
A BET Protein Inhibitor Targeting Mononuclear Myeloid Cells Affects Specific Inflammatory Mediators and Pathways in Crohn’s Disease

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Human Clinical Samples
2.3. Mass Cytometry
2.4. Human PBMCs and Monocyte In Vitro Culture
2.5. Ex Vivo Derived CD Fistula Tract Cells Culture
2.6. RNAseq Transcriptome Analysis
2.7. Statistical Analysis
3. Results
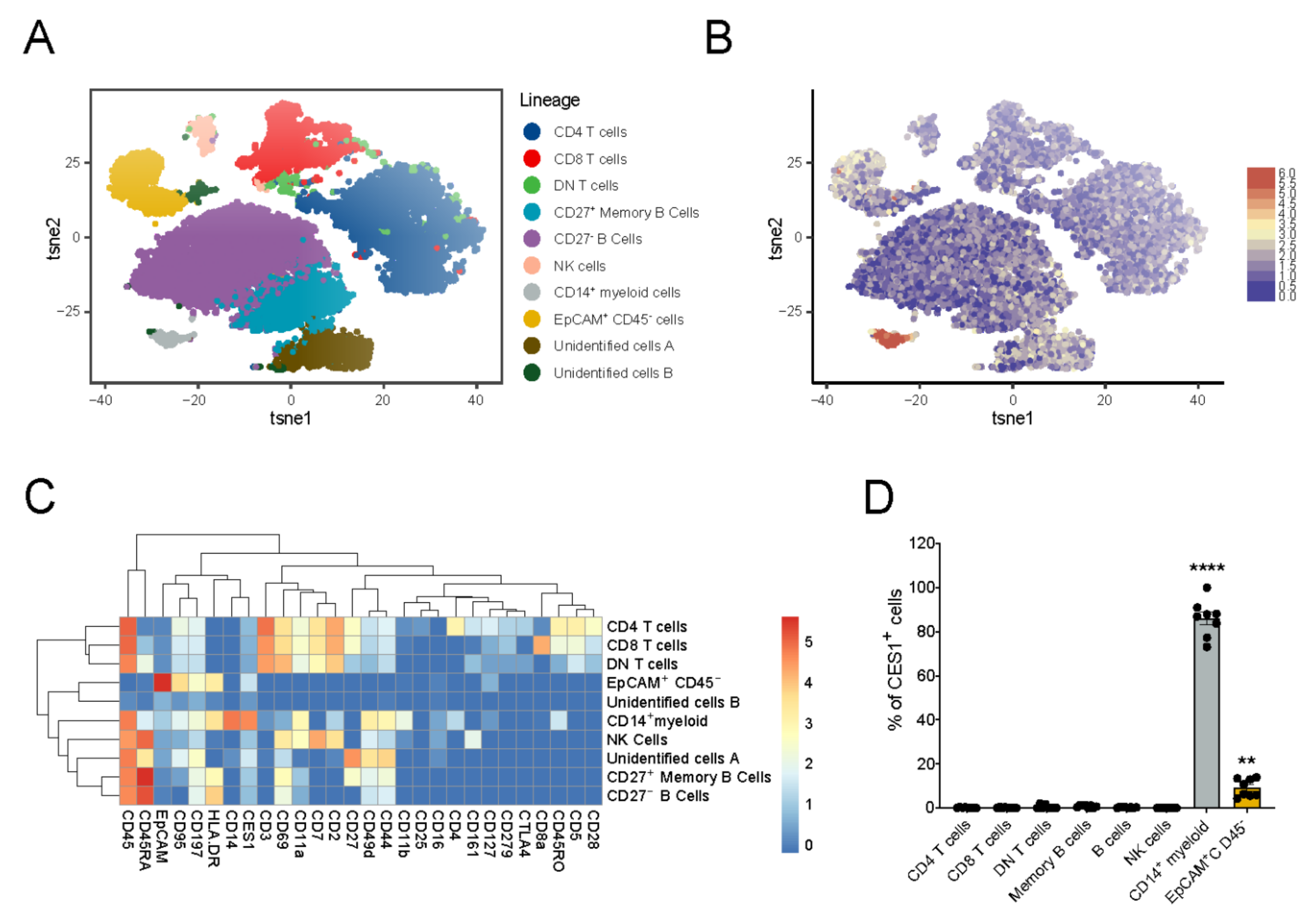
3.1. Immuno-Phenotyping of IBD Intestinal Biopsies Reveals Specific CES1 Expression in CD14+ Myeloid Cell Population
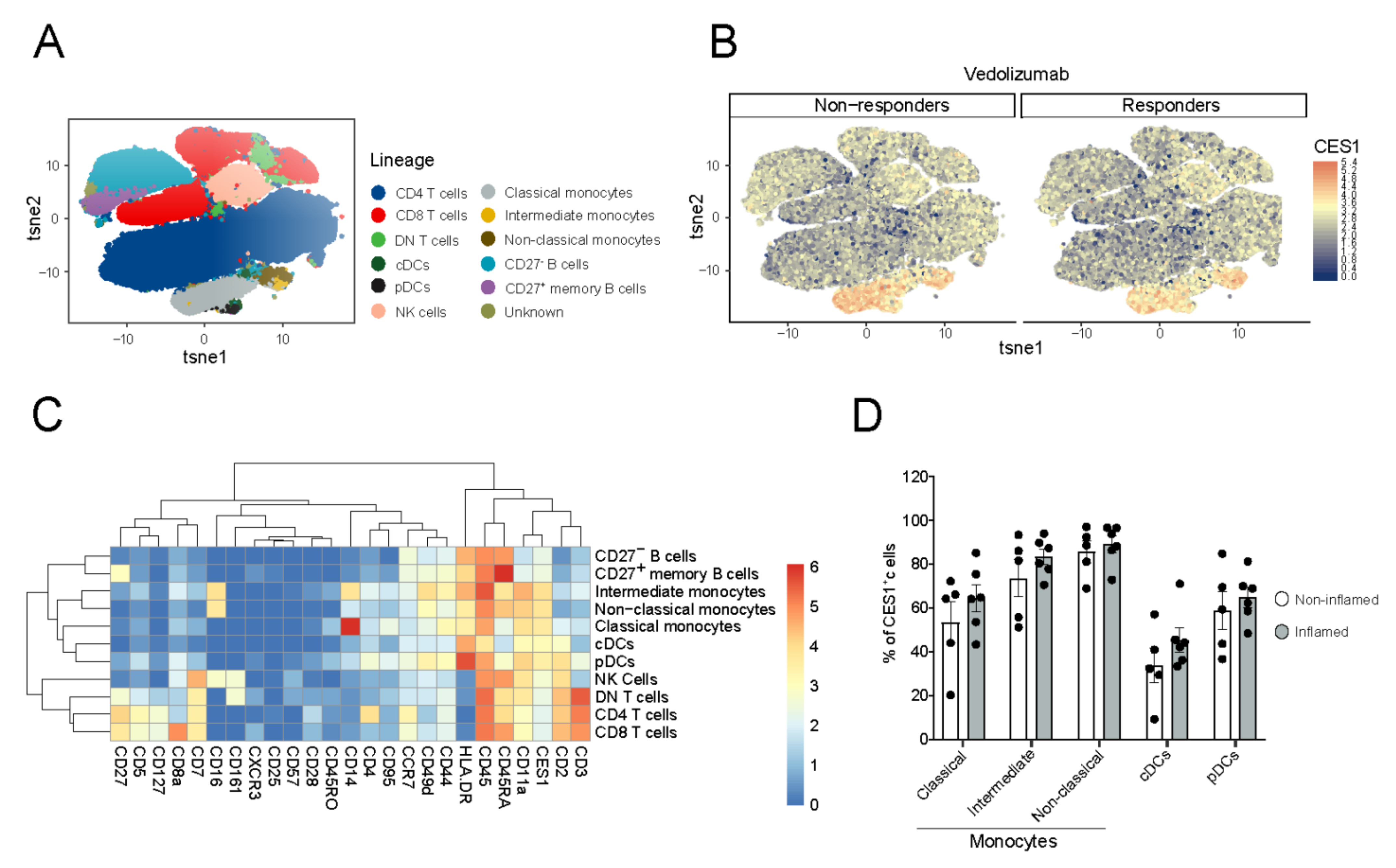
3.2. CES1 Is Expressed in Peripheral Blood Mononuclear Myeloid Cells of CD Patients
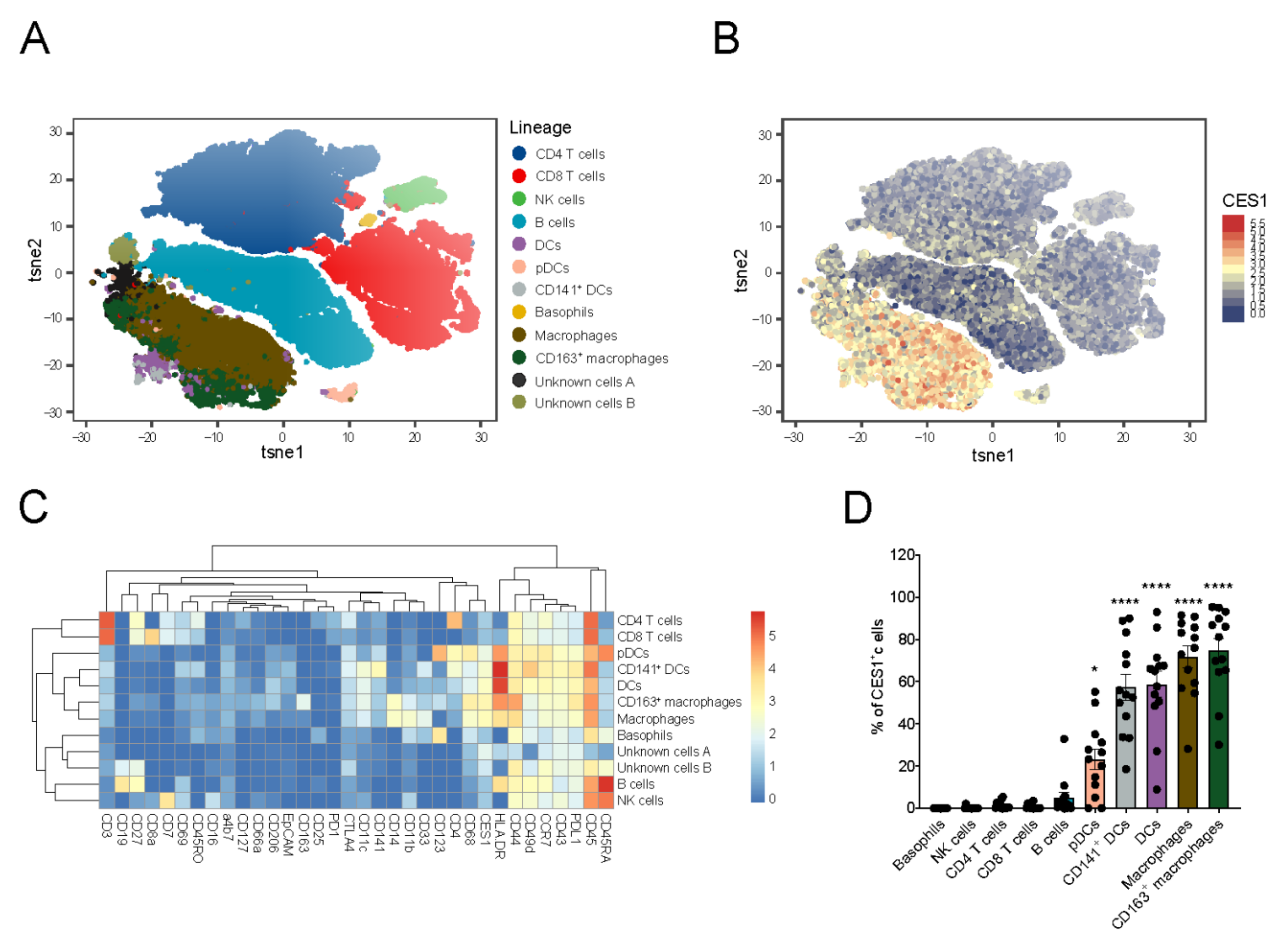
3.3. CES1 Is Expressed within Macrophages and Dendritic Cells Retrieved from Inflamed Fistula Tracts of CD Patients
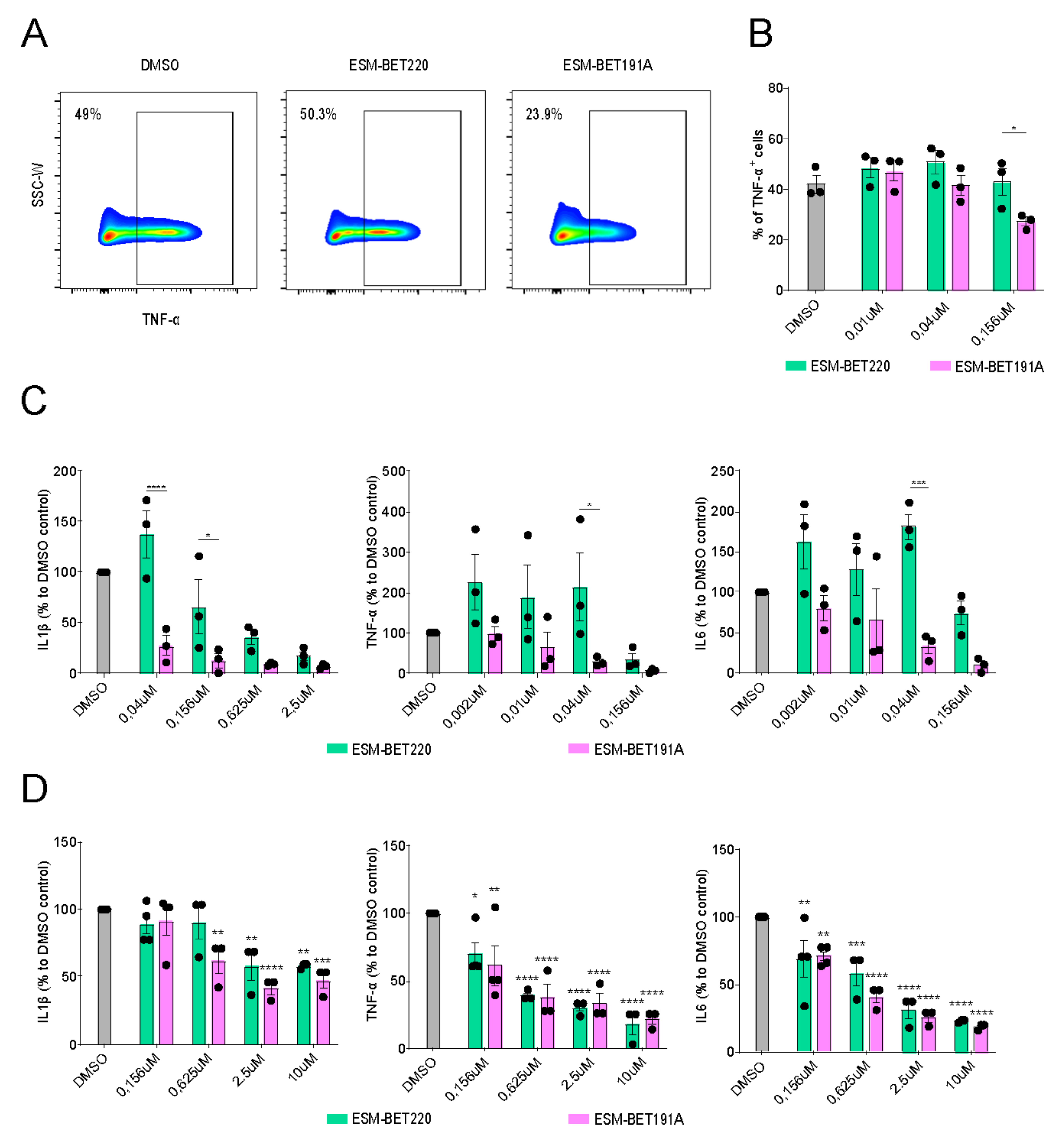
3.4. ESM-iBET Demonstrated an Increased Anti-Inflammatory Effect Compared to Its Non-Hydrolysable Control iBET in Healthy Donors Monocytes
3.5. ESM-iBET and iBET Similarly Inhibited Inflammatory Cytokine Secretion from CD Fistula Tract-Derived Immune Cells in a Dose-Dependent Fashion
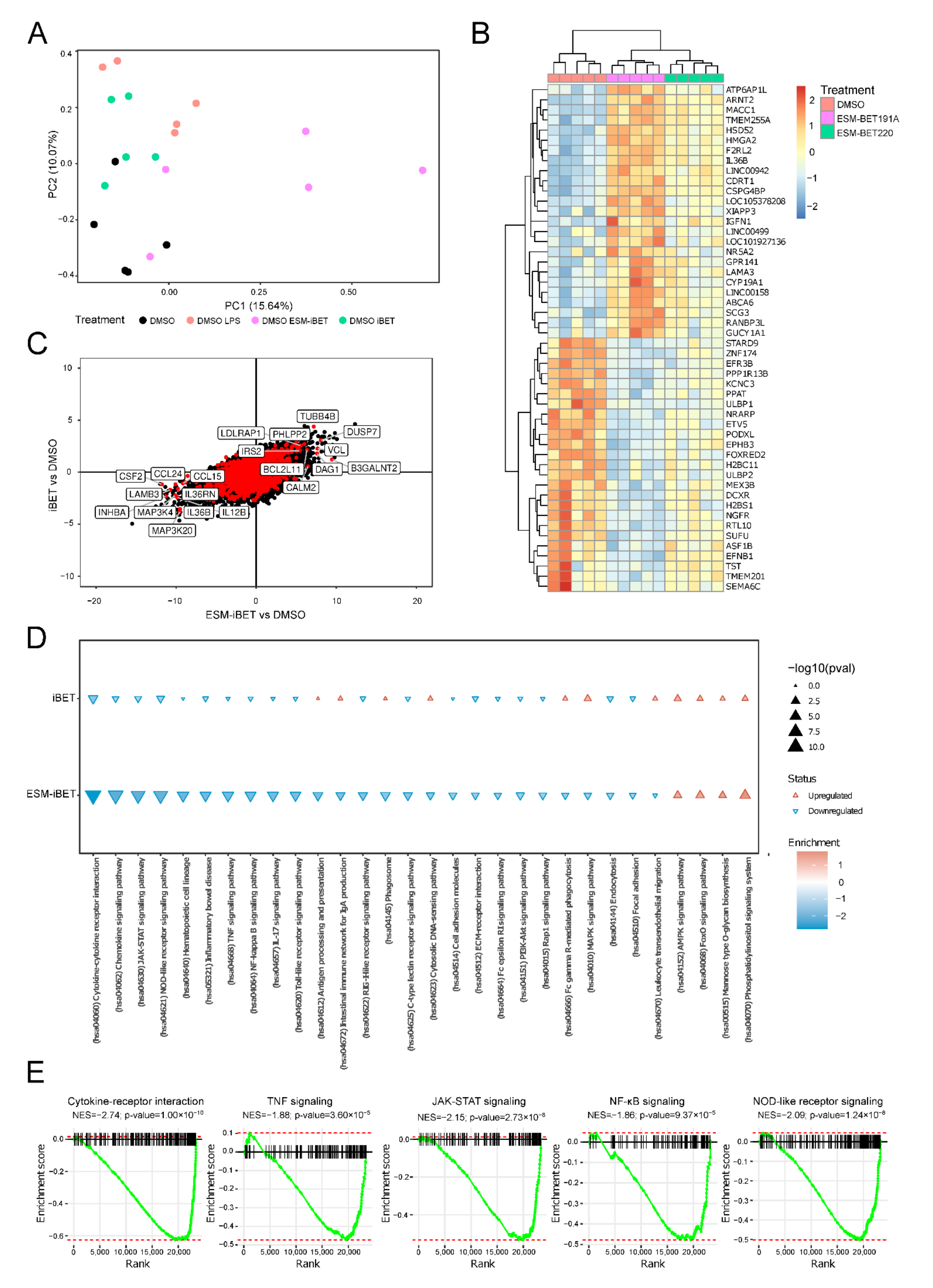
3.6. ESM-iBET Influences the Transcription of Inflammatory Related Genes and Pathways with Increased Potency over iBET in Blood CD14+ Monocytes
