
Serum of Post-COVID-19 Syndrome Patients with or without ME/CFS Differentially Affects Endothelial Cell Function In Vitro

 , , and
, , and
Abstract
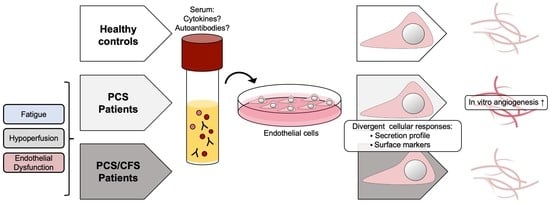
:
1. Introduction
2. Methods
2.1. Patients
2.2. Cell Culture
2.3. Cell-Based ELISA
2.4. Endothelial Cell Small Molecule Release Assay
2.5. Flow Cytometric Detection of Endothelial Cell Activation Markers
2.6. Serum Cytokine/Chemokine Measurement
2.7. Tube Formation Assay
2.8. Statistical Analysis
3. Results
3.1. Serum Factor Profile among the Patients Did Not Indicate Vascular Inflammation
3.2. Serum AECA Detection Revealed Elevated Levels among PCS/CFS Patients
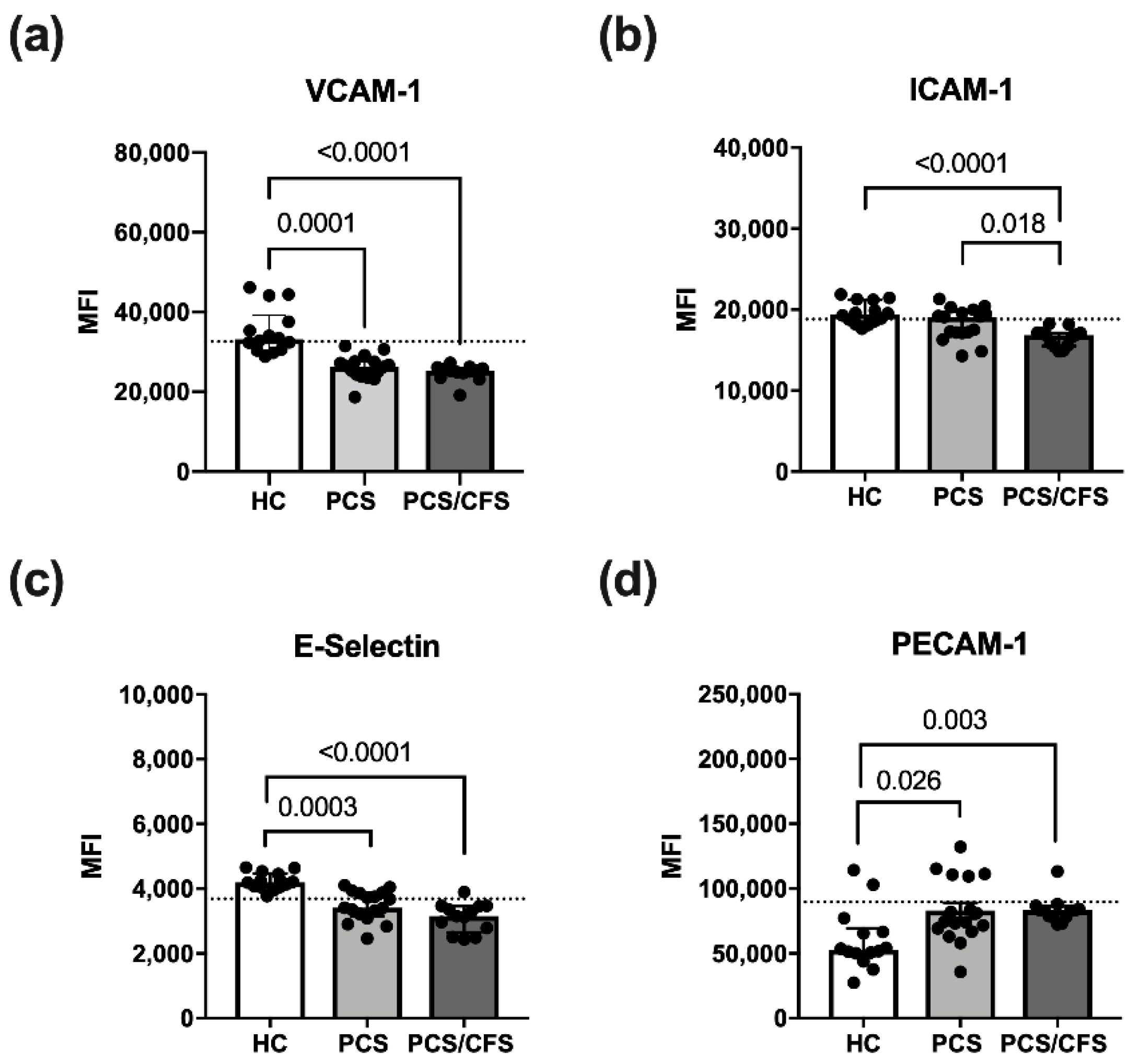
3.3. Reduced Activation Marker Surface Expression on HUVEC Following PCS/CFS and in Parts PCS Serum Incubation
3.4. Altered HUVEC Secretion Profile Differs between PCS and PCS/CFS Serum Incubation
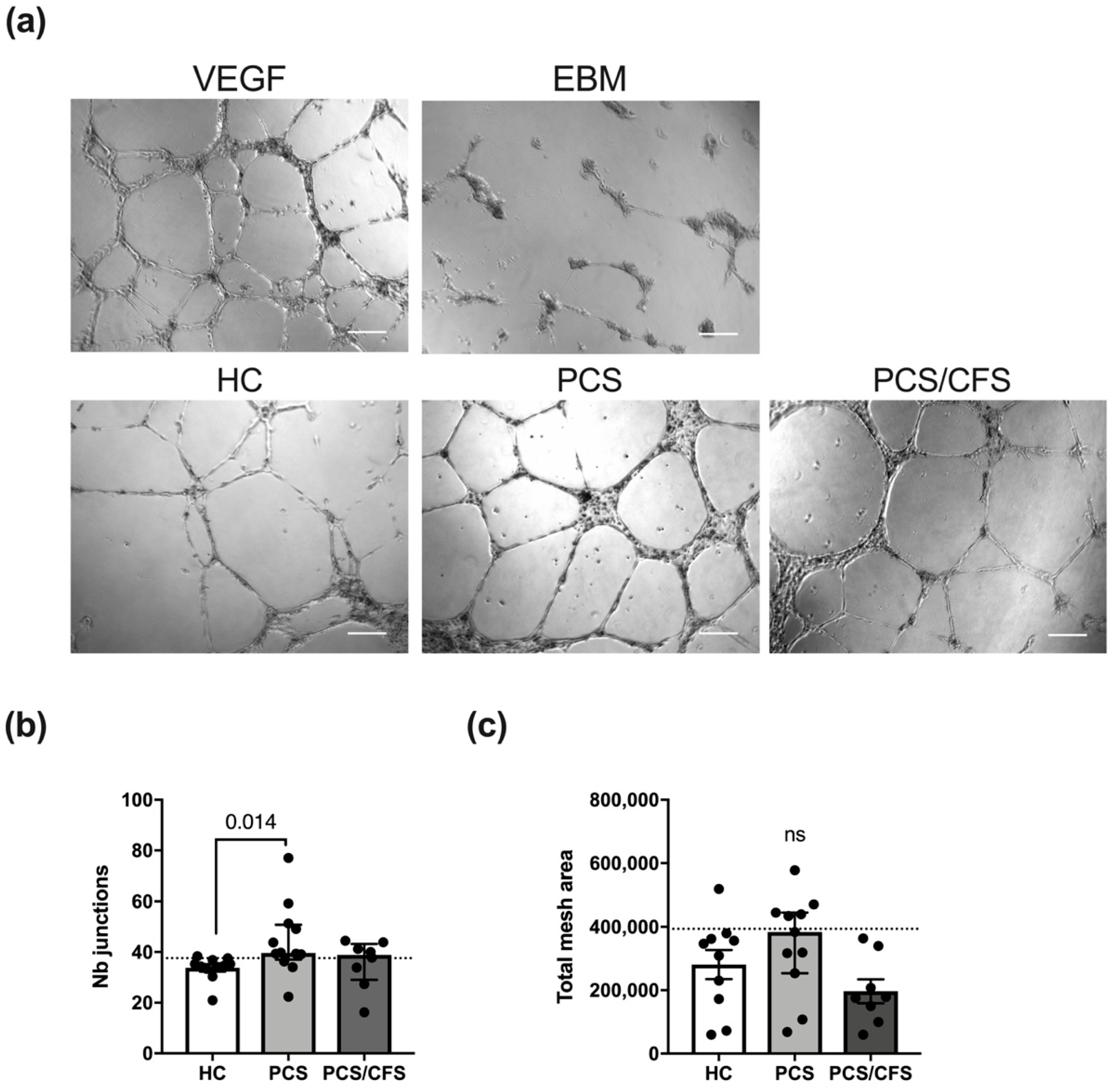
3.5. PCS and PCS/CFS Sera Differed in Their Pro-Angiogenic Potential In Vitro
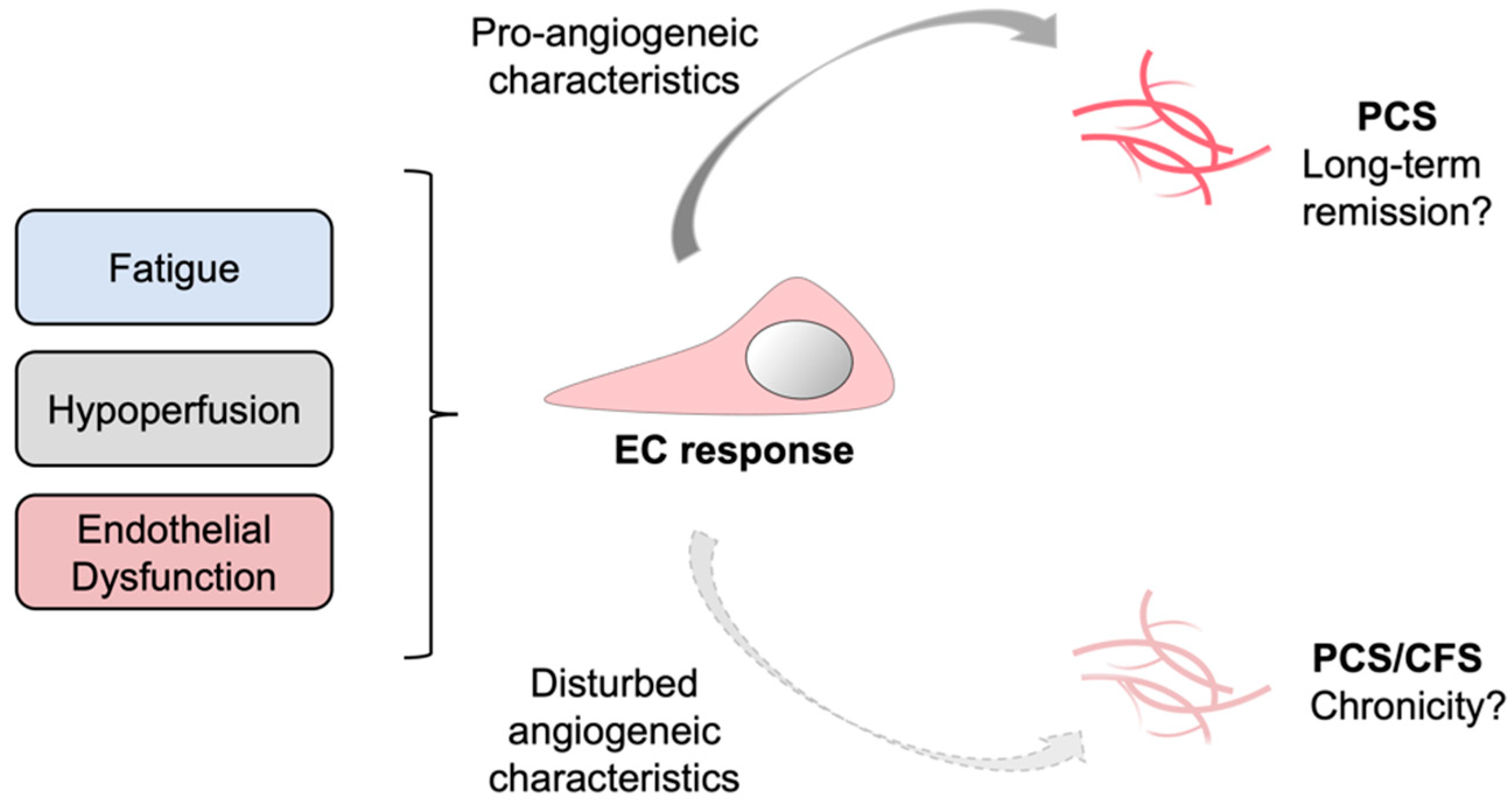
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nacul, L.; Authier, F.J.; Scheibenbogen, C.; Lorusso, L.; Helland, I.B.; Martin, J.A.; Sirbu, C.A.; Mengshoel, A.M.; Polo, O.; Behrends, U.; et al. European Network on Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (EUROMENE): Expert Consensus on the Diagnosis, Service Provision, and Care of People with ME/CFS in Europe. Medicina 2021, 57, 510. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.E.; Assaf, G.S.; McCorkell, L.; Wei, H.; Low, R.J.; Re’em, Y.; Redfield, S.; Austin, J.P.; Akrami, A. Characterizing Long COVID in an International Cohort: 7 Months of Symptoms and Their Impact. eClinicalMedicine 2021, 38, 101019. [Google Scholar] [CrossRef]
- Goërtz, Y.M.J.; Van Herck, M.; Delbressine, J.M.; Vaes, A.W.; Meys, R.; Machado, F.V.C.; Houben-Wilke, S.; Burtin, C.; Posthuma, R.; Franssen, F.M.E.; et al. Persistent Symptoms 3 Months after a SARS-CoV-2 Infection: The Post-COVID-19 Syndrome? ERJ Open Res. 2020, 6, 00542–02020. [Google Scholar] [CrossRef] [PubMed]
- Townsend, L.; Dyer, A.H.; Jones, K.; Dunne, J.; Mooney, A.; Gaffney, F.; O’Connor, L.; Leavy, D.; O’Brien, K.; Dowds, J.; et al. Persistent Fatigue Following SARS-CoV-2 Infection Is Common and Independent of Severity of Initial Infection. PLoS ONE 2020, 15, e0240784. [Google Scholar] [CrossRef] [PubMed]
- Sotzny, F.; Blanco, J.; Capelli, E.; Castro-Marrero, J.; Steiner, S.; Murovska, M.; Scheibenbogen, C. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome—Evidence for an Autoimmune Disease. Autoimmun. Rev. 2018, 17, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Kedor, C.; Freitag, H.; Meyer-Arndt, L.; Wittke, K.; Zoller, T.; Steinbeis, F.; Haffke, M.; Rudolf, G.; Heidecker, B.; Volk, H.; et al. Chronic COVID-19 Syndrome and Chronic Fatigue Syndrome (ME/CFS) Following the First Pandemic Wave in Germany—A First Analysis of a Prospective Observational Study. medRxiv 2021. [Google Scholar] [CrossRef]
