
Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation

 , , , ,
, , , ,
Abstract
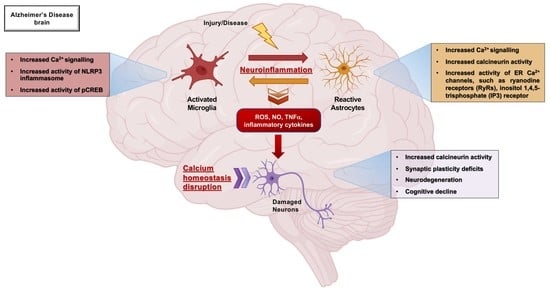
:
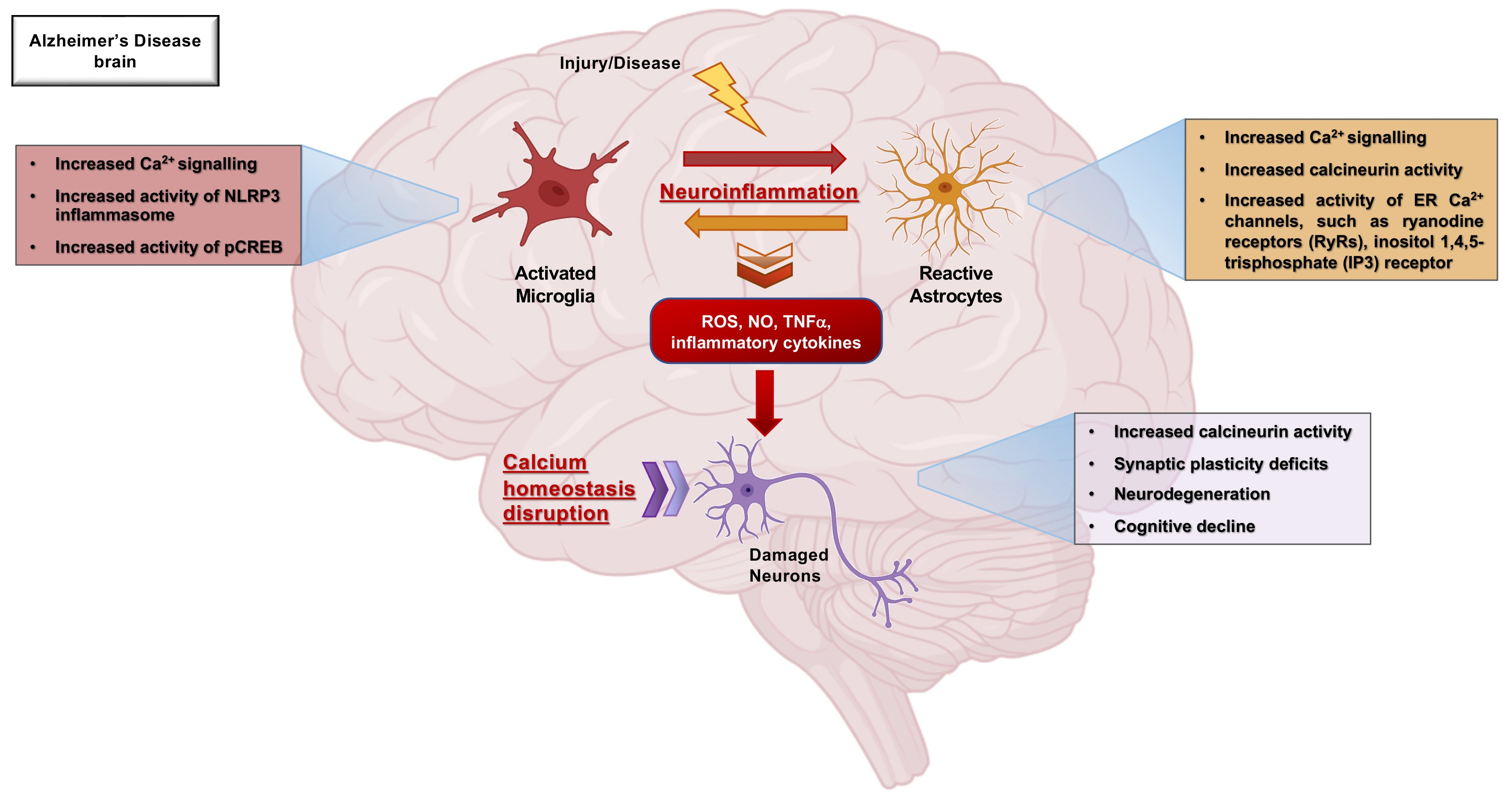{kind=link}
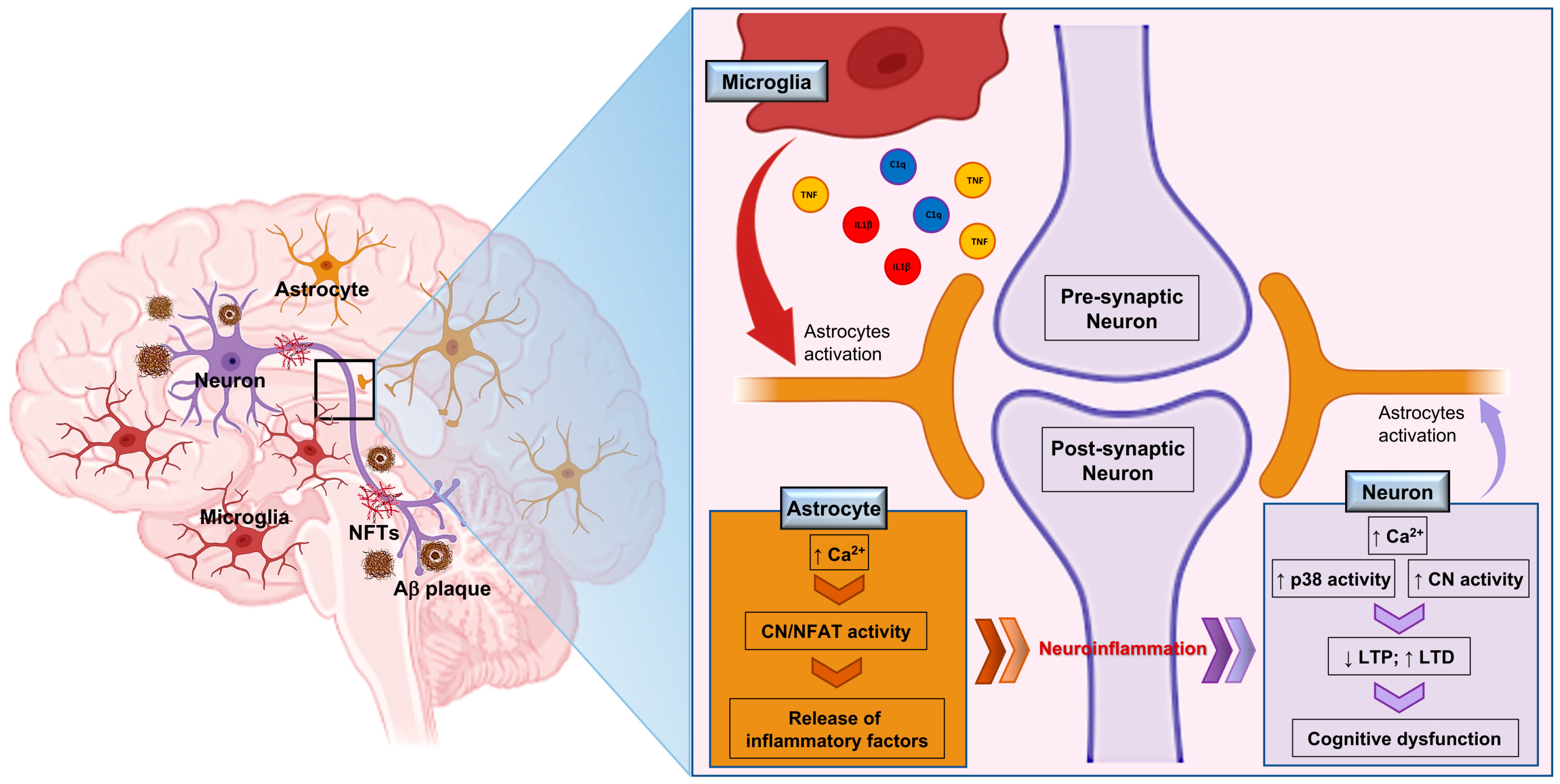{kind=link}
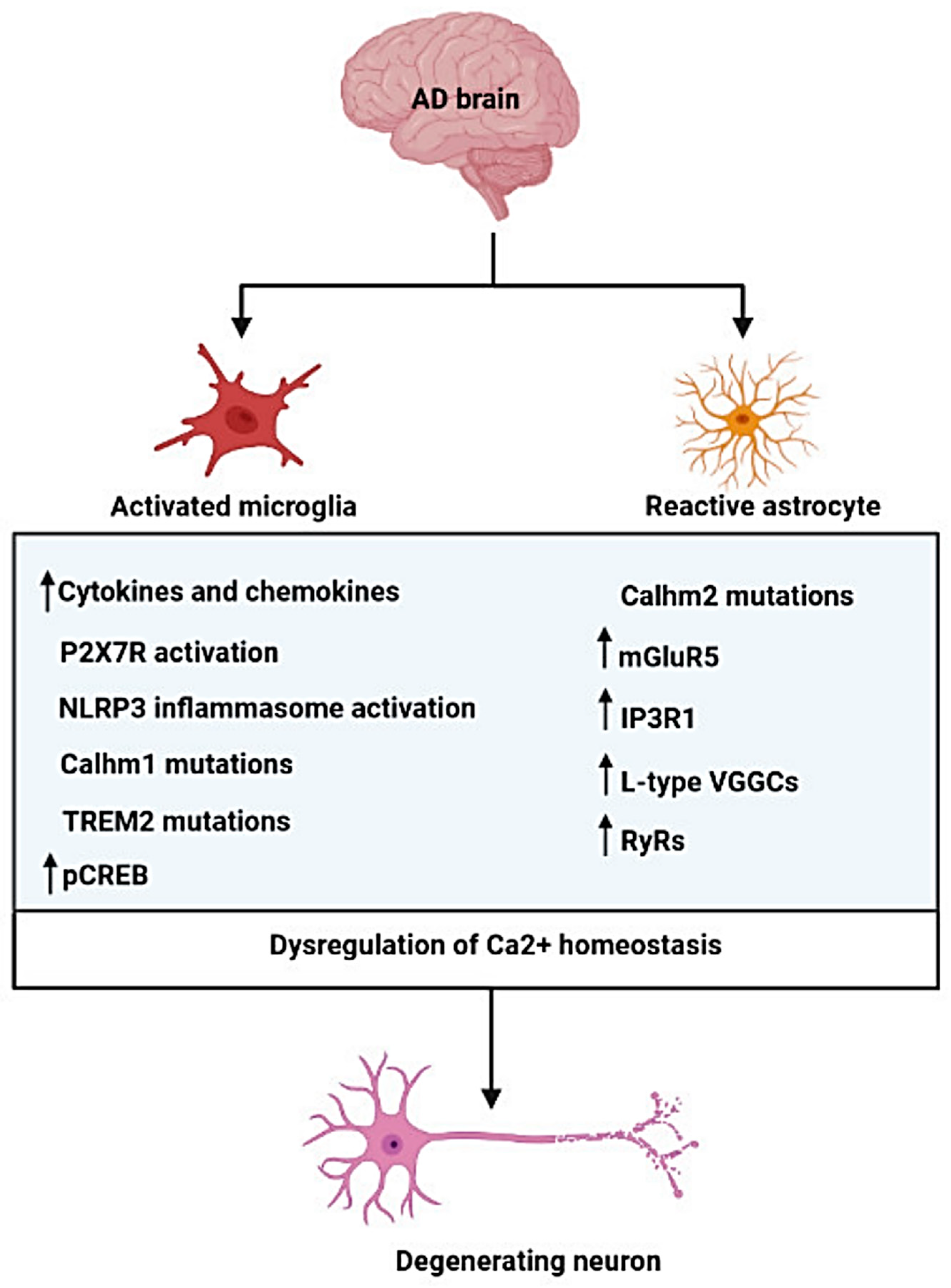{kind=link}
1. Introduction
2. Neuroinflammatory Scenario in Alzheimer’s Disease: The Role of Microglia and Astrocytes
2.1. Microglia and Astrocytes: The Main Actors of the Neuroinflammatory Response
2.2. Microglia: The Sentinels of the Brain
2.3. Astrocyte: A Crucial Regulator of Brain Homeostasis
2.4. Microglia and Astrocytes Activation Profile in Alzheimer’s Disease Is Heterogeneous
2.5. The Neuroinflammatory Cycle: Microglia and Astrocytes Crosstalk in Alzheimer’s Disease and Potential Therapeutic Approaches