3.7. ESM-iBET Potently Modulates Cytokines/Chemokines Transcription in Monocytes
3.8. ESM-iBET Affects Transcription of Key Pathways in CD Pathogenesis, Such as TNFα, JAK-STAT, NF-kB, and NOD2 Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Bonovas, S.; Doherty, G.; Kucharzik, T.; Gisbert, J.P.; Raine, T.; Adamina, M.; Armuzzi, A.; Bachman, O.; Bager, P.; et al. ECCO Guidelines on Therapeutics in Crohn’s Disease: Medical Treatment. J. Crohn’s Colitis 2020, 14, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, G.R.; Loftus, E.V.; Isaacs, K.L.; Regueiro, M.D.; Gerson, L.B.; Sands, B.E. ACG Clinical Guideline: Management of Crohn’s Disease in Adults. Am. J. Gastroenterol. 2018, 113, 481–517. [Google Scholar] [CrossRef] [PubMed]
- Yeshi, K.; Ruscher, R.; Hunter, L.; Daly, N.L.; Loukas, A.; Wangchuk, P. Revisiting Inflammatory Bowel Disease: Pathology, Treatments, Challenges and Emerging Therapeutics Including Drug Leads from Natural Products. J. Clin. Med. 2020, 9, 1273. [Google Scholar] [CrossRef]
- Molendijk, I.; Peeters, K.C.; Baeten, C.I.; Veenendaal, R.A.; van der Meulen-de Jong, A.E. Improving the outcome of fistulising Crohn’s disease. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 505–518. [Google Scholar] [CrossRef]