- Fluge, Ø.; Tronstad, K.J.; Mella, O. Pathomechanisms and Possible Interventions in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). J. Clin. Investig. 2021, 131, e150377. [Google Scholar] [CrossRef]
- Alexander, Y.; Osto, E.; Schmidt-Trucksäss, A.; Shechter, M.; Trifunovic, D.; Duncker, D.J.; Aboyans, V.; Bäck, M.; Badimon, L.; Cosentino, F.; et al. Endothelial Function in Cardiovascular Medicine: A Consensus Paper of the European Society of Cardiology Working Groups on Atherosclerosis and Vascular Biology, Aorta and Peripheral Vascular Diseases, Coronary Pathophysiology and Microcirculation, and Thrombosis. Cardiovasc. Res. 2021, 117, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huertas, A.; Perros, F.; Tu, L.; Cohen-Kaminsky, S.; Montani, D.; Dorfmüller, P.; Guignabert, C.; Humbert, M. Immune Dysregulation and Endothelial Dysfunction in Pulmonary Arterial Hypertension. Circulation 2014, 129, 1332–1340. [Google Scholar] [CrossRef]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spanò, F.; Puppo, F. Endothelial Dysfunction in Rheumatic Autoimmune Diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef]
- Galley, H.F.; Webster, N.R. Physiology of the Endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, D.J.; Kennedy, G.; Chan, K.K.F.; Lang, C.C.; Belch, J.J.F.; Khan, F. Large and Small Artery Endothelial Dysfunction in Chronic Fatigue Syndrome. Int. J. Cardiol. 2012, 154, 335–336. [Google Scholar] [CrossRef]
- Scherbakov, N.; Szklarski, M.; Hartwig, J.; Sotzny, F.; Lorenz, S.; Meyer, A.; Grabowski, P.; Doehner, W.; Scheibenbogen, C. Peripheral Endothelial Dysfunction in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. ESC Heart Fail. 2020, 7, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Sørland, K.; Sandvik, M.K.; Rekeland, I.G.; Ribu, L.; Småstuen, M.C.; Mella, O.; Fluge, Ø. Reduced Endothelial Function in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome-Results From Open-Label Cyclophosphamide Intervention Study. Front. Med. 2021, 8, 642710. [Google Scholar] [CrossRef] [PubMed]
- Haffke, M.; Freitag, H.; Rudolf, G.; Seifert, M.; Doehner, W.; Scherbakov, N.; Hanitsch, L.; Wittke, K.; Bauer, S.; Konietschke, F.; et al. Endothelial Dysfunction and Altered Endothelial Biomarkers in Patients with Post-COVID-19 Syndrome and Chronic Fatigue Syndrome (ME/CFS). J. Transl. Med. 2022, 20, 138. [Google Scholar] [CrossRef] [PubMed]
