
Bilirubin-Induced Neurological Damage: Current and Emerging iPSC-Derived Brain Organoid Models


Institute for Stem Cell Research and Regenerative Medicine, Faculty of Medicine, Heinrich-Heine University, Moorenstrasse 5, 40225 Dusseldorf, Germany
*
Author to whom correspondence should be addressed.
Cells 2022, 11(17), 2647; https://doi.org/10.3390/cells11172647
Submission received: 23 June 2022
/
Revised: 4 August 2022
/
Accepted: 17 August 2022
/
Published: 25 August 2022
(This article belongs to the Special Issue iPS Cells (iPSCs) for Modelling and Treatment of Human Diseases 2022)
Abstract
:Bilirubin-induced neurological damage (BIND) has been a subject of studies for decades, yet the molecular mechanisms at the core of this damage remain largely unknown. Throughout the years, many in vivo chronic bilirubin encephalopathy models, such as the Gunn rat and transgenic mice, have further elucidated the molecular basis of bilirubin neurotoxicity as well as the correlations between high levels of unconjugated bilirubin (UCB) and brain damage. Regardless of being invaluable, these models cannot accurately recapitulate the human brain and liver system; therefore, establishing a physiologically recapitulating in vitro model has become a prerequisite to unveil the breadth of complexities that accompany the detrimental effects of UCB on the liver and developing human brain. Stem-cell-derived 3D brain organoid models offer a promising platform as they bear more resemblance to the human brain system compared to existing models. This review provides an explicit picture of the current state of the art, advancements, and challenges faced by the various models as well as the possibilities of using stem-cell-derived 3D organoids as an efficient tool to be included in research, drug screening, and therapeutic strategies for future clinical applications.
1. Introduction
Bilirubin is an endogenous toxin that results as a by-product of hemoglobin breakdown. It is often used to diagnose liver and blood diseases and has a complicated metabolism, which is significant in relation to various drug metabolism pathways [1]. Bilirubin is metabolized in the liver by the enzyme encoded by the uridine diphosphate glucuronosyltransferase 1A1 gene (UGT1A1), which conjugates bilirubin to glucuronic acid, making it water-soluble [2]. Being lipophilic, unconjugated bilirubin (UCB) cannot take part in the physiological elimination process and starts accumulating. Conjugation of bilirubin is required for increasing its solubility in plasma, thereby enhancing bilirubin elimination from the body. Furthermore, the high level of UCB can become dangerous and cause various complications.
When bilirubin levels in plasma or serum cross the laboratory reference range due to bilirubin metabolism irregularities, it is diagnosed as hyperbilirubinemia, which can be further categorized as conjugated or unconjugated hyperbilirubinemia (UHB) [3]. Clinical jaundice, for instance, which is caused by neonatal UHB, is a commonly occurring, transitional condition that affects about 85% of newborns in their first week of postnatal life [4,5,6]. UHB is a condition regulated by the albumin-bound UCB. As a consequence, there is enhanced UCB production, reduced conjugation and dysfunctional hepatic uptake [3]. On the other hand, at mildly elevated concentrations, bilirubin has a protective antioxidant-like effect on the body [7,8]. It can neutralize reactive oxygen species (ROS), prevent oxidative damage, and is even necessary for newborns when they face high concentrations of oxygen in the air for the first time [9,10,11,12]. It has been shown that UCB also possesses potent anti-oxidant properties, and modest hyperbilirubinemia may even have health benefits [1]. However, high levels of UCB can pose serious threats, such as severe brain injury, with the possibility of progressing into chronic bilirubin encephalopathy (also referred to as kernicterus) in one in every 100,000 cases, if not treated immediately [6,7]. In cases where it does develop into kernicterus, almost 70% of newborns die within the week and the other 30%, suffer irreversible brain damage [13].
Apart from being an adverse effect of spontaneous neonatal hyperbilirubinemia, BIND may also result from a genetic disorder known as Crigler–Najjar Syndrome (CNS). This life-threatening disorder is caused by the mutation in UGT1A1, which causes a complete or partial defect that prevents the liver from metabolizing bilirubin. This hinders bilirubin conjugation, causing UCB to accumulate in serum and eventually cross the blood–brain barrier, proceeding to deposit in the basal ganglia or cerebellum, thereby resulting in BIND [2,3]. Table 1 offers a comprehensive overview of various clinical indications related to hyperbilirubinemia and the respective targets in the brain.
BIND is not only temporarily disabling but also permanent, and it is usually accompanied by movement disorders as well as hearing loss [4,6]. The targeted damage to the central nervous system reflects the regional topography of bilirubin-induced neuropathology, involving the globus pallidus, subthalamic nucleus, metabolic sector of the hippocampus, hippocampal Cornu Ammonis (CA2) neurons, and Purkinje’s cells of the cerebellar cortex and the brainstem, as well as the oculomotor and ventral cochlear nuclei [14,15,16]. However, the key cellular mechanisms accounting for this well-defined regional topography of bilirubin sensitivity are still unclear. One probable reason could be the lack of efficient and suitable in vitro and in vivo models with consistent and comparable findings [12]. Advanced stem-cell-based studies offer great opportunity to establish 3D in vitro model systems to study neurological complications [17,18,19,20]. On that account, human-induced pluripotent stem cell (iPSC)-derived 2D neuronal cell cultures along with 3D brain organoids present convenient and efficient models to enable deciphering the molecular mechanisms underlying BIND.
1.1. UGT1A1
Glucuronidation is a conjugation reaction in which glucuronic acid, which is produced from the cofactor UDP-glucuronic acid, is covalently bound to a nucleophilic functional group on a substrate [28]. The UGT1A1 gene, or uridine diphosphate glucuronosyltransferase 1A1 gene, is part of the UGT1 locus, which encodes the enzymes that glucoronidate a variety of substrates. This gene plays a crucial role in the glucuronidation pathway by converting bilirubin from an unconjugated (toxic) state to a conjugated (nontoxic) state [2]. Bilirubin is formed as a by-product of the heme catabolic pathway. After hemoglobin is broken down into heme, it is then transformed into biliverdin (BLV) and subsequently into bilirubin. UGT1A1 particularly encodes the enzyme that has the ability to convert small lipophilic molecules such as bilirubin into hydrophilic (water-soluble) molecules that can be easily excreted [29,30]. During bilirubin glucuronidation, glucuronic acid is attached (conjugated) to bilirubin through a bilirubin-UDP-glucuronosyltransferase (B-UGT) enzyme-dependent reaction, as B-UGT1 is the only enzyme capable of glucoronating bilirubin [2]. The glucuronidation process takes place in the liver; therefore, liver cells are the primary source of the B-UGT1 enzyme. Thereafter, the water-soluble conjugated version of bilirubin is dissolved in bile and excreted from the body with solid waste.
UGT1A1 was initially cloned by Ritter et al. in 1991 and is located on chromosome 2q37 [30,31]. The UGT1 locus has 13 unique promoters and alternate first exons, followed by four common exons, designated 2, 3, 4 and 5. Before transcription, one of the first exons and its promoter are spliced to the four common exons. This results in 13 different UDP-glucuronosyltransferases being expressed; however, out of 13 possible genes that can be encoded, the only one responsible for bilirubin conjugation is the one containing the alternate exon A1 [2,30].
Reduced expression and partial or total impairment of the B-UGT1 enzyme is caused by mutations in the UGT1A1 gene’s common or bilirubin-specific domains. This can result in inherited unconjugated bilirubinemia disorders, with the most common ones being Gilbert syndrome, Crigler–Najjar syndrome type I (CNS-I) and Crigler–Najjar syndrome type II (CNS-II), also known as Arias syndrome (Table 1) [4,29,30]. The nature of these mutations varies, resulting in phenotypes that range from moderate, in the case of Gilbert syndrome, to severe in CNS-I [4,29,32,33].
In 2000, Kadakol et al. tabulated more than 50 genetic lesions of UGT1A1 that engender CNS-I and II and presented a correlation of structure to function of UGT1A1 [29]. Building upon that research, almost a decade later, Canu et al. published an explicit list of Gilbert and CNS disease, causing mutations including more than 130 cases. Single-nucleotide changes were liable for around 70% of these alterations, whereas deletions, insertions, and polymorphisms attributed for the remaining 30% of alterations [32]. As UGT1A1 is the crucial player in these diseases, being able to alter its expression in customizable in vitro models could help provide more insights into possible translational treatments.
1.2. Crigler–Najjar Syndrome Type I and II
The most serious form of inherited UHB is Crigler–Najjar syndrome Type-I (CNS-I) [34,35,36]. It is the outcome of the complete aberration of UGT1A1, a very rare autosomal recessive disease, only affecting one in a million individuals [37]. The aberration of UGT1A1 leads to high bilirubin plasma levels and severe jaundice in neonates [2]. Increased bilirubin availability in plasma may result in bilirubin accumulation in the brain, turning into a life-threatening condition known as bilirubin encephalopathy. Crigler–Najjar Syndrome Type-II (CNS-II) and Gilbert syndrome are two milder versions of CNS-I, where UGT1A1 is either partially deficient or altered, resulting in a less severe phenotypic manifestation [37].
To lower the plasma bilirubin level and prevent bilirubin encephalopathy, CNS patients rely on 10–12 h of intensive phototherapy treatment every day. Numerous dermatological disorders have been safely and successfully treated with phototherapy for over 40 years [38]. This treatment uses UV radiation to counteract the pathological changes that characterize inflammatory skin diseases through several mechanisms, such as induction of apoptosis, modification of the cytokine milieu, and immunosuppression. Phototherapy is so effective because through UV radiation, bilirubin is irreversibly photo-altered into lumirubin, a structural isomer that is more water-soluble, less dangerous and can be expelled with bile and urine [38,39]. However, the efficiency of the phototherapy can decrease depending on multiple factors, such as age, thickness of the skin, etc. Conversely, skin thickening is one of the effects obtained from the phototherapy itself, which later decreases the therapeutic efficiency. Additionally, extremely low-birth-weight newborns might face potential toxicity due to aggressive phototherapy [40]. A hemolytic process is indicated with the enhancement of total serum bilirubin level despite intensive phototherapy. Exchange transfusions have also been used to control hyperbilirubinemia at a hazardous level and lower the risk of kernicterus. However, phototherapy has greatly reduced the necessity and demand for exchange transfusion [6]. Another approach to control hyperbilirubinemia and prevent acute bilirubin encephalopathy is intravenous immune globulin therapy. Despite the mechanism being unclear, the immune globulin therapy seems to have biological activity against immune-mediated hemolytic diseases associated with the lowering effect of the immune globulin present on the total serum bilirubin level [6]. Pharmacological compounds may provide a direct protection to the neurons from bilirubin toxicity. CNS-II patients respond quite well to the pharmacological therapies, such as treatment with phenobarbital, whereas CNS-I patients do not. Bilirubin conjugation is increased by the activated phenobarbital enhancer module of the UGT1A1 promoter sequence, thus resulting in enhancement in bilirubin clearance. On the other hand, heme oxygenase inhibitors, such as metallophyrins, can be employed to reduce bilirubin production [41]. Minocycline, which is a tetracycline antibiotic, has shown protective effects in Gunn rat pups against bilirubin-induced neurotoxicity, including neuromotor dysfunction, abnormalities in the auditory pathway and cerebellar hypoplasia [6,42]. Finally, liver transplantation remains the only effective treatment for this life-threatening disease (Table 2) [2,36,43].
2. Unravelling the Mechanisms Underlying BIND
To increase our meagre knowledge of BIND, we must understand the pathophysiology underlying high bilirubin neurotoxicity at the molecular level. The brain is a highly specialized and compartmentalized organ with divergent cell populations consisting of neurons and glia, which comprises astrocytes, oligodendrocytes and microglia [48]. Therefore, the location, source and causal agents of BIND are the primary areas worth investigating, along with the cascade of molecular and cellular events that lead to severe damage.
Autopsy results of jaundiced neonates showed disperse yellow spots in the majority of brain areas, except the basal ganglia and medulla oblongata, while intense coloring was observed in those particular areas [49]. These observations indicate that UCB binds to specific types of neurons compared to others and has distinct sensitivities amongst neurons and glia [50]. Microscopic observations of jaundiced brain sections revealed the presence of bilirubin within neurons, neuronal processes and microglia; however, the contribution of individual neuronal cell types and cell-dependent sensitivity towards bilirubin toxicity are still not clarified [48]. In vitro studies have revealed the mechanisms associated with UCB neurotoxicity [48,50]. An increased impairment of cell function has been observed in astrocytes upon high UCB exposure, while neurons show higher susceptibility to cell death [50]. Astrocytes and microglia also seem to play key roles in activating oxidative stress and inflammatory responses. Investigation into intracellular processes of astrocyte and microglia showed that TNF-alpha and IL-1beta pathways as well as MAPK and NF- κB pathways play a key role in cytokine production and cytotoxicity upon UCB stimulation, resulting in UCB-induced neurotoxicity [5,51]. In vivo and in vitro data indicate oxidative stress to play a major role in cytotoxicity upon highly concentrated (toxic) UCB exposure, while increases in oxidative stress and cytotoxicity were observed in synaptic vesicles, tissue culture cells, and primary cell culture of neurons, astrocytes and oligodendrocytes [5,52,53].
The creation and elimination of bilirubin both result from a sequence of metabolic reactions; therefore, there are distinct ways of limiting the production and degradation of UCB [7]. The heme catabolic pathway primarily regulates bilirubin conjugation, as UCB is the consecutive end-product and UCB is endogenously produced by following this pathway within the majority of cells [54]. Briefly, heme is converted into BLV by heme oxygenase enzyme 1 and 2 (HMOX1, HMOX2), and then BLV reductase (BLVR) converts BLV into UCB. Both HMOX1 and HMOX2 reside in mitochondria, endoplasmic reticulum (ER) and caveolae (membrane micro-domains observed at the interface with the extracellular environment), which might have a correlation with BIND-induced neurocytotoxicity, as various molecular pathways become activated in the course of BIND neurotoxicity. These pathways include inflammation, mitochondrial damage and oxidative stress to ER [5,54,55]. The disturbances in mitochondria and ER usually lead to several additional sequelae, such as neuronal excito-toxicity (a complex process triggered by glutamate receptor activation resulting in dendrite degeneration and cell death), mitochondrial energy failure, increased intracellular calcium concentration and deoxyribonucleic acid (DNA) damage (Figure 1) [16,56,57]. All of these factors may subsequently contribute to neuronal death and bilirubin encephalopathy, leading to kernicterus [5,7,16].