- Zaboli, P.; Abdollahi, M.; Mozaffari, S.; Nikfar, S. Tumor Necrosis Factor-alpha Antibodies in Fistulizing Crohn’s Disease: An Updated Systematic Review and Meta-analysis. J. Res. Pharm. Pract. 2017, 6, 135–144. [Google Scholar]
- Shaffer, V.O.; Wexner, S.D. Surgical management of Crohn’s disease. Langenbecks Arch. Surg. 2013, 398, 13–27. [Google Scholar] [CrossRef]
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET family in immunity and disease. Signal Transduct Target Ther. 2021, 6, 23. [Google Scholar] [CrossRef]
- Kulikowski, E.; Rakai, B.D.; Wong, N.C.W. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med. Res. Rev. 2021, 41, 223–245. [Google Scholar] [CrossRef]
- Jahagirdar, R.; Attwell, S.; Marusic, S.; Bendele, A.; Shenoy, N.; McLure, K.G.; Gilham, D.; Norek, K.; Hansen, H.C.; Yu, R.; et al. RVX-297, a BET Bromodomain Inhibitor, Has Therapeutic Effects in Preclinical Models of Acute Inflammation and Autoimmune Disease. Mol. Pharmacol. 2017, 92, 694–706. [Google Scholar] [CrossRef]
- Chen, L.; Zhong, X.; Cao, W.; Mao, M.; Li, W.; Yang, H.; Li, M.; Shi, M.; Zhang, Y.; Deng, Y.; et al. JQ1 as a BRD4 Inhibitor Blocks Inflammatory Pyroptosis-Related Acute Colon Injury Induced by LPS. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Cheung, K.; Lu, G.; Sharma, R.; Vincek, A.; Zhang, R.; Plotnikov, A.N.; Zhang, F.; Zhang, Q.; Ju, Y.; Hu, Y.; et al. BET N-terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 2952–2957. [Google Scholar] [CrossRef]
- Wienerroither, S.; Rauch, I.; Rosebrock, F.; Jamieson, A.M.; Bradner, J.; Muhar, M.; Zuber, J.; Müller, M.; Decker, T. Regulation of NO Synthesis, Local Inflammation, and Innate Immunity to Pathogens by BET Family Proteins. Mol. Cell. Biol. 2014, 34, 415–427. [Google Scholar] [CrossRef]
- Bolden, J.E.; Tasdemir, N.; Dow, L.E.; van Es, J.H.; Wilkinson, J.E.; Zhao, Z.; Clevers, H.; Lowe, S.W. Inducible In Vivo Silencing of Brd4 Identifies Potential Toxicities of Sustained BET Protein Inhibition. Cell Rep. 2014, 8, 1919–1929. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Sun, Y.; Han, J.; Wang, Z.; Li, X.; Sun, Y.; Hu, Z. Safety and Efficacy of Bromodomain and Extra-Terminal Inhibitors for the Treatment of Hematological Malignancies and Solid Tumors: A Systematic Study of Clinical Trials. Front. Pharmacol. 2020, 11, 621093. [Google Scholar] [CrossRef]