3. The Relevance of Ca2+ Homeostasis on Microglia and Astrocytes
3.1. Intracellular Ca2+ Concentration ([Ca2+]i): A Key Read-Out of Glial Activity
3.2. The Versatility of Ca2+ as an Essential Driver of Biological Processes in CNS
3.3. Mechanisms Mediating Ca2+ Exit from the Microglial Cytosol: The Role of Pumps and Exchangers
3.4. Astrocyte Excitability Depends upon Variations of Cytosolic Ca2+ Concentration
4. The Role of Ca2+ Homeostasis Dysfunction in Microglia and Astrocytes in Alzheimer’s Disease
4.1. Microglial Ca2+ Signaling in Alzheimer’s Disease
4.2. Astrocytic Dysregulation of Ca2+ Homeostasis in Alzheimer’s Disease
4.3. How Does Aβ Disrupt Ca2+ Homeostasis?
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Monfared, A.A.T.; Byrnes, M.J.; White, L.A.; Zhang, Q. Alzheimer’s Disease: Epidemiology and Clinical Progression. Neurol. Ther. 2022, 11, 553–569. [Google Scholar] [CrossRef] [PubMed]
- Munafò, A.; Burgaletto, C.; Di Benedetto, G.; Di Mauro, M.; Di Mauro, R.; Bernardini, R.; Cantarella, G. Repositioning of Immunomodulators: A Ray of Hope for Alzheimer’s Disease? Front. Neurosci. 2020, 14, 614643. [Google Scholar] [CrossRef]
- Bonaventura, G.; Munafò, A.; Bellanca, C.; La Cognata, V.; Iemmolo, R.; Attaguile, G.; Di Mauro, R.; Di Benedetto, G.; Cantarella, G.; Barcellona, M.; et al. Stem Cells: Innovative Therapeutic Options for Neurodegenerative Diseases? Cells 2021, 10, 1992. [Google Scholar] [CrossRef]
- Gong, C.-X.; Liu, F.; Iqbal, K. Multifactorial Hypothesis and Multi-Targets for Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, S107–S117. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Onyango, I.; Jauregui, G.; Čarná, M.; Bennett, J.; Stokin, G. Neuroinflammation in Alzheimer’s Disease. Biomedicines 2021, 9, 524. [Google Scholar] [CrossRef]
- Carret-Rebillat, A.-S.; Pace, C.; Gourmaud, S.; Ravasi, L.; Montagne-Stora, S.; Longueville, S.; Tible, M.; Sudol, E.; Chang, R.C.-C.; Paquet, C.; et al. Neuroinflammation and Aβ Accumulation Linked To Systemic Inflammation Are Decreased By Genetic PKR Down-Regulation. Sci. Rep. 2015, 5, srep08489. [Google Scholar] [CrossRef]
- Metcalfe, M.; Figueiredo-Pereira, M.E. Relationship Between Tau Pathology and Neuroinflammation in Alzheimer’s Disease. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2010, 77, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Dá Mesquita, S.; Ferreira, A.C.; Sousa, J.C.; Correia-Neves, M.; Sousa, N.; Marques, F. Insights on the pathophysiology of Alzheimer’s disease: The crosstalk between amyloid pathology, neuroinflammation and the peripheral immune system. Neurosci. Biobehav. Rev. 2016, 68, 547–562. [Google Scholar] [CrossRef]
- Burgaletto, C.; Platania, C.B.M.; Di Benedetto, G.; Munafò, A.; Giurdanella, G.; Federico, C.; Caltabiano, R.; Saccone, S.; Conti, F.; Bernardini, R.; et al. Targeting the miRNA-155/TNFSF10 network restrains inflammatory response in the retina in a mouse model of Alzheimer’s disease. Cell Death Dis. 2021, 12, 905. [Google Scholar] [CrossRef]
- Brawek, B.; Garaschuk, O. Network-wide dysregulation of calcium homeostasis in Alzheimer’s disease. Cell Tissue Res. 2014, 357, 427–438. [Google Scholar] [CrossRef]
- Zotova, E.; Holmes, C.; Johnston, D.; Neal, J.W.; Nicoll, J.A.R.; Boche, D. Microglial alterations in human Alzheimer’s disease following Aβ42 immunization. Neuropathol. Appl. Neurobiol. 2011, 37, 513–524. [Google Scholar] [CrossRef]
- Jung, S.; Schwartz, M. Non-Identical Twins—Microglia and Monocyte-Derived Macrophages in Acute Injury and Autoimmune Inflammation. Front. Immunol. 2012, 3, 89. [Google Scholar] [CrossRef]
- Moreira, E.G.; Boll, K.M.; Correia, D.G.; Soares, J.F.; Rigobello, C.; Maes, M. Why Should Psychiatrists and Neuroscientists Worry about Paraoxonase 1? Curr. Neuropharmacol. 2019, 17, 1004–1020. [Google Scholar] [CrossRef]
- Acosta, C.; Anderson, H.D.; Anderson, C.M. Astrocyte dysfunction in Alzheimer disease. J. Neurosci. Res. 2017, 95, 2430–2447. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Choi, H.B.; Lue, L.-F.; Walker, D.G.; Kim, S.U. Perturbations in calcium-mediated signal transduction in microglia from Alzheimer’s disease patients. J. Neurosci. Res. 2005, 81, 426–435. [Google Scholar] [CrossRef]
- Yu, J.-T.; Chang, R.C.-C.; Tan, L. Calcium dysregulation in Alzheimer’s disease: From mechanisms to therapeutic opportunities. Prog. Neurobiol. 2009, 89, 240–255. [Google Scholar] [CrossRef]
- Sama, D.M.; Norris, C.M. Calcium dysregulation and neuroinflammation: Discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef]
- Price, B.R.; Johnson, L.A.; Norris, C.M. Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101335. [Google Scholar] [CrossRef]
- Tam, K.Y.; Ju, Y. Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2022, 17, 543. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Burgaletto, C.; Munafò, A.; Di Benedetto, G.; De Francisci, C.; Caraci, F.; Di Mauro, R.; Bucolo, C.; Bernardini, R.; Cantarella, G. The immune system on the TRAIL of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 298. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Solfrizzi, V.; Panza, F. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front. Aging Neurosci. 2010, 2, 19. [Google Scholar] [CrossRef]
- E Hickman, S.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.-C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [Green Version]