During moderate to severe neonatal jaundice, pre-term newborns show an accelerated susceptibility to UCB toxic effects, which makes prematurity a significant abrasive factor for UCB encephalopathy [58,59]. The first week of postnatal life might be sensitive due to an increased chance of higher amounts of UCB availability in the circulation due to several factors. Consequently, the conjugation probability of UCB is suppressed and the unbound fraction of UCB (free bilirubin) increases [60]. The entry of UCB in the brain is restricted by the blood–brain barrier (BBB), as BBB is composed of tightly jointed microvascular endothelial cells, forming elaborate junctional complexes and providing unique properties by strictly regulating the ions, molecules and cell movement between blood and brain [60,61]. Lower UCB binding capacity and the higher UCB availability facilitate the entrance of free bilirubin by passive or facilitated diffusion into the brain, thus causing a condition of mild or severe hyperbilirubinemia. Further research is required to increase our meagre understanding of bilirubin entrance into the brain and the resulting cytotoxicity [59].
2.1. Bilirubin-Induced Oxidative Stress
Bilirubin plays a dual role depending on the physiological level of its unconjugated form. At very low levels, it acts beneficially as an antioxidant; however, after attaining a given threshold, it becomes toxic [5,62]. The neuroprotective role of bilirubin within a certain range of concentrations has been known for more than two decades to protect neurons from H2O2-induced toxicity [62]. Furthermore, the role of bilirubin as an anti-inflammatory agent and a scavenger of ROS have been intensively studied for a long time [54,63,64,65,66,67]. With the help of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, bilirubin prevents the generation of superoxides, inhibits ROS production and regulates redox homeostasis. This implies that at lower concentrations, bilirubin is potentially involved in several important cellular signaling pathways, such as cell proliferation, apoptosis, inflammation, and immune system upkeep. Moreover, bilirubin has also been proven to be a powerful signaling molecule that can help guard against a variety of disorders linked to elevated levels of oxidative stress [54,68,69].
On the other hand, bilirubin itself is the cause of oxidative stress. Increased oxidative stress activates transcription factor NF- κB and also increases phosphorylation of mitogen-activated protein kinases (MAPKs), therefore resulting in cytokine production and cell toxicity [70]. It is also clear that neurons are more susceptible to oxidative damage than other cell types in the brain such as astrocytes [71]. Bilirubin-induced DNA damage was found to be significantly increased in vitro, when neuronal and non-neuronal cells were exposed to 140 nM of free bilirubin. As potential adaptive responses to repair the damage, bilirubin therapy triggered primary DNA repair pathways through homologous recombination (HR) and nonhomologous end joining (NHEJ) [7]. These findings add to our understanding of the mechanisms underlying bilirubin toxicity and may have implications for newborns with severe hyperbilirubinemia because DNA damage and oxidative stress may be another significant element causing neuronal death and bilirubin encephalopathy. Studies in Gunn rats and UGT −/− mice have additionally shown high levels of lipid peroxidation by sulfadimethoxine-induced hyperbilirubinemia, as well the activation of key oxidative stress markers [70,72,73,74]. We anticipate further therapeutic discoveries concerning the role of bilirubin in diseases related to oxidative stress, as the breadth of all its biological functions have yet to be fully uncovered.
2.2. Effects of UCB on the Brain
Autopsies of hyperbilirubinemic brains have shown UCB to be localized within neurons and microglia, which results in the loss of neurons, demyelination, and gliosis (Figure 1). On the other hand, along with inducing oxidative stress in cortical neurons, UCB also disrupts the dynamics of the neuronal network in hippocampal neurons or in immature developing neurons, making these early-staged neurons more susceptible to UCB-induced injury [75]. In isolated cell cultures, UCB impairs neuronal arborization and induces the release of pro-inflammatory cytokines from microglia and astrocytes. However, cell-dependent sensitivity to UCB toxicity and the role of each neural cell type are not yet understood [5].
Clinical manifestations of hyperbilirubinemia indicate higher selectivity of bilirubin towards damaged brain regions, which particularly includes its preference for basal ganglia, cerebellum, brainstem nuclei, peripheral and central auditory pathways, and hippocampus [12,21]. The increased selectivity towards these injured brain areas has been well-known to closely correlate with the clinical signs of hyperbilirubinemia. However, it is the impairment of intracellular defense mechanisms in these areas, rather than the accumulation of UCB itself, that plays the primary role in brain damage [12]. As a result, bilirubin may disrupt developmental processes while incorporating multiple overlap and co-morbid neurodevelopmental disorders [21]. The damage to the basal ganglia and cerebellum correlates with movement disorders, athetosis (slow, involuntary, and writhing movements of the limbs, tongue, face, neck, and other muscle groups) and abnormal tone; the damage to the auditory brain nuclei and inferior colliculi is correlated to the auditory dysfunctions and hearing loss; and the damage to the brainstem and hippocampus correlates with the impaired oculomotor brainstem response and impairments in memory and learning (Table 1) [12].
Barateiro and his colleagues [76] used a kernicterus mouse model to display axonal damage as well as myelination deficits and glial activation in brain regions that usually accompany the neurological sequelae observed in severe hyperbilirubinemia such as the pons, medulla oblongata, and cerebellum. The observations from the study indicate the cerebellum as the most affected area, displaying greater myelination impairment and glia burden, as well as a loss of Purkinje cells and a reduced arborization of the remaining ones. The increase in astroglial and microglial reactivity possibly emerges as a response to myelination injury. It has also been hypothesized that excessive accumulation of total serum bilirubin (TSB) in the early neonatal period may promote the activation of the gene responsible for myelin basic protein (MBP). The increase in MBP seems to correlate with the inhibition or lack of myelin sheath formation. This may occur in response to inflammatory insults that affected the brain in the first place, leading to the production of ROS, or it may be a compensatory response to the lack of functional MBP due to the damage [76].
The Brites lab demonstrated that neuronal growth impairment and cell death caused by UCB is mediated by nitric oxide (NO) and glutamate, modulated by microglia, and prevented by glycoursodeoxycholic acid and interleukin-10 (IL-10) [77]. In another study, Falcao et al., created a model where astrocytes abrogated the well-known UCB-induced neurotoxic effects by preventing the loss of cell viability, dysfunction, and death by apoptosis, as well as the impairment of neuronal outgrowth [78]. UCB-induced alterations on neurogenesis, spinogenesis, neuritogenesis and axonal cytoskeleton dynamics indicate the relevance of UCB in synaptic plasticity abnormalities and the long-term neurodevelopmental disabilities, thereby making pre-term infants more vulnerable towards BIND [5]. Ultimately, the critical dual role of UCB in the brain raises questions, such as (1) which exact mechanisms and physiological switches lead to this beneficial–toxic threshold and (2) how can we regulate this duality of UCB to our advantage for future clinical applications? Having a BIND model as close to the clinical manifestation as possible may help to answer these questions by allowing us to investigate the cellular and pathophysiological mechanisms caused by UCB entry and its further effects in the brain.
2.3. Epigenetic Alterations Due to Bilirubin-Induced Neurotoxicity
Epigenetic studies have shown bilirubin neurotoxicity to affect vital regulatory mechanisms by significant modulation of gene expression [79]. Epigenetic processes involve DNA methylation, RNA methylation, histone post-translational modifications, and non-coding RNAs (ncRNAs), among which histone acetylation plays a vital role in gene modulation for several neuro-biological processes, including synaptic plasticity, brain development, differentiation, maintenance, and survival [80,81,82]. Apart from affecting cell fate and behavior, the acetylation/de-acetylation-mediated changes in gene expression induce excitotoxicity, oxidative stress, increased calcium load, inflammation, and apoptosis [83,84] (Figure 1). The observed induced mechanisms indicate a probable link between epigenetic impairment in neurodevelopmental processes and the hyperbilirubinemic phenotype [79]. Following these leads, Vianello et al. used developing and adult Gunn rats to track histone 3 lysine 14 acetylation (H3K14Ac) level in the cerebellum and observed age-dependent alteration of H3K14Ac in hyperbilirubinemic conditions. Gene ontology analysis of H3K14Ac-linked chromatin also revealed 45% of genes to be involved in CNS development. This finding suggests that epigenetic modulation during development and maturation of the brain structure is one of the causes of cerebellum hypoplasia in hyperbilirubinemic Gunn rats. On the other hand, histone acetylation plays a role in controlling oligodendrocyte differentiation and myelin production, and the down-regulation of myelin-associated glycoprotein (Mag) is one of the known repercussions of bilirubin-induced disturbances of oligodendrocyte maturation [81,85]. Studies have reported down-regulation of Mag in vitro along with other BIND models, including in pre-term infants [76,79]. This indicates that oligodendrocyte maturation and myelination can be affected by altered histone acetylation due to bilirubin-induced neurotoxicity, both in physiological CNS development and post-demyelinated repair processes. Remarks from these studies confirm that epigenetically impaired neurodevelopmental processes in hyperbilirubinemia may have a correlation in bilirubin neurotoxicity [79].
3. BIND and CNS Disease Models
Generating and studying model systems that closely recapitulate the main characteristics of BIND and severe UHB, is of high importance for developing effective clinical treatments and therapies to gain a better understanding of the pathophysiological mechanisms underlying this condition. Figure 2 illustrates a general overview of the most common in vivo and in vitro models of BIND and CNS (Figure 2A,B), as well as why having a CNS patient-derived iPSC model would be a better option (Figure 2C).
3.1. Animal Models
Animal models are often able to bridge the gap that in vitro models fail to recapitulate, as they much better resemble the disease features manifested in patients. Bortolussi and Muro rigorously reviewed animal models used to study bilirubin neurotoxicity and metabolism as well as the in vivo mechanisms of hyperbilirubinemia [13]. The most widely used amongst these models is the Gunn rat. This strain of Wistar rats spontaneously developed a one-base deletion of exon 4 in the UGT1 locus, thereby creating an in-frame premature stop codon. Since this codon is translated into a truncated protein lacking the transmembrane domain, it results in the deficiency of all members of the UGT1A1 iso-enzymes. The complete deficiency of UGT1A1 enzymatic activity causes hyperbilirubinemia in the Gunn rat, making it the first hyperbilirubinemia animal model to mimic the CNS-I syndrome. This model has enabled scientists to gather a considerable amount of knowledge on bilirubin metabolism and toxicity in vivo [4,86].
Despite having a mild phenotype, Gunn rats display life-long non-hemolytic UHB, which is an important feature of human CNS-I. In order to develop acute central nervous system dysfunction and recapitulate hyperbilirubinemia more precisely, Gunn rats are often treated with hemolytic drugs or albumin–bilirubin displacers, such as sulphonamides or erythrocyte-lysing agents such as phenylhydrazine. An application of this method to induce hyperbilirubinemia is direct administration of sulfadimethoxine, a displacer of bilirubin from albumin binding sites. This increases the fraction of free bilirubin migrating towards lipophilic tissues such as the brain and is accompanied by a drop of systemic bilirubin [13]. If left untreated, homozygous Gunn rats display abnormalities in the cerebellum and hearing impairments just like the respective human CNS-I phenotype; however, unlike the patient-manifested features, these rats reach adulthood and are fertile.
The UGT1A-null mouse is another popular in vivo model, which presents a much more severe phenotype than the Gunn rat, with aggravated neurological damage and consecutive death [87].
Using genetic tools and technologies enable the creation of the mutation. Constitutive and conditional knockout, knock-in and transgenic strains of mice have been generated by manipulating the mouse genome and have allowed for the further exploration of key aspects of this disease. With the disruption of UGT1 exon 4 by neomycin cassette, scientists were able to generate the first bioengineered mouse model of severe UHB. Mutant mice are a good model to study CNS-I, as they do not express UGT1A1 and display neonatal hyperbilirubinemia. However, these mice die within 11 days after birth, which makes the model inconvenient for broad-spectrum investigations and reproducibility.
These invaluable animal models have provided an undeniable contribution in understanding the mechanisms underlying severe neonatal hyperbilirubinemia. Nevertheless, they still leave an open question regarding the mechanism and pathology in the human brain, which emphasizes the establishment of a human cell-derived model system to provide more insights into the molecular basis of the disease.
3.2. In Vitro Models
Cell types of different origins are also used to model several aspects of bilirubin toxicity and its main sequelae such as oxidative stress, ER stress and DNA damage. In vitro cultures are being applied extensively to study bilirubin neurotoxicity. These cultures mainly include immortalized cell lines such as human neuroblastoma cell lines, HeLa cells, Hepa 1c1c7 mouse hepatoma cells and human U87 astrocytoma cells, as well as primary cultures of rat and mouse neurons, astrocytes, microglia, oligodendrocytes, endothelial cells, and embryonic fibroblasts (Figure 2). Even though these 2D systems do not mimic the in vivo cell–cell interactions nor the morphological and physiological complexities of the whole tissue, they still display various properties of the in vivo situation. Exposing different types of cells to different concentrations of UCB is one of the key methods in exploring BIND [13].
Hippocampal neurons are the most frequently used cell type for testing the response of neuronal cells to bilirubin. When exposed to bilirubin, these cells exhibit a reduction in axons and dendritic processes, increased cell death, oxidation, and mitochondrial dysfunction, as well as overexpression of protection mechanisms. Moreover, newly differentiated neuronal cell types that are less differentiated display higher sensitivity compared to mature differentiated neurons. Similarly, oligodendrocytes also display high bilirubin susceptibility. Oligodendrocytes downregulate MBP production with the consequent impairment of myelin sheath formation and neuronal axonal function.