- Roboz, G.J.; Desai, P.; Lee, S.; Ritchie, E.K.; Winer, E.S.; DeMario, M.; Brenan, B.; Nuesch, E.; Chesne, E.; Brenan, L.; et al. A dose escalation study of RO6870810/TEN-10 in patients with acute myeloid leukemia and myelodysplastic syndrome. Leuk. Lymphoma 2021, 62, 1740–1748. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Herbschleb, K.; Massard, C.; Woodcock, V.; Soria, J.C.; Walter, A.O.; Ewerton, F.; Poelman, M.; Benson, N.; Ocker, M.; et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: Emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur. J. Cancer 2019, 109, 103–110. [Google Scholar] [CrossRef]
- Needham, L.A.; Davidson, A.H.; Bawden, L.J.; Belfield, A.; Bone, E.A.; Brotherton, D.H.; Bryant, S.; Charlton, M.H.; Clark, V.L.; Davies, S.J.; et al. Drug Targeting to Monocytes and Macrophages Using Esterase-Sensitive Chemical Motifs. J. Pharmacol. Exp. Ther. 2011, 339, 132–142. [Google Scholar] [CrossRef]
- Elfiky, A.M.; Ghiboub, M.; Yim, A.Y.F.L.; Hageman, I.L.; Verhoeff, J.; de Krijger, M.; van Hamersveld, P.H.P.; Welting, O.; Admiraal, I.; Rahman, S.; et al. Carboxylesterase-1 Assisted Targeting of HDAC Inhibitors to Mononuclear Myeloid Cells in Inflammatory Bowel Disease. J. Crohn’s Colitis 2021, 16, 668–681. [Google Scholar] [CrossRef]