- Cowan, M.; Petri, W.A.J. Microglia: Immune Regulators of Neurodevelopment. Front. Immunol. 2018, 9, 2576. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Heneka, M.T. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol. 2016, 101, 87–98. [Google Scholar] [CrossRef]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2016, 37, 152–163. [Google Scholar] [CrossRef]
- Simon, E.; Obst, J.; Gomez-Nicola, D. The Evolving Dialogue of Microglia and Neurons in Alzheimer’s Disease: Microglia as Necessary Transducers of Pathology. Neuroscience 2019, 405, 24–34. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; El Khoury, J. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2015, 8, a020560. [Google Scholar] [CrossRef]
- Placone, A.L.; McGuiggan, P.M.; Bergles, D.; Guerrero-Cazares, H.; Quiñones-Hinojosa, A.; Searson, P.C. Human astrocytes develop physiological morphology and remain quiescent in a novel 3D matrix. Biomaterials 2014, 42, 134–143. [Google Scholar] [CrossRef]
- Patabendige, A.; Singh, A.; Jenkins, S.; Sen, J.; Chen, R. Astrocyte Activation in Neurovascular Damage and Repair Following Ischaemic Stroke. Int. J. Mol. Sci. 2021, 22, 4280. [Google Scholar] [CrossRef]
- Pivoriūnas, A.; Verkhratsky, A. Astrocyte–Endotheliocyte Axis in the Regulation of the Blood–Brain Barrier. Neurochem. Res. 2021, 46, 2538–2550. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J. Astrocytes to the rescue! Glia limitans astrocytic endfeet control CNS inflammation. J. Clin. Investig. 2017, 127, 2897–2899. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Hunter, J.; Quadri, Z.; Zamudio, F.; Rocha-Rangel, P.V.; Chan, D.; Kesarwani, A.; Nash, K.; Lee, D.C.; Morgan, D.; et al. CCL2 Overexpression in the Brain Promotes Glial Activation and Accelerates Tau Pathology in a Mouse Model of Tauopathy. Front. Immunol. 2020, 11, 997. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflammation 2011, 8, 150. [Google Scholar] [CrossRef]
- Monterey, M.D.; Wei, H.; Wu, X.; Wu, J.Q. The Many Faces of Astrocytes in Alzheimer’s Disease. Front. Neurol. 2021, 12, 619626. [Google Scholar] [CrossRef]
- Bouvier, D.S.; Jones, E.V.; Quesseveur, G.; Davoli, M.A.; Ferreira, T.; Quirion, R.; Mechawar, N.; Murai, K.K. High Resolution Dissection of Reactive Glial Nets in Alzheimer’s Disease. Sci. Rep. 2016, 6, 24544. [Google Scholar] [CrossRef]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Vandenbark, A.A.; Offner, H.; Matejuk, S.; Matejuk, A. Microglia and astrocyte involvement in neurodegeneration and brain cancer. J. Neuroinflammation 2021, 18, 298. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Thrash, C.; Walter, B. The cellular response in neuroinflammation: The role of leukocytes, microglia and astrocytes in neuronal death and survival. Clin. Neurosci. Res. 2006, 6, 237–245. [Google Scholar] [CrossRef]
- Jha, M.K.; Jo, M.; Kim, J.-H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neurosci. 2018, 25, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, G.; Lempereur, L.; D’Alcamo, M.A.; Risuglia, N.; Cardile, V.; Pennisi, G.; Scoto, G.M.; Bernardini, R. Trail interacts redundantly with nitric oxide in rat astrocytes: Potential contribution to neurodegenerative processes. J. Neuroimmunol. 2007, 182, 41–47. [Google Scholar] [CrossRef]
- Guerriero, F.; Sgarlata, C.; Francis, M.; Maurizi, N.; Faragli, A.; Perna, S.; Rondanelli, M.; Rollone, M.; Ricevuti, G. Neuroinflammation, immune system and Alzheimer disease: Searching for the missing link. Aging Clin. Exp. Res. 2017, 29, 821–831. [Google Scholar] [CrossRef]
- Li, T.; Lu, L.; Pember, E.; Li, X.; Zhang, B.; Zhu, Z. New Insights into Neuroinflammation Involved in Pathogenic Mechanism of Alzheimer’s Disease and Its Potential for Therapeutic Intervention. Cells 2022, 11, 1925. [Google Scholar] [CrossRef]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Price, B.R.; Sudduth, T.L.; Weekman, E.M.; Johnson, S.; Hawthorne, D.; Woolums, A.; Wilcock, D.M. Therapeutic Trem2 activation ameliorates amyloid-beta deposition and improves cognition in the 5XFAD model of amyloid deposition. J. Neuroinflammation 2020, 17, 238. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Sun, C.; Ma, Y.; Wang, S.; Wang, X.; Zhang, Y. Inhibition of TLR4 Induces M2 Microglial Polarization and Provides Neuroprotection via the NLRP3 Inflammasome in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 444. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Araiz, A.; Finucane, O.M.; Keogh, S.; Lynch, M. Anti-TLR2 antibody triggers oxidative phosphorylation in microglia and increases phagocytosis of β-amyloid. J. Neuroinflammation 2018, 15, 247. [Google Scholar] [CrossRef]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Calcium signalling in glial cells. Neurophysiology 1997, 29, 205–212. [Google Scholar] [CrossRef]
- Finkbeiner, S.M. Glial calcium. Glia 1993, 9, 83–104. [Google Scholar] [CrossRef]
- Zündorf, G.; Reiser, G. Calcium Dysregulation and Homeostasis of Neural Calcium in the Molecular Mechanisms of Neurodegenerative Diseases Provide Multiple Targets for Neuroprotection. Antioxidants Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- Lim, D.; Ronco, V.; Grolla, A.A.; Verkhratsky, A.; Genazzani, A.A. Glial Calcium Signalling in Alzheimer’s Disease. Rev. Physiol. Biochem. Pharmacol. 2014, 167, 45–65. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.D.S. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef]