- Castro, P.; Palomo, M.; Moreno-Castaño, A.B.; Fernández, S.; Torramadé-Moix, S.; Pascual, G.; Martinez-Sanchez, J.; Richardson, E.; Téllez, A.; Nicolas, J.M.; et al. Is the Endothelium the Missing Link in the Pathophysiology and Treatment of COVID-19 Complications? Cardiovasc. Drugs 2021, 36, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Georg, P.; Astaburuaga-García, R.; Bonaguro, L.; Brumhard, S.; Michalick, L.; Lippert, L.J.; Kostevc, T.; Gäbel, C.; Schneider, M.; Streitz, M.; et al. Complement Activation Induces Excessive T Cell Cytotoxicity in Severe COVID-19. Cell 2022, 185, 493–512.e25. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.; Distler, O. How Does Endothelial Cell Injury Start? The Role of Endothelin in Systemic Sclerosis. Arthritis Res. Ther. 2007, 9, S2. [Google Scholar] [CrossRef] [Green Version]
- Atehortúa, L.; Rojas, M.; Vásquez, G.M.; Castaño, D. Endothelial Alterations in Systemic Lupus Erythematosus and Rheumatoid Arthritis: Potential Effect of Monocyte Interaction. Mediat. Inflamm. 2017, 2017, 9680729. [Google Scholar] [CrossRef]
- Montoya, J.G.; Holmes, T.H.; Anderson, J.N.; Maecker, H.T.; Rosenberg-Hasson, Y.; Valencia, I.J.; Chu, L.; Younger, J.W.; Tato, C.M.; Davis, M.M. Cytokine Signature Associated with Disease Severity in Chronic Fatigue Syndrome Patients. Proc. Natl. Acad. Sci. USA 2017, 114, E7150–E7158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.C.P.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Null 2003, 11, 7–115. [Google Scholar] [CrossRef]
- Cotler, J.; Holtzman, C.; Dudun, C.; Jason, L.A. A Brief Questionnaire to Assess Post-Exertional Malaise. Diagnostics 2018, 8, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal Keratinization in a Spontaneously Immortalized Aneuploid Human Keratinocyte Cell Line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Yang, Y.; Wang, D.; Li, C.; Qu, Y.; Guo, J.; Shi, T.; Bo, W.; Sun, Z.; Asakawa, T. The Clinical Value of Cytokines in Chronic Fatigue Syndrome. J. Transl. Med. 2019, 17, 213. [Google Scholar] [CrossRef] [PubMed]
- Corbitt, M.; Eaton-Fitch, N.; Staines, D.; Cabanas, H.; Marshall-Gradisnik, S. A Systematic Review of Cytokines in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis/Systemic Exertion Intolerance Disease (CFS/ME/SEID). BMC Neurol. 2019, 19, 207. [Google Scholar] [CrossRef] [PubMed]
- Komaroff, A.L. Inflammation Correlates with Symptoms in Chronic Fatigue Syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 8914–8916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maamar, M.; Artime, A.; Pariente, E.; Fierro, P.; Ruiz, Y.; Gutiérrez, S.; Tobalina, M.; Díaz-Salazar, S.; Ramos, C.; Olmos, J.M.; et al. Post-COVID-19 Syndrome, Low-Grade Inflammation and Inflammatory Markers: A Cross-Sectional Study. Curr. Med. Res. Opin. 2022, 38, 901–909. [Google Scholar] [CrossRef]
- Maltezou, H.C.; Pavli, A.; Tsakris, A. Post-COVID Syndrome: An Insight on Its Pathogenesis. Vaccines 2021, 9, 497. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, C.; Bombardieri, M.; Valesini, G. Pathogenic Mechanisms of Anti-Endothelial Cell Antibodies (AECA): Their Prevalence and Clinical Relevance. Adv. Clin. Chem. 2006, 42, 297–326. [Google Scholar] [CrossRef] [PubMed]