Organotypic cultures are another form of ex vivo model that can be used to study bilirubin toxicity; however, there are limited studies exploiting these models, particularly using hippocampal slices. These models were able to demonstrate the impairment of synaptic plasticity due to bilirubin toxicity as well as the involvement of microglia in the UCB-induced neurotoxicity. Thus, 2D cultures have been employed as in vitro models for decades to study the cellular response in biochemical and biophysical directions and have contributed towards the significant advancement in understanding cell behavior and bioactivities [88]. Despite of being well accepted, it cannot be denied that the cell bioactivities and interactions in 2D cultures deviate remarkably compared to the in vivo responses. Considering the urge of having a model that more efficiently mimics in vivo conditions, 3D culture models such as spheroids or organoids have emerged as a potential platform to study different physiological and pathological processes. Employing another dimension around 2D cells with the extracellular matrix (ECM) markedly impacts the cellular fate with respect to proliferation, differentiation, mechano-response, and cell viability [88,89].
3.3. IPSCs and Organoids as Tools for Disease Modeling
Creating a model that properly recapitulates the molecular events underlying a specific neurological disorder is not an easy task. The majority of studies attempting to model BIND and CNS rely primarily on either mouse models, which poorly represent the human pathogenesis and phenotype, or post-mortem tissues, which usually only reflect the final stages of the disease [20,90]. Embryonic stem cells (ESCs) have indefinite self-renewal capacity and plasticity to differentiate into somatic cell types in the embryo, which makes this cell type valuable for studying the mechanisms involved in specialized cells and organ development. ESCs offer a great opportunity for regenerative medicine by generating specialized cells based on different degenerative diseases and replace those with the damaged tissues [91]. However, derivation and application of ESCs remain ethically controversial, as the derivation process involves the use of human inner-cell-mass cells isolated from blastocysts [92,93]. Moreover, it is not always convenient to obtain samples, and the reproducibility of results is affected. These concerns can be side-lined by using human-induced pluripotent stem cells (hiPSCs), as these cells hold great promise for increasing our fundamental understanding of human biology during early development and pave the way for future regeneration therapies and personalized medicine. Recent advancements in gene editing technologies such as clustered regularly inter-spaced short palindromic repeats (CRISPR) have also made it possible to introduce genetic variants, for example through inducible gene knockout, thus opening new doors for in vitro disease modeling [94,95,96,97,98,99].
Further expansions in iPSC research have increasingly revealed the multifaceted use of these cells in modeling various diseases in vitro [17,18,19,20,100]. With the development of iPSCs, researchers have been able to replicate many diseases, including Parkinson’s disease, Nijmegen Breakage Syndrome, and Alzheimer’s disease, all by generating different types of cells that mimic the in vivo environment very closely [19,101,102,103,104,105,106,107,108,109]. For instance, deriving iPSCs from patients with genetic-based neurological conditions and differentiating them into neurons opens up more possibilities to closely observe the pathological mechanisms underlying the disease in vitro [106,110,111,112].
Shinya Yamanaka and Kazutoshi Takahashi introduced four defined factors; OCT3/4, SOX2, c-MYC, and KLF4, and established the pioneering protocol of generating iPS cells by reprogramming adult human fibroblasts [113,114]. Afterwards, Junying Yu et al. demonstrated another efficient combination of factors with OCT4, SOX2, NANOG, and LIN28 for reprogramming human somatic cells, which specifically exclude c-MYC [92]. Moreover, numerous studies have emerged in recent years, demonstrating the successful generation of iPSCs from different human somatic cells using integrating (retrovirus, lentivirus) and non-integrating (adenovirus, sendai virus, pSin plasmid, episomal plasmids, minicircle DNA) delivery systems [113,115,116,117,118,119]. With the other somatic cell (e.g., blood, urine cells)-derived iPSCs, it has been possible to avoid the invasive approach of skin biopsy, yet some methylation profile differences are still present between iPSCs and ESCs [120,121,122,123,124]. Nevertheless, iPSCs are ethically approved and considered identical regarding cell morphology, proliferation, and differentiation capacity, which also make it possible to generate large quantities of neuronal cultures for disease modeling, drug screening and therapy [19,92,125,126,127]. Additionally, iPSCs enable studying patient-specific disease conditions by reprogramming the cells obtained directly from the patient and therefore increase the scope to attain customized medication and therapy.
The iPSC-derived 2D monolayer model is the classical approach for obtaining specific neural cell types to enable the investigation of cellular and molecular mechanism associated with healthy and disease states. Neural stem cells (NSCs) or neural progenitor cells (NPCs) have a self-renewing capacity and can differentiate into the neuronal lineage, resulting in multiple types of brain cells during mammalian developmental and adult stage (fetal to postnatal, through adulthood) [128,129,130]. However, NSCs show heterogeneity and high regional specificity in adults, while the newly differentiated neurons derived from the primary progenitors migrate and intermingle with specific brain regions [130,131]. The type of generated neurons is determined by the neuroepithelial origin of NSCs, which is linked to NSC localization and developmental timing regions [131]. There are various established protocols for generating NSCs derived from iPSCs (Figure 3). Adherent iPSCs are used to generate embryoid bodies (EBs) and these are then with specific growth factors such as epidermal growth factor (EGF), fibroblast growth factor 2 (FGF-2) along with B27 (without retinoic acid) and N2 in the medium to achieve neural rosettes. Afterwards, these neural rosettes can be re-plated in a monolayer culture to obtain NSCs [132,133]. On the other hand, neural rosettes can also be generated without EB formation by using ESC and iPSC colonies, which are detached and then treated with EGF and FGF-2 to grow as cell aggregates. These cell aggregates have the potential to form neural rosettes and are able to differentiate into a range of both central and peripheral neural lineages [132,134,135,136,137]. However, the NSCs obtained from neural rosettes may provide a heterogeneous and inconsistent proportion of differentiated cells, which can be avoided by deriving pure cultures of specific types of brain cells from iPSCs with specific inductors [138,139,140,141]. Overall, neural rosette formation and differentiating into specific cell types can be employed as a potent in vitro system to study human neurological diseases by uncovering molecular pathways. Nonetheless, some major limitations include distinction among different iPSC lines, batch-to-batch variability, and growth of rosettes in an irregular and non-coordinated manner. Even though it is possible to characterize and measure the quality of individual rosettes using different assays to some extent, the understanding of the dynamics from monolayer cells to a developed rosette is presently limited [137,142]. Conversely, generating 2D monolayered homogenous neuronal cultures by directed differentiations are financially and technically feasible, along with high-resolution cell morphology and great reproducibility. Guided differentiation to specific neuronal subtypes holds the potential for cell therapy or personalized medicine to treat neurodegenerative diseases [143,144]. However, the non-identical cellular age of the cells and the differences in differentiation, culture and maintenance procedure may also affect the comparability of the results [145].
The self-organizing capacity of hiPSCs to form whole tissues of various organ systems have evolved as a great advancement from 2D to 3D in vitro models [146,147,148]. In vivo methods provide complex and three-dimensional spatial arrangement to the cells, where circulating molecules, neighboring cell and the extracellular matrix are surrounding them [149]. Mono-layered mono- or co-culture systems lack this in vivo physiological relevance, which has a vital effect on cellular and physiological responses. In this regard, a three-dimensional system offers more physiological resemblance with respect to structural complexity. hiPSC-derived three-dimensional brain organoids recapitulate the key aspects of neurodevelopment along with reflecting some function of the system [150]. Three-dimensional organoids contain highly divergent cell types and subtypes, providing complex architecture and interplays with spatial organization. Being an intact tissue with spatial organization, organoid models offer the opportunity to observe the dynamic growth and development of the system over time [19]. Genetic mutations affect cell type, cell behavior, their interactions, neuronal network, and components of the various neurodevelopmental and physiological processes. iPSC-derived brain organoids afford studying these genetic mutations and multi-faceted brain diseases [108,148,151,152,153]. Moreover, being cultured in vitro, organoids provide easy accessibility genetically and for live assays [154].
Generally, cerebral organoids are composed of functioning neurons, astrocytes, oligodendrocytes and to some extent microglia [148]. Lancaster et al. established the protocol to generate self-patterned cerebral organoids containing forebrain, midbrain, hindbrain, and choroid plexus identity (non-directed differentiation) [148,155]. They used iPSCs to form EBs, which were gradually directed towards the neuroectodermal lineage, and then maintained these neuroectodermal tissues with extracellular matrix support in a spinning bioreactor to provide nutrition and a three-dimensional environment. With this approach, neural identity can be obtained in 8–10 days, resulting in defined brain regions by 20–30 days of culture, and the organoids can be cultivated for longer period to study later stages of neurodevelopment [154]. As the non-directed protocol relies on the cells’ differentiation and self-organization capacity without providing any inductive signals, it is considered as intrinsic and a non-manipulated system [156]. Cerebral organoid research has significantly expanded within the last decade with the introduction of more complexity and specificity [157]. Relying on small molecules, individual brain-region-specific organoids can be generated by growth-factor-based manipulation, which consequently determine the cellular identities such as cerebral organoids with choroid plexus, hippocampus, retina and striatum [158,159,160,161]. The generation of organoids by co-culturing different cells is another advanced and innovative approach to enhance the model complexity and to investigate the cell–cell and cell–matrix interplays in a 3D environment during human brain development and disease [162,163,164]. The co-culture systems in organoid technology reveals the mechanism of stem cell interactions, which might be useful in regenerative medicine study [165]. For example, iPSC-derived microglia (cells or assembloids) can be integrated into the midbrain organoids (generated from iPSCs), and then neurodegenerative and neuroinflammatory diseases can be investigated using this model. In contrast, microglia differentiation medium lacks neurotrophic factors, which are required for promoting dopaminergic neuron differentiation, resulting in lower numbers of dopaminergic neurons [166]. Consequently, the incompatibility between the small molecules in the medium might interact and affect the quantity and quality of existing cell types. Therefore, it is challenging to compose a perfect culture medium for co-culture organoids, as these comprise different cell types, where individual cell types require distinct medium compositions [165]. Additionally, a major drawback includes different proliferation rates of the co-cultured cells in the system, which might affect the maturity of the organoids and limit the long-term culture. On the other hand, release of paracrine factors by one cell type might affect the other cells either positively or negatively in this system [167,168]. Since both directed and non-directed organoid generation processes have benefits and drawbacks, the application of each should be determined according to the purpose of the study (Figure 3).
Considering various incorporated features in cerebral organoids, these models are closer to the developing human brain and mimic the neural environment much better compared to other in vitro models. This facilitates the understanding of disease pathology, drug mechanism and customized medication [108,153,169,170,171]. Cerebral organoids represent a higher degree of maturation and developmental dynamics mimicking the early second trimester of the fetal brain tissue; nevertheless, the accurate human brain equivalent age of the organoids still remains an unanswered question [172]. Furthermore, the organoid model shows high batch-to-batch variability in common with other iPSC-based models and requires sophisticated methods [19]. During slow development of the organoids, a tissue-degenerated necrotic core tends to form in the center due to the lack of optimal diffusion of nutrients and metabolites. Although the culture medium is oxygenated in the bioreactors, this is not enough to support culture for a longer period [148]. Despite the limitations, iPSC-derived brain organoids are a promising tool for 3D in vitro model systems, as they display functions and circuitry comparable to the human brain [148]. Even though they do not fully recapitulate the complexities of the human brain, they can still be a valuable study tool, as they are composed of distinct neural cell types important for the central nervous system [173]. Furthermore, human brain organoids have revealed useful insights into human brain development and successfully helped to model a variety of neurological disorders such as microcephaly, Timothy syndrome, and Nijmegen Breakage Syndrome, as well as brain tumors such as gliomas [108,148,174,175,176,177,178]. All of these models offer a solid platform for future of brain organoids as a valid tool for studying neurological disorders affecting the human brain [179].
UHB is an ailment observed in the first postnatal week, which can lead to acute or chronic UCB encephalopathy. The neonates show vulnerability towards UCB and have an increased risk associated with particular conditions, such as premature birth, sepsis, and hypoxia. Pre-term and low-birth-weight infants are even more vulnerable towards BIND due to neurodevelopmental immaturity, when sepsis or infection is incorporated [5,180,181]. Since brain organoids recapitulate key aspects of neurodevelopment and reflect certain functions of the system, they can therefore be exposed to UCB for modeling BIND. Both the immature and mature stage of cerebral organoids can be exposed to UCB for shorter (4–5 h) and longer (72 h–several days) periods to mimic the acute and chronic effect of UCB in the CNS. As UCB is a lipophilic compound, it should be able to penetrate the organoids. Additionally, iPSCs derived from CNS-I patients can be used to generate brain organoids, which will model the disease more precisely due to defective UGT1A1, and these organoids can be exposed to UCB to mimic the hyperbilirubinemic condition in the CNS [112]. As autopsy revealed the presence of UCB in neurons, astrocytes, neuronal process and so on, divergent cell types containing organoids will help us to understand the pathophysiology of BIND. Neurons are known to be more susceptible to UCB than astrocytes and generally demonstrate a higher level of ROS, protein oxidation and lipid peroxidation upon UCB exposure [71,182]. On the other hand, astrocytes cause morphological changes in mitochondria and ER when affected by high concentrations of UCB, leading to oxidative stress and cell death [183,184]. Furthermore, high levels of ROS produced by neurons upon UCB exposure results in oxidative stress in microglia [185]. Overall, UCB exposure affects the redox status of neurons and glial cells and induce inflammation with increased ROS, thus resulting in cell death in the CNS [71,182]. As cerebral organoids are composed of neuronal cell types and subtypes with some functionality and network complexity, it is possible to recapitulate the altered redox status induced by UCB toxicity and consecutive inflammatory responses using this model. However, it should be noted that bilirubin might also be considered as a neuroprotective compound when the concentration is below 100 nM [5,62]. In this regard, a kill curve should be performed to identify the suitable concentration for UCB, which can be used to mimic the hyperbilirubinemic condition in the CNS.
