Congestive heart failure | Rapid ventricular pacing | Dog | 5 weeks | (Replacement) fibrosis | Li, Circ, 1999 [23] |
| Valvular insufficiency | Mitral valve avulsion | Dog | 4 weeks | Heterogeneous fibrosis | Verheule, Circ, 2003 [24] |
| Bi-atrial dilation | AV node ablation | Goat | 4 weeks | Myocyte hypertrophy | Neuberger, Circ, 2005 [25] |
| Coronary artery disease | RA artery ligation | Dog | 8 days | Granulation tissue, replacement fibrosis | Nishida, Circ, 2011 [26] |
| Obesity | High-fat diet | Sheep | 8 months | Fibrosis, adipocyte infiltration | Abed, Heart Rhythm, 2013 [27] |
| Sleep apnea | Tracheal occlusion | Pig | 2 min | None; acute model | Linz, Heart Rhythm, 2011 [28] |
| Hypertension | Prenatal corticosteroid exposure | Sheep | 4 years | Myocyte hypertrophy, myolysis, heterogenous fibrosis | Kistler, Eur Heart J, 2006 [29] |
| Ageing | Wait | Dog | 8 years | Endomysial fibrosis and gap junction redistribution | Koura, Circ, 2002 [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Alessandro, E.; Winters, J.; van Nieuwenhoven, F.A.; Schotten, U.; Verheule, S. The Complex Relation between Atrial Cardiomyopathy and Thrombogenesis. Cells 2022, 11, 2963. https://doi.org/10.3390/cells11192963
D’Alessandro E, Winters J, van Nieuwenhoven FA, Schotten U, Verheule S. The Complex Relation between Atrial Cardiomyopathy and Thrombogenesis. Cells. 2022; 11(19):2963. https://doi.org/10.3390/cells11192963
Chicago/Turabian StyleD’Alessandro, Elisa, Joris Winters, Frans A. van Nieuwenhoven, Ulrich Schotten, and Sander Verheule. 2022. "The Complex Relation between Atrial Cardiomyopathy and Thrombogenesis" Cells 11, no. 19: 2963. https://doi.org/10.3390/cells11192963