- Mihai, C.; Tervaert, J.W.C. Anti-Endothelial Cell Antibodies in Systemic Sclerosis. Ann. Rheum. Dis. 2010, 69, 319. [Google Scholar] [CrossRef]
- Liao, J.K. Linking Endothelial Dysfunction with Endothelial Cell Activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef] [PubMed]
- Woodfin, A.; Voisin, M.-B.; Nourshargh, S. PECAM-1: A Multi-Functional Molecule in Inflammation and Vascular Biology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2514–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, H.; Hampton, D.; Marques de Menezes, E.G.; Deng, X.; Montoya, J.G.; Anderson, J.; Norris, P.J. Comparative Analysis of Extracellular Vesicles in Patients with Severe and Mild Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Immunol. 2022, 13, 841910. [Google Scholar] [CrossRef] [PubMed]
- Giloteaux, L.; O’Neal, A.; Castro-Marrero, J.; Levine, S.M.; Hanson, M.R. Cytokine Profiling of Extracellular Vesicles Isolated from Plasma in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: A Pilot Study. J. Transl. Med. 2020, 18, 387. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Kiss, T.; Wren, J.D.; Giles, C.B.; Griffin, C.T.; Murfee, W.L.; Pacher, P.; Csiszar, A. Endothelial Dysfunction and Angiogenesis Impairment in the Ageing Vasculature. Nat. Rev. Cardiol. 2018, 15, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Romano, E.; Rosa, I.; Fioretto, B.S.; Guiducci, S.; Bellando-Randone, S.; Pigatto, E.; Cozzi, F.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Systemic Sclerosis Serum Significantly Impairs the Multi-Step Lymphangiogenic Process: In Vitro Evidence. Int. J. Mol. Sci. 2019, 20, 6189. [Google Scholar] [CrossRef] [Green Version]
- Distler, J.H.W.; Gay, S.; Distler, O. Angiogenesis and Vasculogenesis in Systemic Sclerosis. Rheumatology 2006, 45 (Suppl. 3), iii26–iii27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, K.; Scheibenbogen, C. A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the Finding of Autoantibodies against SS2-Adrenergic Receptors. Autoimmun. Rev. 2020, 19, 102527. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.N.; Davidge, S.T.; Barankiewicz, J.; Roberts, J.M. Plasma of Preeclamptic Women Stimulates and Then Inhibits Endothelial Prostacyclin. Hypertension 1996, 27, 56–61. [Google Scholar] [CrossRef]
- Baker, P.N.; Davidge, S.T.; Roberts, J.M. Plasma From Women With Preeclampsia Increases Endothelial Cell Nitric Oxide Production. Hypertension 1995, 26, 244–248. [Google Scholar] [CrossRef]
- Bertinat, R.; Villalobos-Labra, R.; Hofmann, L.; Blauensteiner, J.; Sepúlveda, N.; Westermeier, F. Decreased NO Production in Endothelial Cells Exposed to Plasma from ME/CFS Patients. Vasc. Pharmacol. 2022, 143, 106953. [Google Scholar] [CrossRef]