Furthermore, co-morbidity along with the degree and timing of UHB can affect an infant to develop one or multiple defects from the BIND spectrum. Therefore, studying the independent correlation of UHB with each neurodevelopmental disorder individually is not sufficient. Appropriate statistical analyses and power can be applied to evaluate possible co-morbidities of multiple neurodevelopmental disorders within the BIND spectrum, which may help us to define the association of each neurodevelopmental disorder with bilirubin-induced neurotoxicity [21,186,187]. Some of the parameters of bilirubin-induced neurotoxicity measurement include assessment of oxidative stress, DNA and RNA damage, post-transcriptional modifications, bilirubin accumulation in the brain and transporters, ER stress, inflammation and autophagy, which are also possible to study in the cerebral organoid model [13,70]. For instance, after UCB exposure to cerebral organoids, oxidative stress or impaired redox status can be monitored by glutathione (GSH) and oxidized glutathione (GSSG) measurements, where a lower ratio of [GSH]/[GSSG] indicates an increased oxidative state [70]. From transcriptome analysis of the treated organoids, bilirubin-induced ER responses can be observed by altered gene expression and regulation of ER stress-related genes (e.g., CHOP, ATF3, FAS) [74]. Gene ontology analysis can also reveal the connection between ER and inflammatory responses through distinct but relevant pathways (e.g., activation of p-ERK, NF-κB pathways) [70,188]. Moreover, assessment of pro-inflammatory mediators such as IL-6, IL-8, TNF-α, IL-1β by cytokine array or ELISA, can help uncover bilirubin-mediated inflammation [189,190]. UCB induced increased oxidative stress and ER stress, and neurodegeneration-mediated inflammation leads to apoptotic cell death, which can be detected by deoxynucleotidyl transferase-mediated deoxyuridine nick-end labeling (TUNEL) assay [70].
4. Concluding Remarks
To date, there are very few effective treatment options for CNS-I. To lower plasma bilirubin levels and prevent bilirubin encephalopathy, patients undergo daily phototherapy treatments, which inevitably become less effective as the patients age. Exchange transfusion is also sometimes used as an emergency treatment for neonates to rapidly lower serum bilirubin concentrations; however, this approach has been associated with serious complications, such as thrombocytopenia, portal vein thrombosis, necrotizing enterocolitis, and sepsis [44]. Liver transplantation remains the only effective treatment for this life-threatening disease, even though it does not reverse or alleviate pre-existing neurological damage [2,36,43,44,45].
Severe neonatal jaundice and hyperbilirubinemia remain a cause of devastating neurological damage in infants. Although this occurrence is rare, it can be completely avoided if the neonates receive treatment on time and the medical professionals prevent early discharge [12]. Currently, there is a clear gap in the knowledge we possess on the molecular mechanisms underlying this neurological damage. Therefore, creating a model of BIND based on genetically inherited disorders of the UGT1A1 gene, such as CNS Type I and II, can help us further understand these mechanisms.
Possible therapeutic approaches include anti-inflammatory-based medicines, gene manipulation and albumin infusion. Some other promising approaches include the modulation of nuclear receptors, cytochromes or BLVR activity, to control bilirubin production or to stimulate alternative bilirubin-disposal pathways. However, further research is needed before these techniques can be applied clinically. To shed light on human biology and health, a thorough understanding of the molecular pathways leading to bilirubin neurotoxicity is critical.
Other potential therapies include hepatocyte transplantation, during which about 5–15% of the liver is replaced by transplanted hepatocytes, as well as gene therapy. Injections of naked plasmid DNA and adeno-associated virus gene therapies are currently being investigated, as preclinical models have been quite promising [44]. The constant hope with new emerging iPSC-derived 3D brain organoid models is that they can help shed light onto developing more effective ways of handling BIND and inherited unconjugated bilirubinemia disorders in the near future. Eventually, this model will enhance our understanding of the etiology underlying BIND and its pathology in the human CNS. This knowledge will aid in the development of drugs and future clinical applications.
Author Contributions
A.I.P. wrote, formulated tables and edited the manuscript. S.S. wrote, edited and helped with the figures. J.A. conceptualized the work, edited the manuscript, acquired funding and supervised the study. All authors have read and agreed to the published version of the manuscript.
Funding
J.A. acknowledges funding from the Medical Faculty of Heinrich Heine University, Duesseldorf, Germany.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors would like to acknowledge Leon-Phillip Szepanowski for providing some of the immunofluorescence images used in this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fevery, J. Bilirubin in clinical practice: A review. Liver Int. 2008, 28, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Memon, N.; Weinberger, B.I.; Hegyi, T.; Aleksunes, L.M. Inherited disorders of bilirubin clearance. Pediatr. Res. 2016, 79, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Jialal, I. Unconjugated Hyperbilirubinemia. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Bortolussi, G.; Muro, A.F. Advances in understanding disease mechanisms and potential treatments for Crigler–Najjar syndrome. Exp. Opin. Orphan Drugs 2018, 6, 425–439. [Google Scholar] [CrossRef]
- Brites, D. The evolving landscape of neurotoxicity by unconjugated bilirubin: Role of glial cells and inflammation. Front. Pharmacol. 2012, 3, 88. [Google Scholar] [CrossRef]
- Watchko, J.F.; Tiribelli, C. Bilirubin-induced neurologic damage--mechanisms and management approaches. N. Engl. J. Med. 2013, 369, 2021–2030. [Google Scholar] [CrossRef]
- Rawat, V.; Bortolussi, G.; Gazzin, S.; Tiribelli, C.; Muro, A.F. Bilirubin-Induced Oxidative Stress Leads to DNA Damage in the Cerebellum of Hyperbilirubinemic Neonatal Mice and Activates DNA Double-Strand Break Repair Pathways in Human Cells. Oxid. Med. Cell Longev. 2018, 2018, 1801243. [Google Scholar] [CrossRef]
- Stocker, R.; Ames, B.N. Potential role of conjugated bilirubin and copper in the metabolism of lipid peroxides in bile. Proc. Natl. Acad. Sci. USA 1987, 84, 8130–8134. [Google Scholar] [CrossRef]
- Bulmer, A.C.; Blanchfield, J.T.; Toth, I.; Fassett, R.G.; Coombes, J.S. Improved resistance to serum oxidation in Gilbert’ss syndrome: A mechanism for cardiovascular protection. Atherosclerosis 2008, 199, 390–396. [Google Scholar] [CrossRef]
- Jangi, S.; Otterbein, L.; Robson, S. The molecular basis for the immunomodulatory activities of unconjugated bilirubin. Int. J. Biochem. Cell Biol. 2013, 45, 2843–2851. [Google Scholar] [CrossRef]
- Vitek, L.; Ostrow, J.D. Bilirubin chemistry and metabolism; harmful and protective aspects. Curr. Pharm. Des. 2009, 15, 2869–2883. [Google Scholar] [CrossRef]
- Gazzin, S.; Tiribelli, C. Bilirubin-induced neurological damage. J. Matern. Fetal. Neonatal. Med. 2011, 24 (Suppl. 1), 154–155. [Google Scholar] [CrossRef]
- Bortolussi, G.; Muro, A.F. Experimental models assessing bilirubin neurotoxicity. Pediatr. Res. 2020, 87, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Ahdab-Barmada, M.; Moossy, J. The neuropathology of kernicterus in the premature neonate: Diagnostic problems. J. Neuropathol. Exp. Neurol. 1984, 43, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Dal Ben, M.; Bottin, C.; Zanconati, F.; Tiribelli, C.; Gazzin, S. Evaluation of region selective bilirubin-induced brain damage as a basis for a pharmacological treatment. Sci. Rep. 2017, 7, 41032. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F. Kernicterus and the molecular mechanisms of bilirubin-induced CNS injury in newborns. Neuromolecular Med. 2006, 8, 513–529. [Google Scholar] [CrossRef]
- Drews, K.; Jozefczuk, J.; Prigione, A.; Adjaye, J. Human induced pluripotent stem cells--from mechanisms to clinical applications. J. Mol. Med. 2012, 90, 735–745. [Google Scholar] [CrossRef]
- Kunkanjanawan, T.; Noisa, P.; Parnpai, R. Modeling neurological disorders by human induced pluripotent stem cells. J. Biomed. Biotechnol. 2011, 2011, 350131. [Google Scholar] [CrossRef]
- Logan, S.; Arzua, T.; Canfield, S.G.; Seminary, E.R.; Sison, S.L.; Ebert, A.D.; Bai, X. Studying Human Neurological Disorders Using Induced Pluripotent Stem Cells: From 2D Monolayer to 3D Organoid and Blood Brain Barrier Models. Compr. Physiol. 2019, 9, 565–611. [Google Scholar] [CrossRef]
- Marchetto, M.C.; Brennand, K.J.; Boyer, L.F.; Gage, F.H. Induced pluripotent stem cells (iPSCs) and neurological disease modeling: Progress and promises. Hum. Mol. Genet. 2011, 20, R109–R115. [Google Scholar] [CrossRef]
- Amin, S.B.; Smith, T.; Timler, G. Developmental influence of unconjugated hyperbilirubinemia and neurobehavioral disorders. Pediatr. Res. 2019, 85, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Jew, J.Y.; Sandquist, D. CNS changes in hyperbilirubinemia. Functional implications. Arch. Neurol. 1979, 36, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Aron, A.R.; Poldrack, R.A. Cortical and subcortical contributions to Stop signal response inhibition: Role of the subthalamic nucleus. J. Neurosci. 2006, 26, 2424–2433. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.J.; Loughry, B.; O’Reilly, R.C. Interactions between frontal cortex and basal ganglia in working memory: A computational model. Cogn. Affect Behav. Neurosci. 2001, 1, 137–160. [Google Scholar] [CrossRef] [PubMed]
- Burgess, N.; Maguire, E.A.; O’Keefe, J. The human hippocampus and spatial and episodic memory. Neuron 2002, 35, 625–641. [Google Scholar] [CrossRef]
- Amin, S.B.; Prinzing, D.; Myers, G. Hyperbilirubinemia and language delay in premature infants. Pediatrics 2009, 123, 327–331. [Google Scholar] [CrossRef]
- Amin, S.B. Clinical assessment of bilirubin-induced neurotoxicity in premature infants. Semin. Perinatol. 2004, 28, 340–347. [Google Scholar] [CrossRef]
- King, C.D.; Rios, G.R.; Green, M.D.; Tephly, T.R. UDP-glucuronosyltransferases. Curr. Drug. Metab. 2000, 1, 143–161. [Google Scholar] [CrossRef]
- Kadakol, A.; Ghosh, S.S.; Sappal, B.S.; Sharma, G.; Chowdhury, J.R.; Chowdhury, N.R. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: Correlation of genotype to phenotype. Hum. Mutat. 2000, 16, 297–306. [Google Scholar] [CrossRef]
- Owens, I.S.; Basu, N.K.; Banerjee, R. UDP-glucuronosyltransferases: Gene structures of UGT1 and UGT2 families. Methods Enzymol. 2005, 400, 1–22. [Google Scholar] [CrossRef]
- Ritter, J.K.; Crawford, J.M.; Owens, I.S. Cloning of two human liver bilirubin UDP-glucuronosyltransferase cDNAs with expression in COS-1 cells. J. Biol. Chem. 1991, 266, 1043–1047. [Google Scholar]
- Canu, G.; Minucci, A.; Zuppi, C.; Capoluongo, E. Gilbert and Crigler Najjar syndromes: An update of the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database. Blood Cells Mol. Dis. 2013, 50, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Skierka, J.M.; Kotzer, K.E.; Lagerstedt, S.A.; O’Kane, D.J.; Baudhuin, L.M. UGT1A1 genetic analysis as a diagnostic aid for individuals with unconjugated hyperbilirubinemia. J. Pediatr. 2013, 162, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Crigler, J.F., Jr.; Najjar, V.A. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics 1952, 10, 169–180. [Google Scholar] [PubMed]
- Ritter, J.K.; Yeatman, M.T.; Ferreira, P.; Owens, I.S. Identification of a genetic alteration in the code for bilirubin UDP-glucuronosyltransferase in the UGT1 gene complex of a Crigler-Najjar type I patient. J. Clin. Investig. 1992, 90, 150–155. [Google Scholar] [CrossRef]
- Sticova, E.; Jirsa, M. New insights in bilirubin metabolism and their clinical implications. World J. Gastroenterol. 2013, 19, 6398–6407. [Google Scholar] [CrossRef]
- Labrune, P.; Myara, A.; Hadchouel, M.; Ronchi, F.; Bernard, O.; Trivin, F.; Chowdhury, N.R.; Chowdhury, J.R.; Munnich, A.; Odievre, M. Genetic heterogeneity of Crigler-Najjar syndrome type I: A study of 14 cases. Hum. Genet. 1994, 94, 693–697. [Google Scholar] [CrossRef]
- Torres, A.E.; Lyons, A.B.; Hamzavi, I.H.; Lim, H.W. Role of phototherapy in the era of biologics. J. Am. Acad. Dermatol. 2021, 84, 479–485. [Google Scholar] [CrossRef]