- Rieder, P.; Gobbo, D.; Stopper, G.; Welle, A.; Damo, E.; Kirchhoff, F.; Scheller, A. Astrocytes and Microglia Exhibit Cell-Specific Ca2+ Signaling Dynamics in the Murine Spinal Cord. Front. Mol. Neurosci. 2022, 15, 840948. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Mattson, M.P. Endoplasmic Reticulum Ca2+ Handling in Excitable Cells in Health and Disease. Pharmacol. Rev. 2011, 63, 700–727. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Parpura, V. Calcium signalling and calcium channels: Evolution and general principles. Eur. J. Pharmacol. 2013, 739, 1–3. [Google Scholar] [CrossRef]
- Perea, G.; Sur, M.; Araque, A. Neuron-glia networks: Integral gear of brain function. Front. Cell. Neurosci. 2014, 8, 378. [Google Scholar] [CrossRef]
- Araque, A.; Navarrete, M. Glial cells in neuronal network function. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2375–2381. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Kettenmann, H. Calcium signalling in glial cells. Trends Neurosci. 1996, 19, 346–352. [Google Scholar] [CrossRef]
- Vernadakis, A. GLIA-NEURON INTERCOMMUNICATIONS AND SYNAPTIC PLASTICITY. Prog. Neurobiol. 1996, 49, 185–214. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Mizoguchi, Y.; Monji, A. Microglial Intracellular Ca2+ Signaling in Synaptic Development and its Alterations in Neurodevelopmental Disorders. Front. Cell. Neurosci. 2017, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Ping, L. Calcium ion influx in microglial cells: Physiological and therapeutic significance. J. Neurosci. Res. 2014, 92, 409–423. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Ruiz, A.; Matute, C.; Alberdi, E. Intracellular Ca2+ release through ryanodine receptors contributes to AMPA receptor-mediated mitochondrial dysfunction and ER stress in oligodendrocytes. Cell Death Dis. 2010, 1, e54. [Google Scholar] [CrossRef] [PubMed]
- Ogunbayo, O.A.; Zhu, Y.; Rossi, D.; Sorrentino, V.; Ma, J.; Zhu, M.X.; Evans, A.M. Cyclic Adenosine Diphosphate Ribose Activates Ryanodine Receptors, whereas NAADP Activates Two-pore Domain Channels. J. Biol. Chem. 2011, 286, 9136–9140. [Google Scholar] [CrossRef] [PubMed]
- Klegeris, A.; Choi, H.B.; McLarnon, J.G.; McGeer, P.L. Functional ryanodine receptors are expressed by human microglia and THP-1 cells: Their possible involvement in modulation of neurotoxicity. J. Neurosci. Res. 2007, 85, 2207–2215. [Google Scholar] [CrossRef]
- Franco, L.; Bodrato, N.; Moreschi, I.; Usai, C.; Bruzzone, S.; Scarfì, S.; Zocchi, E.; De Flora, A. Cyclic ADP-ribose is a second messenger in the lipopolysaccharide-stimulated activation of murine N9 microglial cell line. J. Neurochem. 2006, 99, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H. Linking NAADP to Ion Channel Activity: A Unifying Hypothesis. Sci. Signal. 2012, 5, pe18. [Google Scholar] [CrossRef]
- Galione, A. NAADP Receptors. Cold Spring Harb. Perspect. Biol. 2019, 11, a035071. [Google Scholar] [CrossRef]
- Mayo, L.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Moutin, M.-J.; Lund, F.E.; Stein, R. Dual Role of CD38 in Microglial Activation and Activation-Induced Cell Death. J. Immunol. 2008, 181, 92–103. [Google Scholar] [CrossRef]
- Bodrato, N.; Franco, L.; Fresia, C.; Guida, L.; Usai, C.; Salis, A.; Moreschi, I.; Ferraris, C.; Verderio, C.; Basile, G.; et al. Abscisic Acid Activates the Murine Microglial Cell Line N9 through the Second Messenger Cyclic ADP-ribose. J. Biol. Chem. 2009, 284, 14777–14787. [Google Scholar] [CrossRef] [Green Version]
- Brawek, B.; Schwendele, B.; Riester, K.; Kohsaka, S.; Lerdkrai, C.; Liang, Y.; Garaschuk, O. Impairment of in vivo calcium signaling in amyloid plaque-associated microglia. Acta Neuropathol. 2014, 127, 495–505. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The Plasma Membrane Ca2+ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb. Perspect. Biol. 2010, 3, a004168. [Google Scholar] [CrossRef]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The Plasma Membrane Calcium ATPasesand Their Role as Major New Players in Human Disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef]
- Wu, X.; Weng, L.; Zhang, J.; Liu, X.; Huang, J. The Plasma Membrane Calcium ATPases in Calcium Signaling Network. Curr. Protein Pept. Sci. 2018, 19, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, P.; Pannaccione, A.; Sisalli, M.J.; Secondo, A.; Cuomo, O.; Sirabella, R.; Cantile, M.; Ciccone, R.; Scorziello, A.; di Renzo, G.; et al. A New Cell-penetrating Peptide That Blocks the Autoinhibitory XIP Domain of NCX1 and Enhances Antiporter Activity. Mol. Ther. 2015, 23, 465–476. [Google Scholar] [CrossRef]
- Di Benedetto, G.; Valerio, O.; Lariccia, V.; Burgaletto, C.; Lempereur, L.; Parenti, C.; Zanghì, G.N.; Matteucci, A.; Amoroso, S.; Bernardini, R.; et al. Tumor necrosis factor-related apoptosis-inducing ligand reduces the expression of the neuroprotective Na+/Ca2+ exchanger isoform NCX 3 in human neuroblastoma SH—SY 5Y cells. FEBS J. 2018, 286, 737–749. [Google Scholar] [CrossRef]
- Newell, E.W.; Stanley, E.F.; Schlichter, L.C. Reversed Na+/Ca2+ Exchange Contributes to Ca2+ Influx and Respiratory Burst in Microglia. Channels 2007, 1, 366–376. [Google Scholar] [CrossRef]
- Sontheimer, H. Voltage-dependent ion channels in glial cells. Glia 1994, 11, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Cornell-Bell, A.H.; Finkbeiner, S.M.; Cooper, M.S.; Smith, S.J. Glutamate Induces Calcium Waves in Cultured Astrocytes: Long-Range Glial Signaling. Science 1990, 247, 470–473. [Google Scholar] [CrossRef]