- Sankaralingam, S.; Xu, H.; Davidge, S.T. Arginase Contributes to Endothelial Cell Oxidative Stress in Response to Plasma from Women with Preeclampsia. Cardiovasc. Res. 2010, 85, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Sankaralingam, S.; Xu, Y.; Sawamura, T.; Davidge, S.T. Increased Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 Expression in the Maternal Vasculature of Women With Preeclampsia. Hypertension 2009, 53, 270–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belizna, C.; Duijvestijn, A.; Hamidou, M.; Tervaert, J.W.C. Antiendothelial Cell Antibodies in Vasculitis and Connective Tissue Disease. Ann. Rheum. Dis. 2006, 65, 1545–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillevin, L.; Dörner, T. Vasculitis: Mechanisms Involved and Clinical Manifestations. Arthritis Res. Ther. 2007, 9 (Suppl. 2), S9. [Google Scholar] [CrossRef] [Green Version]
- Castellon, X.; Bogdanova, V. Chronic Inflammatory Diseases and Endothelial Dysfunction. Aging. Dis. 2016, 7, 81–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, L.; Pink, I.; Kühne, J.F.; Beushausen, K.; Keil, J.; Christoph, S.; Sauer, A.; Boblitz, L.; Schmidt, J.; David, S.; et al. Endothelial Dysfunction Contributes to Severe COVID-19 in Combination with Dysregulated Lymphocyte Responses and Cytokine Networks. Signal Transduct. Target. Ther. 2021, 6, 418. [Google Scholar] [CrossRef] [PubMed]
- Gamble, J.R.; Bradley, S.; Noack, L.; Vadas, M.A. TGF-β and Endothelial Cells Inhibit VCAM-1 Expression on Human Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 949–955. [Google Scholar] [CrossRef]
- Gamble, J.R.; Khew-Goodall, Y.; Vadas, M.A. Transforming Growth Factor-Beta Inhibits E-Selectin Expression on Human Endothelial Cells. J. Immunol. 1993, 150, 4494. [Google Scholar] [PubMed]
- Park, S.; Yang, W.S.; Lee, S.K.; Ahn, H.; Park, J.S.; Hwang, O.; Lee, J.D. TGF-β1 Down-regulates Inflammatory Cytokine-induced VCAM-1 Expression in Cultured Human Glomerular Endothelial Cells. Nephrol. Dial. Transplant. 2000, 15, 596–604. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Sun, L.; Tsuruoka, N.; Ishigaki, Y.; Yoshitomi, Y.; Yoshitake, Y.; Yonekura, H. Hypoxia Down-Regulates SFlt-1 (SVEGFR-1) Expression in Human Microvascular Endothelial Cells by a Mechanism Involving MRNA Alternative Processing. Biochem. J. 2011, 436, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Chioh, F.W.; Fong, S.-W.; Young, B.E.; Wu, K.-X.; Siau, A.; Krishnan, S.; Chan, Y.-H.; Carissimo, G.; Teo, L.L.; Gao, F.; et al. Convalescent COVID-19 Patients Are Susceptible to Endothelial Dysfunction Due to Persistent Immune Activation. Elife 2021, 10, e64909. [Google Scholar] [CrossRef] [PubMed]