- Luque-Martin, R.; Bossche, J.V.D.; Furze, R.C.; Neele, A.E.; van der Velden, S.; Gijbels, M.J.; van Roomen, C.P.; Bernard, S.G.; de Jonge, W.J.; Rioja, I.; et al. Targeting Histone Deacetylases in Myeloid Cells Inhibits Their Maturation and Inflammatory Function with Limited Effects on Atherosclerosis. Front. Pharmacol. 2019, 10, 1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.A.; Bal, J.; Simeoni, M.; Williams, P.; Mander, P.K.; Soden, P.E.; Daga, S.; Fahy, W.A.; Wong, G.K.; Bloomer, J.C.; et al. A randomized study of the safety and pharmacokinetics of GSK3358699, a mononuclear myeloid-targeted bromodomain and extra-terminal domain inhibitor. Br. J. Clin. Pharmacol. 2022, 88, 2140–2155. [Google Scholar] [CrossRef] [PubMed]
- Konnikova, L.; Boschetti, G.; Rahman, A.; Mitsialis, V.; Lord, J.; Richmond, C.; Tomov, V.T.; Gordon, W.; Jelinsky, S.; Canavan, J.; et al. High-dimensional immune phenotyping and transcriptional analyses reveal robust recovery of viable human immune and epithelial cells from frozen gastrointestinal tissue. Mucosal Immunol. 2018, 11, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. A 2015, 87, 636–645. [Google Scholar] [CrossRef]
- El Amir, A.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [PubMed]
- A language and Environment for Statistical Computing. R Foundation for Statistical. Computing. In Team RC, editor. Vienna, Austria. Available online: http://www.R-project.org/2014 (accessed on 12th March 2021).