- Di Castro, M.A.; Chuquet, J.; Liaudet, N.; Bhaukaurally, K.; Santello, M.; Bouvier, D.; Tiret, P.; Volterra, A. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat. Neurosci. 2011, 14, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gonzalo, M.; Navarrete, M.; Perea, G.; Covelo, A.; Martín-Fernández, M.; Shigemoto, R.; Luján, R.; Araque, A. Endocannabinoids Induce Lateral Long-Term Potentiation of Transmitter Release by Stimulation of Gliotransmission. Cereb. Cortex 2014, 25, 3699–3712. [Google Scholar] [CrossRef] [PubMed]
- Panatier, A.; Theodosis, D.T.; Mothet, J.-P.; Touquet, B.; Pollegioni, L.; Poulain, D.A.; Oliet, S.H. Glia-Derived d-Serine Controls NMDA Receptor Activity and Synaptic Memory. Cell 2006, 125, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Takata, N.; Mishima, T.; Hisatsune, C.; Nagai, T.; Ebisui, E.; Mikoshiba, K.; Hirase, H. Astrocyte Calcium Signaling Transforms Cholinergic Modulation to Cortical Plasticity In Vivo. J. Neurosci. 2011, 31, 18155–18165. [Google Scholar] [CrossRef] [PubMed]
- Metea, M.R. Glial Cells Dilate and Constrict Blood Vessels: A Mechanism of Neurovascular Coupling. J. Neurosci. 2006, 26, 2862–2870. [Google Scholar] [CrossRef]
- Mulligan, S.J.; MacVicar, B. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nat. 2004, 431, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Tian, G.-F.; Peng, W.; Lou, N.; Libionka, W.; Han, X.; Nedergaard, M. Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 2005, 9, 260–267. [Google Scholar] [CrossRef]
- Zonta, M.; Sebelin, A.; Gobbo, S.; Fellin, T.; Pozzan, T.; Carmignoto, G. Glutamate-mediated cytosolic calcium oscillations regulate a pulsatile prostaglandin release from cultured rat astrocytes. J. Physiol. 2003, 553, 407–414. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Fiacco, T.A.; McCarthy, K.D. Multiple Lines of Evidence Indicate That Gliotransmission Does Not Occur under Physiological Conditions. J. Neurosci. 2018, 38, 3–13. [Google Scholar] [CrossRef]
- Savtchouk, I.; Volterra, A. Gliotransmission: Beyond Black-and-White. J. Neurosci. 2018, 38, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Fellin, T.; Pascual, O.; Gobbo, S.; Pozzan, T.; Haydon, P.G.; Carmignoto, G. Neuronal Synchrony Mediated by Astrocytic Glutamate through Activation of Extrasynaptic NMDA Receptors. Neuron 2004, 43, 729–743. [Google Scholar] [CrossRef] [PubMed]
- Perea, G. Properties of Synaptically Evoked Astrocyte Calcium Signal Reveal Synaptic Information Processing by Astrocytes. J. Neurosci. 2005, 25, 2192–2203. [Google Scholar] [CrossRef] [PubMed]
- Morquette, P.; Verdier, D.; Kadala, A.; Féthière, J.; Philippe, A.G.; Robitaille, R.; Kolta, A. An astrocyte-dependent mechanism for neuronal rhythmogenesis. Nat. Neurosci. 2015, 18, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lou, N.; Xu, Q.; Tian, G.-F.; Peng, W.G.; Han, X.; Kang, J.; Takano, T.; Nedergaard, M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 2006, 9, 816–823. [Google Scholar] [CrossRef]
- Asada, A.; Ujita, S.; Nakayama, R.; Oba, S.; Ishii, S.; Matsuki, N.; Ikegaya, Y. Subtle modulation of ongoing calcium dynamics in astrocytic microdomains by sensory inputs. Physiol. Rep. 2015, 3, e12454. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Semyanov, A.; Zorec, R. Physiology of Astroglial Excitability. Function 2020, 1, zqaa016. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Anderova, M.; Chvatal, A. Differential calcium signalling in neuronal-glial networks. Front. Biosci. 2009, 14, 2004–2016. [Google Scholar] [CrossRef]
- Shigetomi, E.; Saito, K.; Sano, F.; Koizumi, S. Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 996. [Google Scholar] [CrossRef]
- Korvers, L.; Costa, A.D.A.; Mersch, M.; Matyash, V.; Kettenmann, H.; Semtner, M. Spontaneous Ca2+ transients in mouse microglia. Cell Calcium 2016, 60, 396–406. [Google Scholar] [CrossRef]
- Heo, D.K.; Lim, H.M.; Nam, J.H.; Lee, M.G.; Kim, J.Y. Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell. Signal. 2015, 27, 177–186. [Google Scholar] [CrossRef]
- McLarnon, J.G. Purinergic mediated changes in Ca2+ mobilization and functional responses in microglia: Effects of low levels of ATP. J. Neurosci. Res. 2005, 81, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Combs, C.K.; Johnson, D.E.; Cannady, S.B.; Lehman, T.M.; Landreth, G.E. Identification of Microglial Signal Transduction Pathways Mediating a Neurotoxic Response to Amyloidogenic Fragments of β-Amyloid and Prion Proteins. J. Neurosci. 1999, 19, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Hemonnot, A.-L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef]
- Färber, K.; Kettenmann, H. Functional role of calcium signals for microglial function. Glia 2006, 54, 656–665. [Google Scholar] [CrossRef]
- Pan, K.; Garaschuk, O. The role of intracellular calcium-store-mediated calcium signals in in vivo sensor and effector functions of microglia. J. Physiol. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Hoffmann, A.; Kann, O.; Ohlemeyer, C.; Hanisch, U.-K.; Kettenmann, H. Elevation of Basal Intracellular Calcium as a Central Element in the Activation of Brain Macrophages (Microglia): Suppression of Receptor-Evoked Calcium Signaling and Control of Release Function. J. Neurosci. 2003, 23, 4410–4419. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Ryu, M.J.K.; Walker, D.G.; Choi, B.H.B. Upregulated Expression of Purinergic P2X7 Receptor in Alzheimer Disease and Amyloid-β Peptide-Treated Microglia and in Peptide-Injected Rat Hippocampus. J. Neuropathol. Exp. Neurol. 2006, 65, 1090–1097. [Google Scholar] [CrossRef]