- Freitag, H.; Szklarski, M.; Lorenz, S.; Sotzny, F.; Bauer, S.; Philippe, A.; Kedor, C.; Grabowski, P.; Lange, T.; Riemekasten, G.; et al. Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Clin. Med. 2021, 10, 3675. [Google Scholar] [CrossRef] [PubMed]
- Byrne, G.J.; Ghellal, A.; Iddon, J.; Blann, A.D.; Venizelos, V.; Kumar, S.; Howell, A.; Bundred, N.J. Serum Soluble Vascular Cell Adhesion Molecule-1: Role as a Surrogate Marker of Angiogenesis. JNCI J. Natl. Cancer Inst. 2000, 92, 1329–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gho, Y.S.; Kleinman, H.K.; Sosne, G. Angiogenic Activity of Human Soluble Intercellular Adhesion Molecule-1. Cancer Res. 1999, 59, 5128–5132. [Google Scholar] [PubMed]
- Haarmann, A.; Nowak, E.; Deiß, A.; van der Pol, S.; Monoranu, C.-M.; Kooij, G.; Müller, N.; van der Valk, P.; Stoll, G.; de Vries, H.E.; et al. Soluble VCAM-1 Impairs Human Brain Endothelial Barrier Integrity via Integrin α-4-Transduced Outside-in Signalling. Acta. Neuropathol. 2015, 129, 639–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakao, S.; Kuwano, T.; Ishibashi, T.; Kuwano, M.; Ono, M. Synergistic Effect of TNF-α in Soluble VCAM-1-Induced Angiogenesis Through α4 Integrins. J. Immunol. 2003, 170, 5704. [Google Scholar] [CrossRef] [Green Version]
- Dirkx, A.; Oude Egbrink, M.; Castermans, K.; Schaft, D.; Thijssen, V.; Dings, R.; Kwee, L.; Mayo, K.; Wagstaff, J.; Steege, J.; et al. Anti-Angiogenesis Therapy Can Overcome Endothelial Cell Anergy and Promote Leukocyte-Endothelium Interactions and Infiltration in Tumors. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, G.; O’Brien, C.D.; Zhou, Z.; Sanders, S.M.; Greenbaum, J.N.; Makrigiannakis, A.; DeLisser, H.M. Involvement of Human PECAM-1 in Angiogenesis and in Vitro Endothelial Cell Migration. Am. J. Physiol. Cell Physiol. 2002, 282, C1181–C1190. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, H.; Yan, L.; Du, W.; Zhang, M.; Chen, H.; Zhang, L.; Li, G.; Li, J.; Dong, Y.; et al. MMP-2 and MMP-9 Contribute to the Angiogenic Effect Produced by Hypoxia/15-HETE in Pulmonary Endothelial Cells. J. Mol. Cell. Cardiol. 2018, 121, 36–50. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [Green Version]
- DI Carlo, A. Evaluation of Neutrophil Gelatinase-Associated Lipocalin (NGAL), Matrix Metalloproteinase-9 (MMP-9) and Their Complex MMP-9/NGAL in Sera and Urine of Patients with Kidney Tumors. Oncol. Lett. 2013, 5, 1677–1681. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Borregaard, N.; Kjeldsen, L.; Moses, M.A. The High Molecular Weight Urinary Matrix Metalloproteinase (MMP) Activity Is a Complex of Gelatinase B/MMP-9 and Neutrophil Gelatinase-Associated Lipocalin (NGAL): MODULATION OF MMP-9 ACTIVITY BY NGAL. J. Biol. Chem. 2001, 276, 37258–37265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, A.; Medfai, H.; Poelvoorde, P.; Kazan, M.F.; Delporte, C.; Van Antwerpen, P.; EL-Makhour, Y.; Biston, P.; Delrée, P.; Badran, B.; et al. Myeloperoxidase Promotes Tube Formation, Triggers ERK1/2 and Akt Pathways and Is Expressed Endogenously in Endothelial Cells. Arch. Biochem. Biophys. 