- Vreman, H.J.; Wong, R.J.; Stevenson, D.K. Phototherapy: Current methods and future directions. Semin. Perinatol. 2004, 28, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Morris, B.H.; Oh, W.; Tyson, J.E.; Stevenson, D.K.; Phelps, D.L.; O’Shea, T.M.; McDavid, G.E.; Perritt, R.L.; Van Meurs, K.P.; Vohr, B.R.; et al. Aggressive vs. conservative phototherapy for infants with extremely low birth weight. N. Engl. J. Med. 2008, 359, 1885–1896. [Google Scholar] [CrossRef]
- Schulz, S.; Wong, R.J.; Vreman, H.J.; Stevenson, D.K. Metalloporphyrins - an update. Front. Pharmacol. 2012, 3, 68. [Google Scholar] [CrossRef]
- Lin, S.; Wei, X.; Bales, K.R.; Paul, A.B.; Ma, Z.; Yan, G.; Paul, S.M.; Du, Y. Minocycline blocks bilirubin neurotoxicity and prevents hyperbilirubinemia-induced cerebellar hypoplasia in the Gunn rat. Eur. J. Neurosci. 2005, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Labrune, P.; Odievre, M. Toward a treatment of Crigler-Najjar syndrome? Arch. Fr. Pediatr. 1992, 49, 467–468. [Google Scholar] [PubMed]
- Dhawan, A.; Lawlor, M.W.; Mazariegos, G.V.; McKiernan, P.; Squires, J.E.; Strauss, K.A.; Gupta, D.; James, E.; Prasad, S. Disease burden of Crigler-Najjar syndrome: Systematic review and future perspectives. J. Gastroenterol. Hepatol. 2020, 35, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, E.; Mtegha, M.; Dhawan, A. Crigler-Najjar syndrome: Therapeutic options and consequences of mutations in the UGT1A1 complex. Expert Rev. Endocrinol. Metab. 2008, 3, 725–737. [Google Scholar] [CrossRef]
- Maines, M.D.; Trakshel, G.M. Tin-protoporphyrin: A potent inhibitor of hemoprotein-dependent steroidogenesis in rat adrenals and testes. J. Pharmacol. Exp. Ther. 1992, 260, 909–916. [Google Scholar]
- Kappas, A.; Drummond, G.S.; Galbraith, R.A. Prolonged clinical use of a heme oxygenase inhibitor: Hematological evidence for an inducible but reversible iron-deficiency state. Pediatrics 1993, 91, 537–539. [Google Scholar] [CrossRef]
- Brites, D. Bilirubin injury to neurons and glial cells: New players, novel targets, and newer insights. Semin. Perinatol. 2011, 35, 114–120. [Google Scholar] [CrossRef]
- Hansen, T.W. Pioneers in the scientific study of neonatal jaundice and kernicterus. Pediatrics 2000, 106, E15. [Google Scholar] [CrossRef]
- Brites, D.; Fernandes, A.; Falcao, A.S.; Gordo, A.C.; Silva, R.F.; Brito, M.A. Biological risks for neurological abnormalities associated with hyperbilirubinemia. J. Perinatol. 2009, 29, S8–S13. [Google Scholar] [CrossRef]
- Fernandes, A.; Falcao, A.S.; Abranches, E.; Bekman, E.; Henrique, D.; Lanier, L.M.; Brites, D. Bilirubin as a determinant for altered neurogenesis, neuritogenesis, and synaptogenesis. Dev. Neurobiol. 2009, 69, 568–582. [Google Scholar] [CrossRef]
- Genc, S.; Genc, K.; Kumral, A.; Baskin, H.; Ozkan, H. Bilirubin is cytotoxic to rat oligodendrocytes in vitro. Brain Res. 2003, 985, 135–141. [Google Scholar] [CrossRef]
- Deganuto, M.; Cesaratto, L.; Bellarosa, C.; Calligaris, R.; Vilotti, S.; Renzone, G.; Foti, R.; Scaloni, A.; Gustincich, S.; Quadrifoglio, F.; et al. A proteomic approach to the bilirubin-induced toxicity in neuronal cells reveals a protective function of DJ-1 protein. Proteomics 2010, 10, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, S.; Vitek, L.; Watchko, J.; Shapiro, S.M.; Tiribelli, C. A Novel Perspective on the Biology of Bilirubin in Health and Disease. Trends Mol. Med. 2016, 22, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Dudau, M.; Codrici, E.; Tanase, C.; Gherghiceanu, M.; Enciu, A.M.; Hinescu, M.E. Caveolae as Potential Hijackable Gates in Cell Communication. Front. Cell Dev. Biol. 2020, 8, 581732. [Google Scholar] [CrossRef]
- Choi, D.W. Excitotoxicity: Still Hammering the Ischemic Brain in 2020. Front. Neurosci. 2020, 14, 579953. [Google Scholar] [CrossRef]
- Mattson, M.P. Excitotoxicity. In Stress: Physiology, Biochemistry, and Pathology; Academic Press: Cambridge, MA, USA, 2019; pp. 125–134. [Google Scholar]
- Dennery, P.A.; Seidman, D.S.; Stevenson, D.K. Neonatal hyperbilirubinemia. N. Engl. J. Med. 2001, 344, 581–590. [Google Scholar] [CrossRef]
- Gourley, G.R. Bilirubin metabolism and kernicterus. Adv. Pediatr. 1997, 44, 173–229. [Google Scholar]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood-brain barrier: Molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Doré, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef]
- Brito, M.A.; Brites, D.; Butterfield, D.A. A link between hyperbilirubinemia, oxidative stress and injury to neocortical synaptosomes. Brain Res. 2004, 1026, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Asad, S.F.; Singh, S.; Ahmad, A.; Khan, N.U.; Hadi, S.M. Prooxidant and antioxidant activities of bilirubin and its metabolic precursor biliverdin: A structure-activity study. Chem. Biol. Interact. 2001, 137, 59–74. [Google Scholar] [CrossRef]
- Dani, C.; Martelli, E.; Bertini, G.; Pezzati, M.; Filippi, L.; Rossetti, M.; Rizzuti, G.; Rubaltelli, F.F. Plasma bilirubin level and oxidative stress in preterm infants. Arch. Dis. Child Fetal. Neonatal. Ed. 2003, 88, F119–F123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dani, C.; Poggi, C.; Pratesi, S. Bilirubin and oxidative stress in term and preterm infants. Free Radic. Res. 2019, 53, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Midan, D.A.R.; Bahbah, W.A.; Bayomy, N.R.; Ashour, N.M. Clinical Assessment of Neuroinflammatory Markers and Antioxidants in Neonates with Hyperbilirubinemia and Their Association with Acute Bilirubin Encephalopathy. Children 2022, 9, 559. [Google Scholar] [CrossRef]
- Vitek, L. Bilirubin as a signaling molecule. Med. Res. Rev. 2020, 40, 1335–1351. [Google Scholar] [CrossRef]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef]
- Yueh, M.F.; Chen, S.; Nguyen, N.; Tukey, R.H. Developmental onset of bilirubin-induced neurotoxicity involves Toll-like receptor 2-dependent signaling in humanized UDP-glucuronosyltransferase1 mice. J. Biol. Chem. 2014, 289, 4699–4709. [Google Scholar] [CrossRef]
- Brito, M.A.; Rosa, A.I.; Falcao, A.S.; Fernandes, A.; Silva, R.F.; Butterfield, D.A.; Brites, D. Unconjugated bilirubin differentially affects the redox status of neuronal and astroglial cells. Neurobiol. Dis. 2008, 29, 30–40. [Google Scholar] [CrossRef]
- Daood, M.J.; Hoyson, M.; Watchko, J.F. Lipid peroxidation is not the primary mechanism of bilirubin-induced neurologic dysfunction in jaundiced Gunn rat pups. Pediatr. Res. 2012, 72, 455–459. [Google Scholar] [CrossRef]
- Bortolussi, G.; Codarin, E.; Antoniali, G.; Vascotto, C.; Vodret, S.; Arena, S.; Cesaratto, L.; Scaloni, A.; Tell, G.; Muro, A.F. Impairment of enzymatic antioxidant defenses is associated with bilirubin-induced neuronal cell death in the cerebellum of Ugt1 KO mice. Cell Death Dis. 2015, 6, e1739. [Google Scholar] [CrossRef] [PubMed]
- Vodret, S.; Bortolussi, G.; Jasprova, J.; Vitek, L.; Muro, A.F. Inflammatory signature of cerebellar neurodegeneration during neonatal hyperbilirubinemia in Ugt1 (-/-) mouse model. J. Neuroinflamm. 2017, 14, 64. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.R.; Silva, S.L.; Barateiro, A.; Falcão, A.S.; Fernandes, A.; Brito, M.A.; Brites, D. Selective vulnerability of rat brain regions to unconjugated bilirubin. Mol. Cell Neurosci. 2011, 48, 82–93. [Google Scholar] [CrossRef]
- Barateiro, A.; Chen, S.; Yueh, M.F.; Fernandes, A.; Domingues, H.S.; Relvas, J.; Barbier, O.; Nguyen, N.; Tukey, R.H.; Brites, D. Reduced Myelination and Increased Glia Reactivity Resulting from Severe Neonatal Hyperbilirubinemia. Mol. Pharmacol. 2016, 89, 84–93. [Google Scholar] [CrossRef]
- Silva, S.L.; Vaz, A.R.; Diogenes, M.J.; van Rooijen, N.; Sebastiao, A.M.; Fernandes, A.; Silva, R.F.; Brites, D. Neuritic growth impairment and cell death by unconjugated bilirubin is mediated by NO and glutamate, modulated by microglia, and prevented by glycoursodeoxycholic acid and interleukin-10. Neuropharmacology 2012, 62, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Falcao, A.S.; Silva, R.F.; Vaz, A.R.; Silva, S.L.; Fernandes, A.; Brites, D. Cross-talk between neurons and astrocytes in response to bilirubin: Early beneficial effects. Neurochem. Res. 2013, 38, 644–659. [Google Scholar] [CrossRef] [PubMed]
- Vianello, E.; Zampieri, S.; Marcuzzo, T.; Tordini, F.; Bottin, C.; Dardis, A.; Zanconati, F.; Tiribelli, C.; Gazzin, S. Histone acetylation as a new mechanism for bilirubin-induced encephalopathy in the Gunn rat. Sci. Rep. 2018, 8, 13690. [Google Scholar] [CrossRef] [PubMed]
- Lilja, T.; Heldring, N.; Hermanson, O. Like a rolling histone: Epigenetic regulation of neural stem cells and brain development by factors controlling histone acetylation and methylation. Biochim. Biophys. Acta 2013, 1830, 2354–2360. [Google Scholar] [CrossRef]
- Fagiolini, M.; Jensen, C.L.; Champagne, F.A. Epigenetic influences on brain development and plasticity. Curr. Opin. Neurobiol. 2009, 19, 207–212. [Google Scholar] [CrossRef]
- Ma, L.H.; Yan, J.; Jiao, X.H.; Zhou, C.H.; Wu, Y.Q. The Role of Epigenetic Modifications in Neurotoxicity Induced by Neonatal General Anesthesia. Front. Mol. Neurosci. 2022, 15, 877263. [Google Scholar] [CrossRef]
- Shein, N.A.; Shohami, E. Histone deacetylase inhibitors as therapeutic agents for acute central nervous system injuries. Mol. Med. 2011, 17, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Covington, H.E., 3rd; Maze, I.; Sun, H.; Bomze, H.M.; DeMaio, K.D.; Wu, E.Y.; Dietz, D.M.; Lobo, M.K.; Ghose, S.; Mouzon, E.; et al. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron 2011, 71, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Brito, M.A.; Zurolo, E.; Pereira, P.; Barroso, C.; Aronica, E.; Brites, D. Cerebellar axon/myelin loss, angiogenic sprouting, and neuronal increase of vascular endothelial growth factor in a preterm infant with kernicterus. J. Child Neurol. 2012, 27, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Gunn, C.K. Hereditary Acholuric Jaundice in the Rat. Can. Med. Assoc. J. 1944, 50, 230–237. [Google Scholar] [PubMed]
- Nguyen, N.; Bonzo, J.A.; Chen, S.; Chouinard, S.; Kelner, M.J.; Hardiman, G.; Belanger, A.; Tukey, R.H. Disruption of the ugt1 locus in mice resembles human Crigler-Najjar type I disease. J. Biol. Chem. 2008, 283, 7901–7911. [Google Scholar] [CrossRef]
- Duval, K.; Grover, H.; Han, L.H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef]
- Bonnier, F.; Keating, M.E.; Wróbel, T.P.; Majzner, K.; Baranska, M.; Garcia-Munoz, A.; Blanco, A.; Byrne, H.J. Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell culture models. Toxicol. In Vitro 2015, 29, 124–131. [Google Scholar] [CrossRef]
- McKinney, C.E. Using induced pluripotent stem cells derived neurons to model brain diseases. Neural Regen. Res. 2017, 12, 1062–1067. [Google Scholar] [CrossRef]
- Vazin, T.; Freed, W.J. Human embryonic stem cells: Derivation, culture, and differentiation: A review. Restor. Neurol. Neurosci. 2010, 28, 589–603. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, Z.; Sun, Z. The potential and challenges of using stem cells for cardiovascular repair and regeneration. Genes Dis. 2014, 1, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cao, J.; Xiong, M.; Petersen, A.J.; Dong, Y.; Tao, Y.; Huang, C.T.; Du, Z.; Zhang, S.C. Engineering Human Stem Cell Lines with Inducible Gene Knockout using CRISPR/Cas9. Cell Stem Cell 2015, 17, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Waddington, S.N.; Privolizzi, R.; Karda, R.; O’Neill, H.C. A Broad Overview and Review of CRISPR-Cas Technology and Stem Cells. Curr. Stem Cell Rep. 2016, 2, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreccio, A.; Mathieu, J.; Detraux, D.; Somasundaram, L.; Cavanaugh, C.; Sopher, B.; Fischer, K.; Bello, T.; A, M.H.; Levy, S.; et al. Inducible CRISPR genome editing platform in naive human embryonic stem cells reveals JARID2 function in self-renewal. Cell Cycle 2018, 17, 535–549. [Google Scholar] [CrossRef]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and Rescue of RP2 Retinitis Pigmentosa Using iPSC-Derived Retinal Organoids. Stem Cell Rep. 2020, 15, 67–79. [Google Scholar] [CrossRef]
- Dell’ Amico, C.; Tata, A.; Pellegrino, E.; Onorati, M.; Conti, L. Genome editing in stem cells for genetic neurodisorders. Prog. Mol. Biol. Transl. Sci. 2021, 182, 403–438. [Google Scholar] [CrossRef]
- Nishiga, M.; Qi, L.S.; Wu, J.C. CRISPRi/a Screening with Human iPSCs. Methods Mol. Biol. 2021, 2320, 261–281. [Google Scholar] [CrossRef]
- Spitzhorn, L.S.; Kordes, C.; Megges, M.; Sawitza, I.; Gotze, S.; Reichert, D.; Schulze-Matz, P.; Graffmann, N.; Bohndorf, M.; Wruck, W.; et al. Transplanted Human Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Support Liver Regeneration in Gunn Rats. Stem Cells Dev. 2018, 27, 1702–1714. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef]