- Sanz, J.M.; Chiozzi, P.; Ferrari, D.; Colaianna, M.; Idzko, M.; Falzoni, S.; Fellin, R.; Trabace, L.; Di Virgilio, F. Activation of Microglia by Amyloid β Requires P2X7 Receptor Expression. J. Immunol. 2009, 182, 4378–4385. [Google Scholar] [CrossRef]
- Chiozzi, P.; Sarti, A.C.; Sanz, J.M.; Giuliani, A.L.; Adinolfi, E.; Vultaggio-Poma, V.; Falzoni, S.; Di Virgilio, F. Amyloid β-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci. Rep. 2019, 9, 6475. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Dong, Z.; Song, W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct. Target. Ther. 2020, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; Van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Van Zeller, M.; Dias, D.M.; Sebastião, A.M.; Valente, C.A. NLRP3 Inflammasome: A Starring Role in Amyloid-β- and Tau-Driven Pathological Events in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 83, 939–961. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kanneganti, T.-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Syrjanen, J.L.; Michalski, K.; Chou, T.-H.; Grant, T.; Rao, S.; Simorowski, N.; Tucker, S.J.; Grigorieff, N.; Furukawa, H. Structure and assembly of calcium homeostasis modulator proteins. Nat. Struct. Mol. Biol. 2020, 27, 150–159. [Google Scholar] [CrossRef]
- Dreses-Werringloer, U.; Lambert, J.-C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A Polymorphism in CALHM1 Influences Ca2+ Homeostasis, Aβ Levels, and Alzheimer’s Disease Risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [Green Version]
- Jun, M.; Xiaolong, Q.; Chaojuan, Y.; Ruiyuan, P.; Shukun, W.; Junbing, W.; Liao, Y.; Hong, C.; Jinbo, C.; Rong, W.; et al. Calhm2 governs astrocytic ATP releasing in the development of depression-like behaviors. Mol. Psychiatry 2017, 23, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Dong, Y.; Ma, J.; Pan, R.; Liao, Y.; Kong, X.; Li, X.; Li, S.; Chen, P.; Wang, L.; et al. Microglial Calhm2 regulates neuroinflammation and contributes to Alzheimer’s disease pathology. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- McLarnon, J.G. Microglial Store-operated Calcium Signaling in Health and in Alzheimer’s Disease. Curr. Alzheimer Res. 2021, 17, 1057–1064. [Google Scholar] [CrossRef]
- Deming, Y.; Li, Z.; Benitez, B.A.; Cruchaga, C. Triggering receptor expressed on myeloid cells 2 (TREM2): A potential therapeutic target for Alzheimer disease? Expert Opin. Ther. Targets 2018, 22, 587–598. [Google Scholar] [CrossRef]
- Jairaman, A.; McQuade, A.; Granzotto, A.; Kang, Y.J.; Chadarevian, J.P.; Gandhi, S.; Parker, I.; Smith, I.; Cho, H.; Sensi, S.L.; et al. TREM2 regulates purinergic receptor-mediated calcium signaling and motility in human iPSC-derived microglia. eLife 2022, 11, e73021. [Google Scholar] [CrossRef] [PubMed]
- Vardjan, N.; Verkhratsky, A.; Zorec, R. Astrocytic Pathological Calcium Homeostasis and Impaired Vesicle Trafficking in Neurodegeneration. Int. J. Mol. Sci. 2017, 18, 358. [Google Scholar] [CrossRef] [PubMed]
- Grolla, A.A.; Fakhfouri, G.; Balzaretti, G.; Marcello, E.; Gardoni, F.; Canonico, P.L.; DiLuca, M.; Genazzani, A.A.; Lim, D. Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cells. Neurobiol. Aging 2013, 34, 511–522. [Google Scholar] [CrossRef]
- O Jalonen, T.; Charniga, C.J.; Wielt, D.B. β-Amyloid peptide-induced morphological changes coincide with increased K+ and Cl− channel activity in rat cortical astrocytes. Brain Res. 1997, 746, 85–97. [Google Scholar] [CrossRef]
- Abramov, A.; Canevari, L.; Duchen, M. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef]
- Alberdi, E.; Wyssenbach, A.; Alberdi, M.; Sánchez-Gómez, M.V.; Cavaliere, F.; Rodríguez, J.J.; Verkhratsky, A.; Matute, C. Ca2+-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 2013, 12, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Takano, T.; Han, X.; Deane, R.; Zlokovic, B.; Nedergaard, M. Two-Photon Imaging of Astrocytic Ca2+ Signaling and the Microvasculature in Experimental Mice Models of Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2007, 1097, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Ahmadpour, N.; Kantroo, M.; Stobart, J.L. Extracellular Calcium Influx Pathways in Astrocyte Calcium Microdomain Physiology. Biomolecules 2021, 11, 1467. [Google Scholar] [CrossRef]
- Cho, J.; Huh, Y. Astrocytic Calcium Dynamics Along the Pain Pathway. Front. Cell. Neurosci. 2020, 14, 594216. [Google Scholar] [CrossRef] [PubMed]
- Anekonda, T.S.; Quinn, J.; Harris, C.; Frahler, K.; Wadsworth, T.L.; Woltjer, R.L. L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol. Dis. 2011, 41, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium Signaling and Amyloid Toxicity in Alzheimer Disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar] [CrossRef]
- Guan, P.-P.; Cao, L.-L.; Wang, P. Elevating the Levels of Calcium Ions Exacerbate Alzheimer’s Disease via Inducing the Production and Aggregation of β-Amyloid Protein and Phosphorylated Tau. Int. J. Mol. Sci. 2021, 22, 5900. [Google Scholar] [CrossRef]
- Supnet, C.; Grant, J.; Kong, H.; Westaway, D.; Mayne, M. Amyloid-β-(1-42) Increases Ryanodine Receptor-3 Expression and Function in Neurons of TgCRND8 Mice. J. Biol. Chem. 2006, 281, 38440–38447. [Google Scholar] [CrossRef]