2018, 654, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, V.; Zinonos, I.; Leach, D.A.; Hay, S.J.; Liapis, V.; Zysk, A.; Ingman, W.V.; DeNichilo, M.O.; Evdokiou, A. Uncovering a New Role for Peroxidase Enzymes as Drivers of Angiogenesis. Int. J. Biochem. Cell Biol. 2015, 68, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Talib, J.; Stanley, C.P.; Rashid, I.; Michaëlsson, E.; Lindstedt, E.-L.; Croft, K.D.; Kettle, A.J.; Maghzal, G.J.; Stocker, R. Inhibition of MPO (Myeloperoxidase) Attenuates Endothelial Dysfunction in Mouse Models of Vascular Inflammation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- La Rocca, G.; Di Stefano, A.; Eleuteri, E.; Anzalone, R.; Magno, F.; Corrao, S.; Loria, T.; Martorana, A.; Di Gangi, C.; Colombo, M.; et al. Oxidative Stress Induces Myeloperoxidase Expression in Endocardial Endothelial Cells from Patients with Chronic Heart Failure. Basic Res. Cardiol. 2009, 104, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Benndorf, R.A. Renal Biomarker and Angiostatic Mediator? Cystatin C as a Negative Regulator of Vascular Endothelial Cell Homeostasis and Angiogenesis. J. Am. Heart Assoc. 2018, 7, e010997. [Google Scholar] [CrossRef] [Green Version]
- Hessian, P.; Edgeworth, J.; Hogg, N.; Hessian, P.A.; Edgeworth, J. Hogg NMRP-8 and MRP-14, Two Abundant Ca(2+)-Binding Proteins of Neutrophils and Monocytes. J. Leukoc. Biol. 1993, 53, 197–204. [Google Scholar] [CrossRef]
- Ehrchen, J.M.; Sunderkötter, C.; Foell, D.; Vogl, T.; Roth, J. The Endogenous Toll-like Receptor 4 Agonist S100A8/S100A9 (Calprotectin) as Innate Amplifier of Infection, Autoimmunity, and Cancer. J. Leukoc. Biol. 2009, 86, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.L.; Garrie, K.; Turner, M.D. Role of S100 Proteins in Health and Disease. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118677. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, S.; Jia, C.; Yang, L.; Song, Z.; Wang, Y. Low Concentration of S100A8/9 Promotes Angiogenesis-Related Activity of Vascular Endothelial Cells: Bridges among Inflammation, Angiogenesis, and Tumorigenesis? Mediat. Inflamm. 2012, 2012, 248574. [Google Scholar] [CrossRef]
- Dobrucki, L.W.; Tsutsumi, Y.; Kalinowski, L.; Dean, J.; Gavin, M.; Sen, S.; Mendizabal, M.; Sinusas, A.J.; Aikawa, R. Analysis of Angiogenesis Induced by Local IGF-1 Expression after Myocardial Infarction Using MicroSPECT-CT Imaging. J. Mol. Cell Cardiol. 2010, 48, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.J.; Ball, M.; Rukhlova, M.; Slinn, J.; L’abbe, D.; Iqbal, U.; Monette, R.; Hagedorn, M.; O’Connor-McCourt, M.D.; Durocher, Y.; et al. IGFBP-4 Anti-Angiogenic and Anti-Tumorigenic Effects Are Associated with Anti-Cathepsin B Activity. Neoplasia 2013, 15, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Delafontaine, P.; Song, Y.-H.; Li, Y. Expression, Regulation, and Function of IGF-1, IGF-1R, and IGF-1 Binding Proteins in Blood Vessels. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 435–444. [Google Scholar] [CrossRef]