- Hossini, A.M.; Megges, M.; Prigione, A.; Lichtner, B.; Toliat, M.R.; Wruck, W.; Schroter, F.; Nuernberg, P.; Kroll, H.; Makrantonaki, E.; et al. Erratum: Induced pluripotent stem cell-derived neuronal cells from a sporadic Alzheimer’s disease donor as a model for investigating AD-associated gene regulatory networks. BMC Genom. 2015, 16, 433. [Google Scholar] [CrossRef]
- Halevy, T.; Akov, S.; Bohndorf, M.; Mlody, B.; Adjaye, J.; Benvenisty, N.; Goldberg, M. Chromosomal Instability and Molecular Defects in Induced Pluripotent Stem Cells from Nijmegen Breakage Syndrome Patients. Cell Rep. 2016, 16, 2499–2511. [Google Scholar] [CrossRef] [PubMed]
- Flamier, A.; El Hajjar, J.; Adjaye, J.; Fernandes, K.J.; Abdouh, M.; Bernier, G. Modeling Late-Onset Sporadic Alzheimer’s Disease through BMI1 Deficiency. Cell Rep. 2018, 23, 2653–2666. [Google Scholar] [CrossRef]
- Pomeshchik, Y.; Klementieva, O.; Gil, J.; Martinsson, I.; Hansen, M.G.; de Vries, T.; Sancho-Balsells, A.; Russ, K.; Savchenko, E.; Collin, A.; et al. Human iPSC-Derived Hippocampal Spheroids: An Innovative Tool for Stratifying Alzheimer Disease Patient-Specific Cellular Phenotypes and Developing Therapies. Stem Cell Rep. 2020, 15, 256–273. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Muller-Schiffmann, A.; Erichsen, L.; Bohndorf, M.; Wruck, W.; Sleegers, K.; Van Broeckhoven, C.; Korth, C.; Adjaye, J. IPSC-Derived Neuronal Cultures Carrying the Alzheimer’s Disease Associated TREM2 R47H Variant Enables the Construction of an Abeta-Induced Gene Regulatory Network. Int. J. Mol. Sci. 2020, 21, 4516. [Google Scholar] [CrossRef] [PubMed]
- Graffmann, N.; Ncube, A.; Martins, S.; Fiszl, A.R.; Reuther, P.; Bohndorf, M.; Wruck, W.; Beller, M.; Czekelius, C.; Adjaye, J. A stem cell based in vitro model of NAFLD enables the analysis of patient specific individual metabolic adaptations in response to a high fat diet and AdipoRon interference. Biol. Open 2021, 10. [Google Scholar] [CrossRef]
- Martins, S.; Erichsen, L.; Datsi, A.; Wruck, W.; Goering, W.; Chatzantonaki, E.; de Amorim, V.C.M.; Rossi, A.; Chrzanowska, K.H.; Adjaye, J. Impaired p53-Mediated DNA Damage Response Contributes to Microcephaly in Nijmegen Breakage Syndrome Patient-Derived Cerebral Organoids. Cells 2022, 11, 802. [Google Scholar] [CrossRef]
- Mlody, B.; Wruck, W.; Martins, S.; Sperling, K.; Adjaye, J. Nijmegen Breakage Syndrome fibroblasts and iPSCs: Cellular models for uncovering disease-associated signaling pathways and establishing a screening platform for anti-oxidants. Sci. Rep. 2017, 7, 7516. [Google Scholar] [CrossRef]
- Yamanaka, S. Patient-specific pluripotent stem cells become even more accessible. Cell Stem Cell 2010, 7, 1–2. [Google Scholar] [CrossRef]
- Yap, M.S.; Nathan, K.R.; Yeo, Y.; Lim, L.W.; Poh, C.L.; Richards, M.; Lim, W.L.; Othman, I.; Heng, B.C. Neural Differentiation of Human Pluripotent Stem Cells for Nontherapeutic Applications: Toxicology, Pharmacology, and In Vitro Disease Modeling. Stem Cells Int. 2015, 2015, 105172. [Google Scholar] [CrossRef]
- Graffmann, N.; Martins, S.; Ljubikj, T.; Matte, J.C.; Bohndorf, M.; Wruck, W.; Adjaye, J. Generation of a Crigler-Najjar Syndrome Type I patient-derived induced pluripotent stem cell line CNS705 (HHUUKDi005-A). Stem Cell Res. 2021, 51, 102167. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Abu-Dawud, R.; Graffmann, N.; Ferber, S.; Wruck, W.; Adjaye, J. Pluripotent stem cells: Induction and self-renewal. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Wang, Y.; Mah, N.; Prigione, A.; Wolfrum, K.; Andrade-Navarro, M.A.; Adjaye, J. A transcriptional roadmap to the induction of pluripotency in somatic cells. Stem Cell Rev. Rep. 2010, 6, 282–296. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 2009, 27, 2667–2674. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.H.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef]
- Kim, K.; Doi, A.; Wen, B.; Ng, K.; Zhao, R.; Cahan, P.; Kim, J.; Aryee, M.J.; Ji, H.; Ehrlich, L.I.; et al. Epigenetic memory in induced pluripotent stem cells. Nature 2010, 467, 285–290. [Google Scholar] [CrossRef]
- Bohndorf, M.; Ncube, A.; Spitzhorn, L.S.; Enczmann, J.; Wruck, W.; Adjaye, J. Derivation and characterization of integration-free iPSC line ISRM-UM51 derived from SIX2-positive renal cells isolated from urine of an African male expressing the CYP2D6 *4/*17 variant which confers intermediate drug metabolizing activity. Stem Cell Res. 2017, 25, 18–21. [Google Scholar] [CrossRef]
- Nguyen, L.; Wruck, W.; Erichsen, L.; Graffmann, N.; Adjaye, J. Correction: Nguyen et al. The Nephrotoxin Puromycin Aminonucleoside Induces Injury in Kidney Organoids Differentiated from Induced Pluripotent Stem Cells. Cells 2022, 11, 635, Erratum in: Cells 2022, 11, 1980. [Google Scholar] [CrossRef] [PubMed]
- Paulitschek, C.; Schulze-Matz, P.; Hesse, J.; Schmidt, T.; Wruck, W.; Adjaye, J.; Schrader, J. Generation and characterization of two iPSC lines from human epicardium-derived cells. Stem Cell Res. 2017, 20, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Okita, K.; Nakagawa, M.; Yamanaka, S. Induction of pluripotent stem cells from fibroblast cultures. Nat. Protoc. 2007, 2, 3081–3089. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Yan, Y.; Canfield, S.; Muravyeva, M.Y.; Kikuchi, C.; Zaja, I.; Corbett, J.A.; Bosnjak, Z.J. Ketamine enhances human neural stem cell proliferation and induces neuronal apoptosis via reactive oxygen species-mediated mitochondrial pathway. Anesth. Analg. 2013, 116, 869–880. [Google Scholar] [CrossRef]
- Kikuchi, C.; Bienengraeber, M.; Canfield, S.; Koopmeiner, A.; Schafer, R.; Bosnjak, Z.J.; Bai, X. Comparison of Cardiomyocyte Differentiation Potential Between Type 1 Diabetic Donor- and Nondiabetic Donor-Derived Induced Pluripotent Stem Cells. Cell Transplant. 2015, 24, 2491–2504. [Google Scholar] [CrossRef]
- Temple, S. The development of neural stem cells. Nature 2001, 414, 112–117. [Google Scholar] [CrossRef]
- Behnan, J.; Stangeland, B.; Langella, T.; Finocchiaro, G.; Tringali, G.; Meling, T.R.; Murrell, W. Identification and characterization of a new source of adult human neural progenitors. Cell Death Dis. 2017, 8, e2991. [Google Scholar] [CrossRef]
- Kriegstein, A.; Alvarez-Buylla, A. The glial nature of embryonic and adult neural stem cells. Ann. Rev. Neurosci. 2009, 32, 149–184. [Google Scholar] [CrossRef]
- Obernier, K.; Tong, C.K.; Alvarez-Buylla, A. Restricted nature of adult neural stem cells: Re-evaluation of their potential for brain repair. Front. Neurosci. 2014, 8, 162. [Google Scholar] [CrossRef]
- Elkabetz, Y.; Panagiotakos, G.; Al Shamy, G.; Socci, N.D.; Tabar, V.; Studer, L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008, 22, 152–165. [Google Scholar] [CrossRef]
- Yuan, T.; Liao, W.; Feng, N.H.; Lou, Y.L.; Niu, X.; Zhang, A.J.; Wang, Y.; Deng, Z.F. Human induced pluripotent stem cell-derived neural stem cells survive, migrate, differentiate, and improve neurologic function in a rat model of middle cerebral artery occlusion. Stem Cell Res. Ther. 2013, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Falk, A.; Koch, P.; Kesavan, J.; Takashima, Y.; Ladewig, J.; Alexander, M.; Wiskow, O.; Tailor, J.; Trotter, M.; Pollard, S.; et al. Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS ONE 2012, 7, e29597. [Google Scholar] [CrossRef]
- Ebert, A.D.; Shelley, B.C.; Hurley, A.M.; Onorati, M.; Castiglioni, V.; Patitucci, T.N.; Svendsen, S.P.; Mattis, V.B.; McGivern, J.V.; Schwab, A.J.; et al. EZ spheres: A stable and expandable culture system for the generation of pre-rosette multipotent stem cells from human ESCs and iPSCs. Stem Cell Res. 2013, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Nemati, S.; Hatami, M.; Kiani, S.; Hemmesi, K.; Gourabi, H.; Masoudi, N.; Alaei, S.; Baharvand, H. Long-term self-renewable feeder-free human induced pluripotent stem cell-derived neural progenitors. Stem Cells Dev. 2011, 20, 503–514. [Google Scholar] [CrossRef]
- Townshend, R.F.; Shao, Y.; Wang, S.; Cortez, C.L.; Esfahani, S.N.; Spence, J.R.; O’Shea, K.S.; Fu, J.; Gumucio, D.L.; Taniguchi, K. Effect of Cell Spreading on Rosette Formation by Human Pluripotent Stem Cell-Derived Neural Progenitor Cells. Front. Cell Dev Biol. 2020, 8, 588941. [Google Scholar] [CrossRef]
- Qi, Y.; Zhang, X.J.; Renier, N.; Wu, Z.; Atkin, T.; Sun, Z.; Ozair, M.Z.; Tchieu, J.; Zimmer, B.; Fattahi, F.; et al. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 2017, 35, 154–163. [Google Scholar] [CrossRef]
- Roybon, L.; Lamas, N.J.; Garcia, A.D.; Yang, E.J.; Sattler, R.; Lewis, V.J.; Kim, Y.A.; Kachel, C.A.; Rothstein, J.D.; Przedborski, S.; et al. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep. 2013, 4, 1035–1048. [Google Scholar] [CrossRef]
- Douvaras, P.; Wang, J.; Zimmer, M.; Hanchuk, S.; O’Bara, M.A.; Sadiq, S.; Sim, F.J.; Goldman, J.; Fossati, V. Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 250–259. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e279. [Google Scholar] [CrossRef]
- Sabitha, K.R.; Shetty, A.K.; Upadhya, D. Patient-derived iPSC modeling of rare neurodevelopmental disorders: Molecular pathophysiology and prospective therapies. Neurosci. Biobehav. Rev. 2021, 121, 201–219. [Google Scholar] [CrossRef]
- Han, F.; Hu, B. Stem Cell Therapy for Parkinson’s Disease. Adv. Exp. Med. Biol. 2020, 1266, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Koo, S.Y.; Studer, L. Pluripotent Stem Cell Therapies for Parkinson Disease: Present Challenges and Future Opportunities. Front. Cell Dev. Biol. 2020, 8, 729. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, J.; Millischer, V.; Wotawa, C.; Sawa, A.; Lanzenberger, R. Making Sense of Patient-Derived iPSCs, Transdifferentiated Neurons, Olfactory Neuronal Cells, and Cerebral Organoids as Models for Psychiatric Disorders. Int. J. Neuropsychopharmacol. 2021, 24, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef]
- Eiraku, M.; Takata, N.; Ishibashi, H.; Kawada, M.; Sakakura, E.; Okuda, S.; Sekiguchi, K.; Adachi, T.; Sasai, Y. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011, 472, 51–56. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Simian, M.; Bissell, M.J. Organoids: A historical perspective of thinking in three dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef]
- Yoon, S.J.; Elahi, L.S.; Pasca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of human cortical organoid generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef]
- Gabriel, E.; Ramani, A.; Karow, U.; Gottardo, M.; Natarajan, K.; Gooi, L.M.; Goranci-Buzhala, G.; Krut, O.; Peters, F.; Nikolic, M.; et al. Recent Zika Virus Isolates Induce Premature Differentiation of Neural Progenitors in Human Brain Organoids. Cell Stem Cell 2017, 20, 397–406.e395. [Google Scholar] [CrossRef]
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Giandomenico, S.L.; Sutcliffe, M.; Lancaster, M.A. Generation and long-term culture of advanced cerebral organoids for studying later stages of neural development. Nat. Protoc. 2021, 16, 579–602. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.J.; Do, J.T. Neural Lineage Differentiation From Pluripotent Stem Cells to Mimic Human Brain Tissues. Front. Bioeng. Biotechnol. 2019, 7, 400. [Google Scholar] [CrossRef] [PubMed]
- Kelava, I.; Lancaster, M.A. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev. Biol. 2016, 420, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Albecka, A.; Mallery, D.L.; Kellner, M.J.; Paul, D.; Carter, A.P.; James, L.C.; Lancaster, M.A. SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids. Cell Stem Cell 2020, 27, 951–961.e955. [Google Scholar] [CrossRef] [PubMed]
- Ciarpella, F.; Zamfir, R.G.; Campanelli, A.; Ren, E.; Pedrotti, G.; Bottani, E.; Borioli, A.; Caron, D.; Di Chio, M.; Dolci, S.; et al. Murine cerebral organoids develop network of functional neurons and hippocampal brain region identity. iScience 2021, 24, 103438. [Google Scholar] [CrossRef]
- Afanasyeva, T.A.V.; Corral-Serrano, J.C.; Garanto, A.; Roepman, R.; Cheetham, M.E.; Collin, R.W.J. A look into retinal organoids: Methods, analytical techniques, and applications. Cell Mol. Life Sci. 2021, 78, 6505–6532. [Google Scholar] [CrossRef]
- Miura, Y.; Li, M.Y.; Birey, F.; Ikeda, K.; Revah, O.; Thete, M.V.; Park, J.Y.; Puno, A.; Lee, S.H.; Porteus, M.H.; et al. Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 1421–1430. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sa, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Kim, H.; Jiang, P. Generation of human pluripotent stem cell-derived fused organoids with oligodendroglia and myelin. STAR Protoc. 2021, 2, 100443. [Google Scholar] [CrossRef] [PubMed]