- Jaatinen, T.; Laine, J. Isolation of Mononuclear Cells from Human Cord Blood by Ficoll-Paque Density Gradient. Curr. Protoc. Stem Cell Biol. 2007, 1, 2A.1.1–2A.1.4. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Sergushichev, A.A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 2016, 60012, 1–9. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Li, H.; Tsokos, G.C. Double-negative T cells in autoimmune diseases. Curr. Opin. Rheumatol. 2020, 33, 163–172. [Google Scholar] [CrossRef]
- See, P.; Dutertre, C.-A.; Chen, J.; Günther, P.; McGovern, N.; Irac, S.E.; Gunawan, M.; Beyer, M.; Händler, K.; Duan, K.; et al. Mapping the human DC lineage through the integration of high-dimensional techniques. Science 2017, 356, eaag3009. [Google Scholar] [CrossRef]
- Newton, A.C.; Trotman, L.C. Turning Off AKT: PHLPP as a Drug Target. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 537–558. [Google Scholar] [CrossRef]
- Ham, J.E.; Oh, E.K.; Kim, D.H.; Choi, S.H. Differential expression profiles and roles of inducible DUSPs and ERK1/2-specific constitutive DUSP6 and DUSP7 in microglia. Biochem. Biophys. Res. Commun. 2015, 467, 254–260. [Google Scholar] [CrossRef]
- Seternes, O.-M.; Kidger, A.M.; Keyse, S.M. Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Et Biophys. Acta 2018, 1866, 124–143. [Google Scholar] [CrossRef]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef]
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S.; Vetrano, S.; Casteele, N.V. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337. [Google Scholar] [CrossRef]
- Schreiner, P.; Neurath, M.F.; Ng, S.C.; El-Omar, E.M.; Sharara, A.I.; Kobayashi, T.; Hisamatsu, T.; Hibi, T.; Rogler, G. Mechanism-Based Treatment Strategies for IBD: Cytokines, Cell Adhesion Molecules, JAK Inhibitors, Gut Flora, and More. Inflamm. Intest. Dis. 2019, 4, 79–96. [Google Scholar] [CrossRef]
- Li, R.; Zhu, S. NLRP6 inflammasome. Mol. Asp. Med. 2020, 76, 100859. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2019, 16, 38–51. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 1–33. [Google Scholar] [CrossRef]
- Jones, G.R.; Bain, C.C.; Fenton, T.M.; Kelly, A.; Brown, S.L.; Ivens, A.C.; Travis, M.A.; Cook, P.C.; MacDonald, A.S. Dynamics of Colon Monocyte and Macrophage Activation during Colitis. Front. Immunol. 2018, 9, 2764. [Google Scholar] [CrossRef]
- Thiesen, S.; Janciauskiene, S.; Uronen-Hansson, H.; Agace, W.; Högerkorp, C.-M.; Spee, P.; Håkansson, K.; Grip, O. CD14hiHLA-DRdimmacrophages, with a resemblance to classical blood monocytes, dominate inflamed mucosa in Crohnˈs disease. J. Leukoc. Biol. 2013, 95, 531–541. [Google Scholar] [CrossRef]
- Wasiak, S.; Dzobo, K.E.; Rakai, B.D.; Kaiser, Y.; Versloot, M.; Bahjat, M.; Stotz, S.C.; Fu, L.; Sweeney, M.; Johansson, J.O.; et al. BET protein inhibitor apabetalone (RVX-208) suppresses pro-inflammatory hyper-activation of monocytes from patients with cardiovascular disease and type 2 diabetes. Clin. Epigenetics 2020, 12, 1–19. [Google Scholar] [CrossRef]
- Jung, K.H.; Das, A.; Chai, J.C.; Kim, S.H.; Morya, N.; Park, K.S.; Lee, Y.S.; Chai, Y.G. RNA sequencing reveals distinct mechanisms underlying BET inhibitor JQ1-mediated modulation of the LPS-induced activation of BV-2 microglial cells. J. Neuroinflammation 2015, 12, 36. [Google Scholar] [CrossRef]
- Baek, M.; Yoo, E.; Choi, H.I.; An, G.Y.; Chai, J.C.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. The BET inhibitor attenuates the inflammatory response and cell migration in human microglial HMC3 cell line. Sci. Rep. 2021, 11, 1–13. [Google Scholar] [CrossRef]