- Skowrońska, K.; Kozłowska, H.; Albrecht, J. Neuron-derived factors negatively modulate ryanodine receptor-mediated calcium release in cultured mouse astrocytes. Cell Calcium 2020, 92, 102304. [Google Scholar] [CrossRef]
- Oules, B.; Del Prete, D.; Greco, B.; Zhang, X.X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Bréchot, P.; Trebak, M.; Checler, F.; et al. Ryanodine Receptor Blockade Reduces Amyloid- Load and Memory Impairments in Tg2576 Mouse Model of Alzheimer Disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef]
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging pathways driving early synaptic pathology in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2016, 483, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Checler, F.; Chami, M. Ryanodine receptors: Physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 2014, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Kulasiri, D.; Samarasinghe, S. Ca2+ dysregulation in the endoplasmic reticulum related to Alzheimer’s disease: A review on experimental progress and computational modeling. Biosystems 2015, 134, 1–15. [Google Scholar] [CrossRef]
- Minicucci, J.; Alfond, M.; Demuro, A.; Gerberry, D.; Latulippe, J. Quantifying the dose-dependent impact of intracellular amyloid beta in a mathematical model of calcium regulation in xenopus oocyte. PLoS ONE 2021, 16, e0246116. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.K.B.; Wu, A.J.; Li, M.; Cheung, K.-H. Calcium signaling in Alzheimer’s disease & therapies. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1865, 1745–1760. [Google Scholar] [CrossRef]
- DeMuro, A.; Parker, I. Cytotoxicity of Intracellular A 42 Amyloid Oligomers Involves Ca2+ Release from the Endoplasmic Reticulum by Stimulated Production of Inositol Trisphosphate. J. Neurosci. 2013, 33, 3824–3833. [Google Scholar] [CrossRef]
- Green, K.N.; LaFerla, F.M. Linking Calcium to Aβ and Alzheimer’s Disease. Neuron 2008, 59, 190–194. [Google Scholar] [CrossRef]
- Paula-Lima, A.C.; Adasme, T.; SanMartín, C.; Sebollela, A.; Hetz, C.; Carrasco, M.A.; Ferreira, S.T.; Hidalgo, C. Amyloid β-Peptide Oligomers Stimulate RyR-Mediated Ca2+ Release Inducing Mitochondrial Fragmentation in Hippocampal Neurons and Prevent RyR-Mediated Dendritic Spine Remodeling Produced by BDNF. Antioxid. Redox Signal. 2011, 14, 1209–1223. [Google Scholar] [CrossRef]
- Shtifman, A.; Ward, C.W.; Laver, D.R.; Bannister, M.L.; Lopez, J.R.; Kitazawa, M.; LaFerla, F.M.; Ikemoto, N.; Querfurth, H.W. Amyloid-β protein impairs Ca2+ release and contractility in skeletal muscle. Neurobiol. Aging 2010, 31, 2080–2090. [Google Scholar] [CrossRef]
- DeMuro, A.; Smith, M.; Parker, I. Single-channel Ca2+ imaging implicates Aβ1–42 amyloid pores in Alzheimer’s disease pathology. J. Cell Biol. 2011, 195, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Ullah, G.; DeMuro, A.; Parker, I.; Pearson, J.E. Analyzing and Modeling the Kinetics of Amyloid Beta Pores Associated with Alzheimer’s Disease Pathology. PLoS ONE 2015, 10, e0137357. [Google Scholar] [CrossRef] [PubMed]
- Fani, G.; Mannini, B.; Vecchi, G.; Cascella, R.; Cecchi, C.; Dobson, C.M.; Vendruscolo, M.; Chiti, F. Aβ Oligomers Dysregulate Calcium Homeostasis by Mechanosensitive Activation of AMPA and NMDA Receptors. ACS Chem. Neurosci. 2021, 12, 766–781. [Google Scholar] [CrossRef] [PubMed]
- Pannuzzo, M. Beta-amyloid pore linked to controlled calcium influx into the cell: A new paradigm for Alzheimer’s Disease. Alzheimer’s Dement. 2021, 18, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Latulippe, J.; Lotito, D.; Murby, D. A mathematical model for the effects of amyloid beta on intracellular calcium. PLoS ONE 2018, 13, e0202503. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.-Y.; Hyman, B.T.; Bacskai, B.J. Aβ Plaques Lead to Aberrant Regulation of Calcium Homeostasis In Vivo Resulting in Structural and Functional Disruption of Neuronal Networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Fontana, I.C.; Zimmer, A.R.; Rocha, A.S.; Gosmann, G.; Souza, D.O.; Lourenco, M.V.; Ferreira, S.T.; Zimmer, E.R. Amyloid-β oligomers in cellular models of Alzheimer’s disease. J. Neurochem. 2020, 155, 348–369. [Google Scholar] [CrossRef]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 4914. [Google Scholar] [CrossRef]
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J. Alzheimer’s Dis. Park. 2017, 7, 374. [Google Scholar] [CrossRef]
- Ge, M.; Zhang, J.; Chen, S.; Huang, Y.; Chen, W.; He, L.; Zhang, Y. Role of Calcium Homeostasis in Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2022, 18, 487–498. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Benedetto, G.; Burgaletto, C.; Bellanca, C.M.; Munafò, A.; Bernardini, R.; Cantarella, G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells 2022, 11, 2728. https://doi.org/10.3390/cells11172728
Di Benedetto G, Burgaletto C, Bellanca CM, Munafò A, Bernardini R, Cantarella G. Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation. Cells. 2022; 11(17):2728. https://doi.org/10.3390/cells11172728
Chicago/Turabian StyleDi Benedetto, Giulia, Chiara Burgaletto, Carlo Maria Bellanca, Antonio Munafò, Renato Bernardini, and Giuseppina Cantarella. 2022. "Role of Microglia and Astrocytes in Alzheimer’s Disease: From Neuroinflammation to Ca2+ Homeostasis Dysregulation" Cells 11, no. 17: 2728. https://doi.org/10.3390/cells11172728