- Shaker, M.R.; Pietrogrande, G.; Martin, S.; Lee, J.H.; Sun, W.; Wolvetang, E.J. Rapid and Efficient Generation of Myelinating Human Oligodendrocytes in Organoids. Front. Cell Neurosci. 2021, 15, 631548. [Google Scholar] [CrossRef] [PubMed]
- Zahmatkesh, E.; Khoshdel-Rad, N.; Mirzaei, H.; Shpichka, A.; Timashev, P.; Mahmoudi, T.; Vosough, M. Evolution of organoid technology: Lessons learnt in Co-Culture systems from developmental biology. Dev. Biol. 2021, 475, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Cukier, J.; Batzofin, J.H.; Conner, J.S.; Franklin, R.R. Genital tract reconstruction in a patient with congenital absence of the vagina and hypoplasia of the cervix. Obstet. Gynecol. 1986, 68, 32S–36S. [Google Scholar] [PubMed]
- Frohlich, E. Comparison of conventional and advanced in vitro models in the toxicity testing of nanoparticles. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1091–1107. [Google Scholar] [CrossRef]
- Kook, Y.M.; Jeong, Y.; Lee, K.; Koh, W.G. Design of biomimetic cellular scaffolds for co-culture system and their application. J. Tissue Eng. 2017, 8, 2041731417724640. [Google Scholar] [CrossRef]
- Nascimento, J.M.; Saia-Cereda, V.M.; Sartore, R.C.; da Costa, R.M.; Schitine, C.S.; Freitas, H.R.; Murgu, M.; de Melo Reis, R.A.; Rehen, S.K.; Martins-de-Souza, D. Human Cerebral Organoids and Fetal Brain Tissue Share Proteomic Similarities. Front. Cell Dev. Biol. 2019, 7, 303. [Google Scholar] [CrossRef]
- Liu, C.; Oikonomopoulos, A.; Sayed, N.; Wu, J.C. Modeling human diseases with induced pluripotent stem cells: From 2D to 3D and beyond. Development 2018, 145, dev156166. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef]
- Krencik, R.; Ullian, E.M. A cellular star atlas: Using astrocytes from human pluripotent stem cells for disease studies. Front. Cell Neurosci. 2013, 7, 25. [Google Scholar] [CrossRef]
- van den Hurk, M.; Bardy, C. Single-cell multimodal transcriptomics to study neuronal diversity in human stem cell-derived brain tissue and organoid models. J. Neurosci. Methods 2019, 325, 108350. [Google Scholar] [CrossRef] [PubMed]
- Goranci-Buzhala, G.; Mariappan, A.; Gabriel, E.; Ramani, A.; Ricci-Vitiani, L.; Buccarelli, M.; D’Alessandris, Q.G.; Pallini, R.; Gopalakrishnan, J. Rapid and Efficient Invasion Assay of Glioblastoma in Human Brain Organoids. Cell Rep. 2020, 31, 107738. [Google Scholar] [CrossRef] [PubMed]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Tanaka, Y.; Patterson, B.; Kang, Y.J.; Govindaiah, G.; Roselaar, N.; Cakir, B.; Kim, K.Y.; Lombroso, A.P.; Hwang, S.M.; et al. Fusion of Regionally Specified hPSC-Derived Organoids Models Human Brain Development and Interneuron Migration. Cell Stem Cell 2017, 21, 383–398.e387. [Google Scholar] [CrossRef] [PubMed]
- Pasca, S.P. Building three-dimensional human brain organoids. Nat. Neurosci. 2018, 1. [Google Scholar] [CrossRef]
- Sidhaye, J.; Knoblich, J.A. Brain organoids: An ensemble of bioassays to investigate human neurodevelopment and disease. Cell Death Differ 2021, 28, 52–67. [Google Scholar] [CrossRef]
- Koo, B.; Choi, B.; Park, H.; Yoon, K.J. Past, Present, and Future of Brain Organoid Technology. Mol. Cells 2019, 42, 617–627. [Google Scholar] [CrossRef]
- Bhutani, V.K.; Johnson, L.H.; Shapiro, S.M. Kernicterus in sick and preterm infants (1999–2002): A need for an effective preventive approach. Semin. Perinatol. 2004, 28, 319–325. [Google Scholar] [CrossRef]
- Moll, M.; Goelz, R.; Naegele, T.; Wilke, M.; Poets, C.F. Are recommended phototherapy thresholds safe enough for extremely low birth weight (ELBW) infants? A report on 2 ELBW infants with kernicterus despite only moderate hyperbilirubinemia. Neonatology 2011, 99, 90–94. [Google Scholar] [CrossRef]
- Conforti, L.; Adalbert, R.; Coleman, M.P. Neuronal death: Where does the end begin? Trends Neurosci. 2007, 30, 159–166. [Google Scholar] [CrossRef]
- Silva, R.F.; Mata, L.M.; Gulbenkian, S.; Brites, D. Endocytosis in rat cultured astrocytes is inhibited by unconjugated bilirubin. Neurochem. Res. 2001, 26, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.C.; Beart, P.M.; Shin, Y.S.; Chen, M.J.; Cheung, N.S.; Nagley, P. Oxidative stress: Emerging mitochondrial and cellular themes and variations in neuronal injury. J. Alzheimers Dis. 2010, 20, S453–S473. [Google Scholar] [CrossRef]
- Floyd, R.A.; Hensley, K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol. Aging 2002, 23, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Bhutani, V.K. The clinical syndrome of bilirubin-induced neurologic dysfunction. Semin. Perinatol. 2011, 35, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Wusthoff, C.J.; Loe, I.M. Impact of bilirubin-induced neurologic dysfunction on neurodevelopmental outcomes. Semin. Fetal Neonatal. Med. 2015, 20, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Qaisiya, M.; Brischetto, C.; Jasprova, J.; Vitek, L.; Tiribelli, C.; Bellarosa, C. Bilirubin-induced ER stress contributes to the inflammatory response and apoptosis in neuronal cells. Arch. Toxicol. 2017, 91, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Silva, R.F.; Falcao, A.S.; Brito, M.A.; Brites, D. Cytokine production, glutamate release and cell death in rat cultured astrocytes treated with unconjugated bilirubin and LPS. J. Neuroimmunol. 2004, 153, 64–75. [Google Scholar] [CrossRef]
- Fernandes, A.; Falcao, A.S.; Silva, R.F.; Gordo, A.C.; Gama, M.J.; Brito, M.A.; Brites, D. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J. Neurochem. 2006, 96, 1667–1679. [Google Scholar] [CrossRef]
Figure 1.
Schematic of the mechanisms involved in BIND induced neurotoxicity. Left: Metabolic pathway leading to unconjugated bilirubin (UCB) production. Heme is converted into biliverdin by heme oxygenase (HMOX-1 and 2), located in mitochondria, endoplasmic reticulum and caveolae. Biliverdin is then converted into unconjugated bilirubin by biliverdin reductase. Right: Neurons are depicted in this scheme to represent the toxic effects of UCB in brain cells. Neurons are known to be the most affected cell type in UCB toxicity, which involves multiple pathways leading to distinct toxic events, including disruption of the mitochondrial energetic breakdown, ionic imbalance, extracellular accumulation of glutamate, release of inflammatory cytokines by glial cells (here depicted as microglia and astrocytes), as well as increase in reactive oxygen species (ROS) production and oxidative stress. This UCB-induced cytotoxicity can result in apoptosis (the different cell type sizes are not depicted to scale, but rather schematically to simplify the view of the mechanisms) (Created with BioRender.com, accessed on 23 August 2022).
Figure 1.
Schematic of the mechanisms involved in BIND induced neurotoxicity. Left: Metabolic pathway leading to unconjugated bilirubin (UCB) production. Heme is converted into biliverdin by heme oxygenase (HMOX-1 and 2), located in mitochondria, endoplasmic reticulum and caveolae. Biliverdin is then converted into unconjugated bilirubin by biliverdin reductase. Right: Neurons are depicted in this scheme to represent the toxic effects of UCB in brain cells. Neurons are known to be the most affected cell type in UCB toxicity, which involves multiple pathways leading to distinct toxic events, including disruption of the mitochondrial energetic breakdown, ionic imbalance, extracellular accumulation of glutamate, release of inflammatory cytokines by glial cells (here depicted as microglia and astrocytes), as well as increase in reactive oxygen species (ROS) production and oxidative stress. This UCB-induced cytotoxicity can result in apoptosis (the different cell type sizes are not depicted to scale, but rather schematically to simplify the view of the mechanisms) (Created with BioRender.com, accessed on 23 August 2022).

Figure 2.
Overview of current models of BIND and Crigler–Najjar Syndrome. (A) In vivo models include the classical Gunn rat model, as well as knockout and transgenic mouse lines; (B) Some examples of in vitro models are primary cultures, mixed neuronal co-cultures as well as neuroblastoma cultures; (C) iPSCs can be generated by reprogramming of distinct types of human cells, such as fibroblasts, blood and urine-derived cells. The cells can be reprogrammed using the Yamanaka transcription factors (OCT4, SOX2, KLF4 and cMYC). A Crigler–Najjar Syndrome patient-derived iPSC model will enable future personalized medicine applications such as organoid cultures, organ-on-chip models and can be used for high throughput drug screenings specific to the patient’s needs (Created with BioRender.com, accessed on 21 August 2022).
Figure 2.
Overview of current models of BIND and Crigler–Najjar Syndrome. (A) In vivo models include the classical Gunn rat model, as well as knockout and transgenic mouse lines; (B) Some examples of in vitro models are primary cultures, mixed neuronal co-cultures as well as neuroblastoma cultures; (C) iPSCs can be generated by reprogramming of distinct types of human cells, such as fibroblasts, blood and urine-derived cells. The cells can be reprogrammed using the Yamanaka transcription factors (OCT4, SOX2, KLF4 and cMYC). A Crigler–Najjar Syndrome patient-derived iPSC model will enable future personalized medicine applications such as organoid cultures, organ-on-chip models and can be used for high throughput drug screenings specific to the patient’s needs (Created with BioRender.com, accessed on 21 August 2022).

Figure 3.
An overview of iPSC-derived 2D and 3D in vitro model generation with examples. Various protocols are available for generating iPSC-derived 2D and 3D in vitro models. (Ai) Schematic of iPSCs differentiation into neuronal cells. iPSC-derived neural progenitor cells (NPCs) have the capacity to differentiate into mature neuronal cells such as neurons, astrocytes, and oligodendrocytes. (Aii) NPCs can be obtained from 2D mono-layered iPSCs cultured as embryoid bodies (EBs) or cell aggregation in suspension via neural rosettes formation, or by direct induction from iPSCs to NPCs in 2D. NPCs generated in both approaches can be used for mono- or co-culture of distinct types of neuronal cells. Three-dimensional organoid cultures can be generated in a directed or non-directed manner, depending on their application purpose. (B) CNS-I patient-derived iPSCs 2D monolayer culture. (Bi) EBs generated from CNS-I patient-derived iPSCs. (Bii) Neural rosettes formation by replating EBs in 2D. (C) iPSCs are dissociated from the 2D culture to generate aggregates. (Ci) Cell aggregates generate spheroids in a shaking incubator. (D) Monolayered NPC culture. Immunofluorescence staining shows. (Di) Nestin-positive NPC cultures. (Dii) GFAP positive astrocytes. (Diii) MAP2-positive neurons (red) and GFAP-positive astrocytes (green) co-culture. (E) iPSC-derived 3D organoid culture in spinner flask. (Ei,Eii) Organoids at different time points of culturing. (Scales in 100 μm, bright-field and immunofluorescent staining images are taken from unpublished work in our lab) (Created with BioRender.com, accessed on 21 August 2022).
Figure 3.
An overview of iPSC-derived 2D and 3D in vitro model generation with examples. Various protocols are available for generating iPSC-derived 2D and 3D in vitro models. (Ai) Schematic of iPSCs differentiation into neuronal cells. iPSC-derived neural progenitor cells (NPCs) have the capacity to differentiate into mature neuronal cells such as neurons, astrocytes, and oligodendrocytes. (Aii) NPCs can be obtained from 2D mono-layered iPSCs cultured as embryoid bodies (EBs) or cell aggregation in suspension via neural rosettes formation, or by direct induction from iPSCs to NPCs in 2D. NPCs generated in both approaches can be used for mono- or co-culture of distinct types of neuronal cells. Three-dimensional organoid cultures can be generated in a directed or non-directed manner, depending on their application purpose. (B) CNS-I patient-derived iPSCs 2D monolayer culture. (Bi) EBs generated from CNS-I patient-derived iPSCs. (Bii) Neural rosettes formation by replating EBs in 2D. (C) iPSCs are dissociated from the 2D culture to generate aggregates. (Ci) Cell aggregates generate spheroids in a shaking incubator. (D) Monolayered NPC culture. Immunofluorescence staining shows. (Di) Nestin-positive NPC cultures. (Dii) GFAP positive astrocytes. (Diii) MAP2-positive neurons (red) and GFAP-positive astrocytes (green) co-culture. (E) iPSC-derived 3D organoid culture in spinner flask. (Ei,Eii) Organoids at different time points of culturing. (Scales in 100 μm, bright-field and immunofluorescent staining images are taken from unpublished work in our lab) (Created with BioRender.com, accessed on 21 August 2022).