- Das, A.; Chai, J.C.; Yang, C.S.; Lee, Y.S.; Das, N.D.; Jung, K.H.; Chai, Y.G. Dual transcriptome sequencing reveals resistance of TLR4 ligand-activated bone marrow-derived macrophages to inflammation mediated by the BET inhibitor JQ1. Sci. Rep. 2015, 5, 16932. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Gortz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, C.; Fan, C.; Yang, Y.; Yang, X.; Lu, H.; Lu, Q.; Zhu, F.; Xiang, C.; Zhang, Z.; et al. Intervention of oncostatin M-driven mucosal inflammation by berberine exerts therapeutic property in chronic ulcerative colitis. Cell Death Dis. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordon, Y. Cytokines: Oncostatin M—A new target in IBD? Nat. Rev. Immunol. 2017, 17, 280. [Google Scholar] [CrossRef]
- Fenton, T.M.; Kelly, A.; Shuttleworth, E.E.; Smedley, C.; Atakilit, A.; Powrie, F.; Campbell, S.; Nishimura, S.L.; Sheppard, D.; Levison, S.; et al. Inflammatory cues enhance TGFβ activation by distinct subsets of human intestinal dendritic cells via integrin αvβ8. Mucosal Immunol. 2017, 10, 624–634. [Google Scholar] [CrossRef]
- Hilt, Z.T.; Maurya, P.; Tesoro, L.; Pariser, D.N.; Ture, S.K.; Cleary, S.J.; Looney, M.R.; McGrath, K.E.; Morrell, C.N. β2M Signals Monocytes Through Non-Canonical TGFβ Receptor Signal Transduction. Circ. Res. 2021, 128, 655–669. [Google Scholar] [CrossRef]
- Ihara, S.; Hirata, Y.; Koike, K. TGF-β in inflammatory bowel disease: A key regulator of immune cells, epithelium, and the intestinal microbiota. J. Gastroenterol. 2017, 52, 777–787. [Google Scholar] [CrossRef]
- Toniolo, P.A.; Liu, S.; Yeh, J.E.; Moraes-Vieira, P.M.; Walker, S.R.; Vafaizadeh, V.; Barbuto, J.A.M.; Frank, D.A. Inhibiting STAT5 by the BET bromodomain inhibitor JQ1 disrupts human dendritic cell maturation. J. Immunol. 2015, 194, 3180–3190. [Google Scholar] [CrossRef]
- Pinz, S.; Unser, S.; Buob, D.; Fischer, P.; Jobst, B.; Rascle, A. Deacetylase inhibitors repress STAT5-mediated transcription by interfering with bromodomain and extra-terminal (BET) protein function. Nucleic Acids Res. 2015, 43, 3524–3545. [Google Scholar] [CrossRef]
- De Krijger, M.; Hageman, I.L.; Li Yim, A.Y.F.; Verhoeff, J.; Garcia Vallejo, J.J.; van Hamersveld, P.H.P.; Levin, E.; Hakvoort, T.B.M.; Wildenberg, M.E.; Henneman, P.; et al. Epigenetic Signatures Discriminate Patients with Primary Sclerosing Cholangitis and Ulcerative Colitis from Patients with Ulcerative Colitis. Front. Immunol. 2022, 13, 840935. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elfiky, A.M.I.; Hageman, I.L.; Becker, M.A.J.; Verhoeff, J.; Li Yim, A.Y.F.; Joustra, V.W.; Mulders, L.; Fung, I.; Rioja, I.; Prinjha, R.K.; et al. A BET Protein Inhibitor Targeting Mononuclear Myeloid Cells Affects Specific Inflammatory Mediators and Pathways in Crohn’s Disease. Cells 2022, 11, 2846. https://doi.org/10.3390/cells11182846
Elfiky AMI, Hageman IL, Becker MAJ, Verhoeff J, Li Yim AYF, Joustra VW, Mulders L, Fung I, Rioja I, Prinjha RK, et al. A BET Protein Inhibitor Targeting Mononuclear Myeloid Cells Affects Specific Inflammatory Mediators and Pathways in Crohn’s Disease. Cells. 2022; 11(18):2846. https://doi.org/10.3390/cells11182846
Chicago/Turabian StyleElfiky, Ahmed M. I., Ishtu L. Hageman, Marte A. J. Becker, Jan Verhoeff, Andrew Y. F. Li Yim, Vincent W. Joustra, Lieven Mulders, Ivan Fung, Inmaculada Rioja, Rab K. Prinjha, and et al. 2022. "A BET Protein Inhibitor Targeting Mononuclear Myeloid Cells Affects Specific Inflammatory Mediators and Pathways in Crohn’s Disease" Cells 11, no. 18: 2846. https://doi.org/10.3390/cells11182846