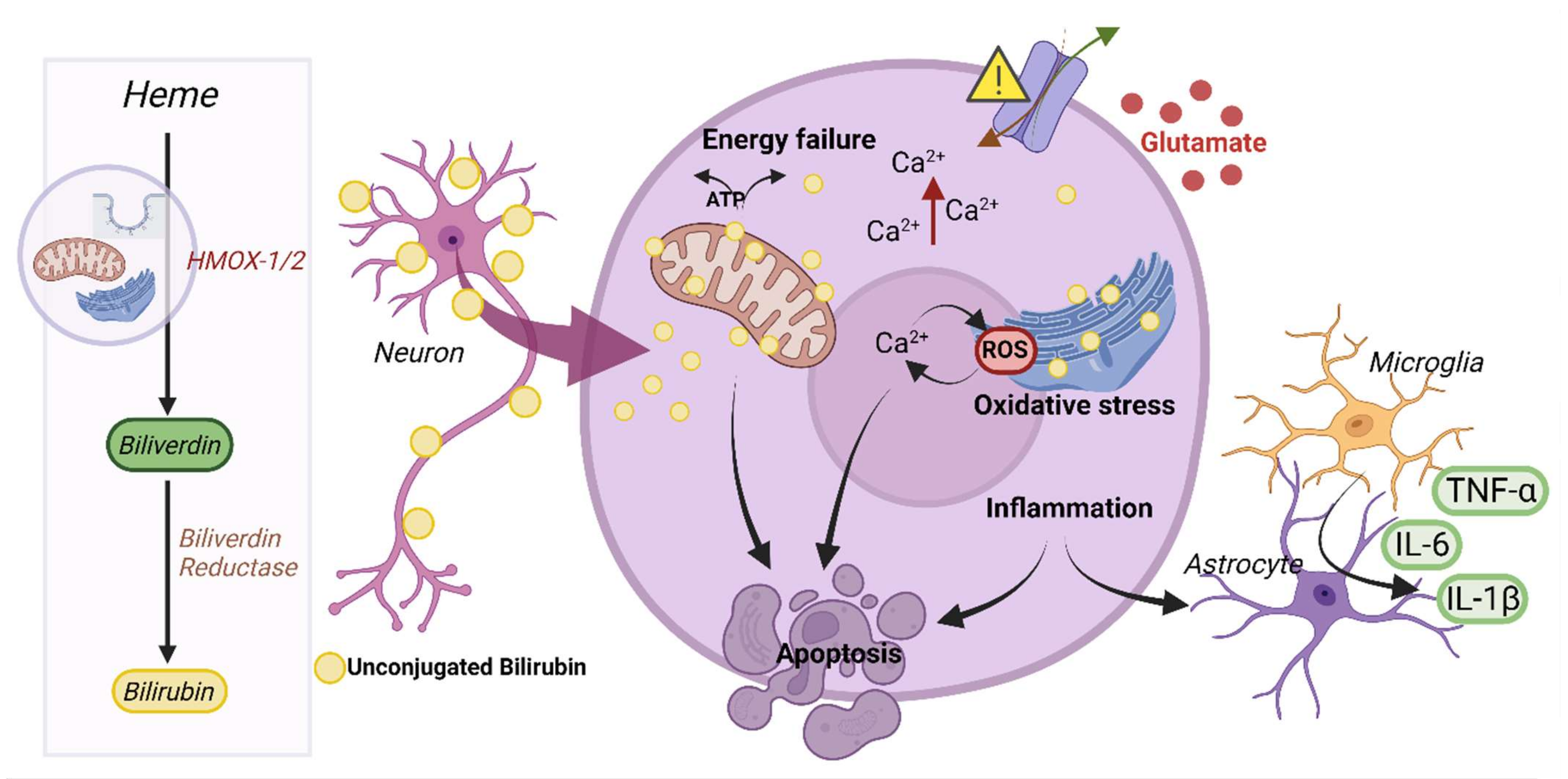{kind=link}
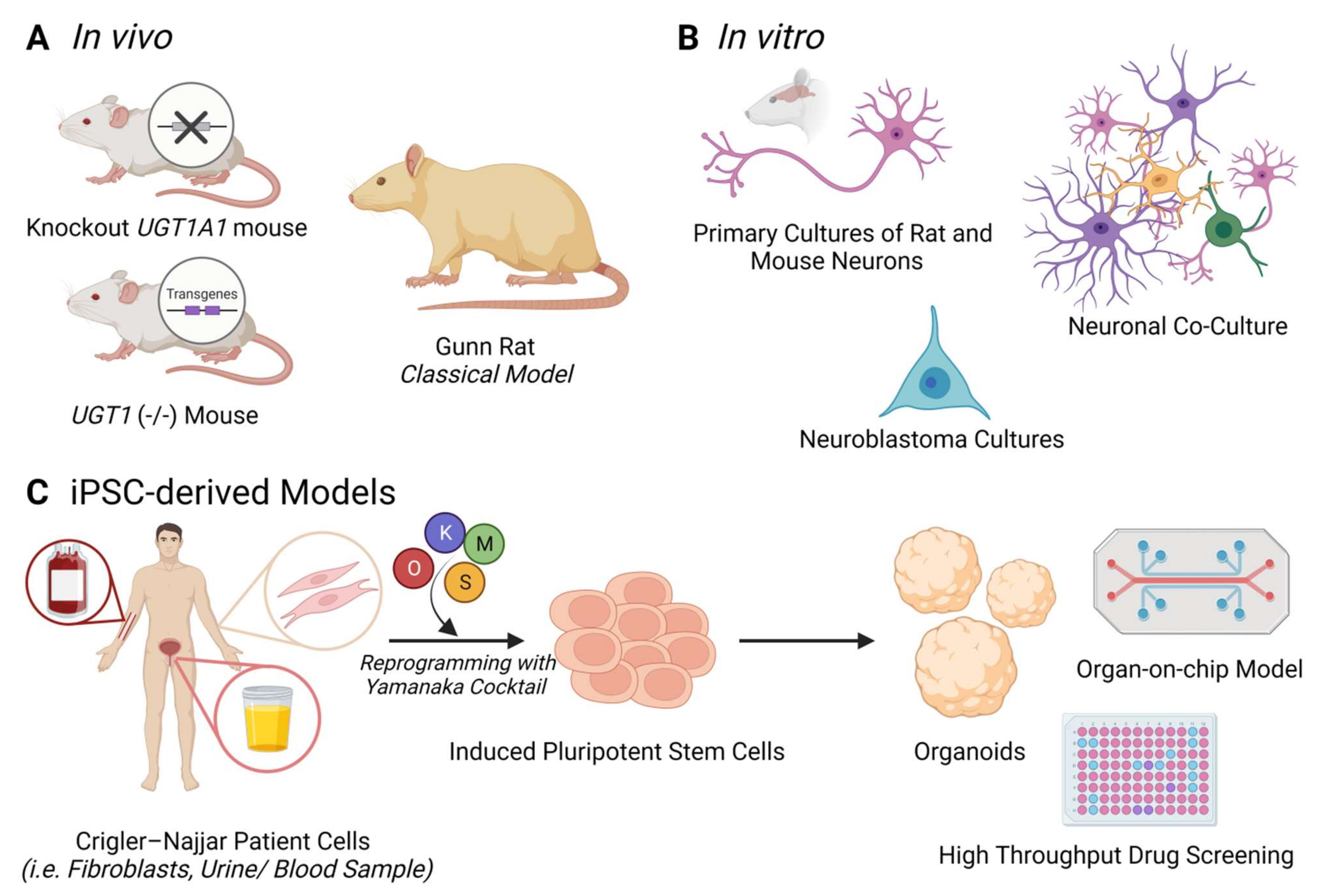{kind=link}
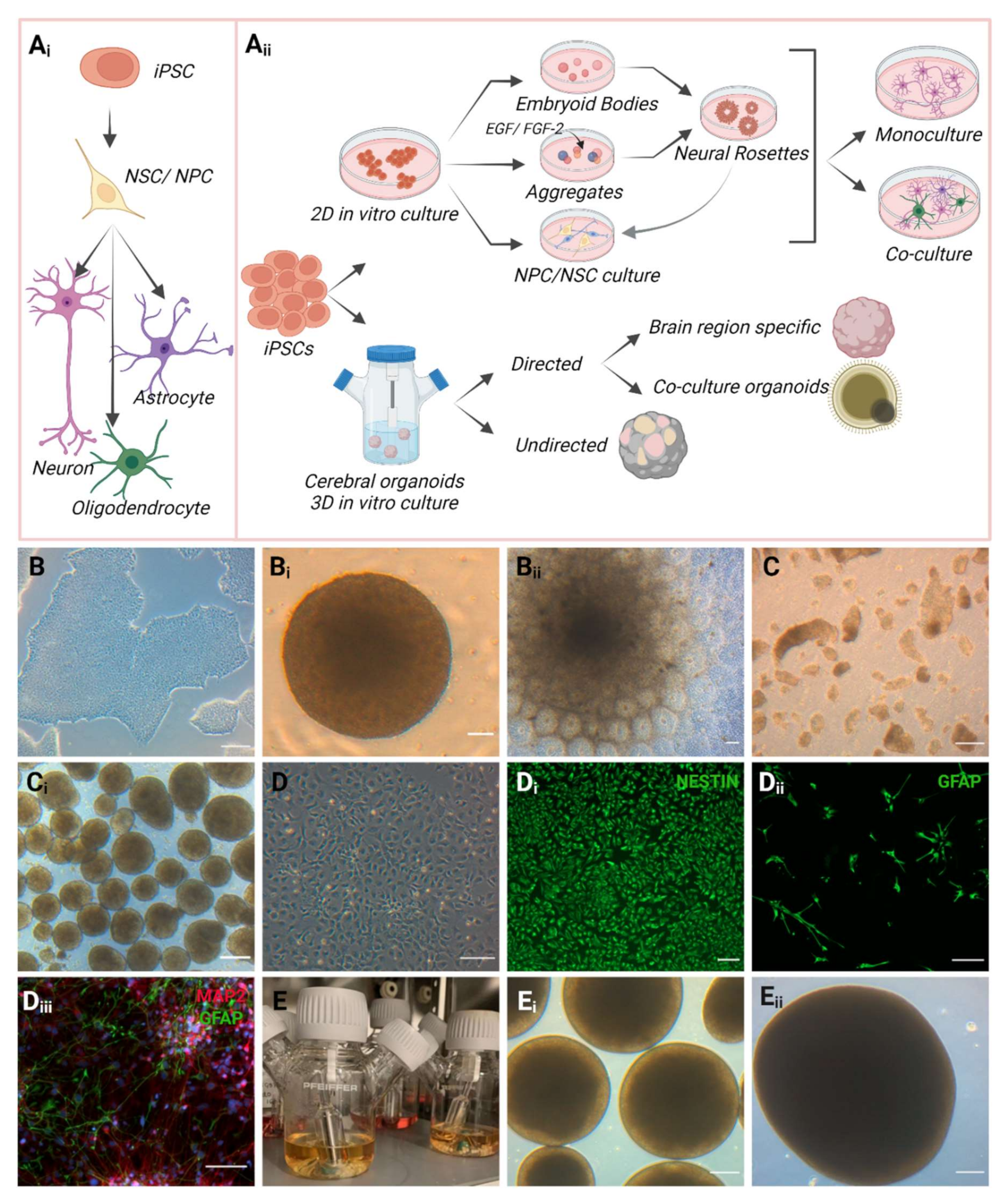{kind=link}
Table 1.
Overview of bilirubin-related diseases and clinical manifestations in the brain.
| Clinical Indication | Brain Target | Clinical Symptoms | Reference |
|---|---|---|---|
| Bilirubin-induced cerebral cortex injury |
|
| [5,21,22] |
| Basal ganglia injury |
|
| [21,23,24] |
| Bilirubin-induced cerebellar injury |
|
| [21] |
| Bilirubin-induced hippocampal injury |
|
| [5,21,25] |
| Bilirubin-induced auditory nervous system injury |
|
| [26,27] |
| Crigler–Najjar Syndrome Type I |
|
| [3] |
| Crigler–Najjar Syndrome Type II |
|
| [3] |
Table 2.
Existing treatments and therapies for CNS.
| Treatment | Advantages | Disadvantages | Reference |
|---|---|---|---|
| Phototherapy |
|
| [6,44] |
| Exchange transfusion |
|
| [44] |
| Intravenous immune globulin therapy |
|
| [6,41] |
| Liver transplantation |
|
| [2,44,45] |
| Phenobarbital |
|
| [6] |
| Metallophyrins |
|
| [41,46,47] |
| Minocycline |
|
| [6] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pranty, A.I.; Shumka, S.; Adjaye, J. Bilirubin-Induced Neurological Damage: Current and Emerging iPSC-Derived Brain Organoid Models. Cells 2022, 11, 2647. https://doi.org/10.3390/cells11172647
AMA Style
Pranty AI, Shumka S, Adjaye J. Bilirubin-Induced Neurological Damage: Current and Emerging iPSC-Derived Brain Organoid Models. Cells. 2022; 11(17):2647. https://doi.org/10.3390/cells11172647
Chicago/Turabian StylePranty, Abida Islam, Sara Shumka, and James Adjaye. 2022. "Bilirubin-Induced Neurological Damage: Current and Emerging iPSC-Derived Brain Organoid Models" Cells 11, no. 17: 2647. https://doi.org/10.3390/cells11172647
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.