- Gatenby, V.K.; Imrie, H.; Kearney, M. The IGF-1 Receptor and Regulation of Nitric Oxide Bioavailability and Insulin Signalling in the Endothelium. Pflügers Arch. Eur. J. Physiol. 2013, 465, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.J.; Krishnan, L.; Sullivan, C.J.; Williams, S.K.; Hoying, J.B. Microvascular Repair: Post-Angiogenesis Vascular Dynamics. Microcirculation 2012, 19, 676–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Angiogenesis Dysregulation in the Pathogenesis of Systemic Sclerosis. Biomed Res. Int. 2017, 2017, 5345673. [Google Scholar] [CrossRef] [PubMed]
- Matucci-Cerinic, M.; Manetti, M.; Bruni, C.; Chora, I.; Bellando-Randone, S.; Lepri, G.; De Paulis, A.; Guiducci, S. The “Myth” of Loss of Angiogenesis in Systemic Sclerosis: A Pivotal Early Pathogenetic Process or Just a Late Unavoidable Event? Arthritis Res. Ther. 2017, 19, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, L.; Valencia, I.J.; Garvert, D.W.; Montoya, J.G. Onset Patterns and Course of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Pediatr. 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HC (n = 14) | PCS (n = 17) | PCS/CFS (n = 13) | |
| Age, Mean (Range) | 45 (31–58) | 42 (27–66) | 43 (24–59) |
| Sex (f/m) | (12/2) | (16/1) | (11/2) |
| Months Since COVID-19 Infection, Mean (Range) | n/a | 8.3 (4.3–11.6) | 9.4 (8.2–11.1) |
| Bell Disability Scale, Mean (Range) | n/a | 48.24 (10–80) | 45.38 (20–80) |
| Chalder Fatigue Scale, Mean (Range) | n/a | 24.76 (15–32) | 26 (20–33) |
| PEM Score, Mean (Range) | n/a | 25.88 (17–46) | 30.92 (16–44) |
| Serum Cytokine | HC (n = 14) Median (IQR) (pg/mL) | PCS (n = 17) Median (IQR) (pg/mL) | PCS/CFS (n = 13) Median (IQR) (pg/mL) | p Value |
|---|---|---|---|---|
| sCD40L | 3583 (2650–4269) | 3570 (2349–4414) | 3256 (2478–4219) | p1: >0.999 p2: >0.999 |
| IL-6 | 6.9 (5.2–10.7) | 5.9 (5.4–9.7) | 6 (4.9–8.1) | p1: >0.999 p2: >0.999 |
| IL-10 | 2 (1.5–3.2) | 2.4 (2.1–3) | 2 (1.6–2.3) | p1: 0.3500 p2: >0.999 |
| IL-18 | 315.7 (263.6–341) | 235.4 (167.3–260.4) | 235.7 (180.6–395.5) | p1: 0.0104 p2: 0.4822 |
| MCP-1 | 67.8 (55.9–100.8) | 57.4 (45.1–85.8) | 56.69 (46.9–67.2) | p1: 0.4611 p2: 0.4597 |
| PIGF | 10.8 (7.8–15) | 10.5 (7–15.4) | 11.7 (8.3–14.3) | p1: >0.999 p2: >0.999 |
| sRAGE | 410.9 (349.5–735.2) | 475.8 (235.1–748.1) | 345.8 (248.5–538.6) | p1: >0.999 p2: 0.6656 |
| sST2 | 114.5 (71.2–213) | 62.5 (42.5–196.5) | 88.9 (43.3–209.1) | p1: 0.9186 p2: >0.999 |
| sVEGFR | 1983 (1728–2112) | 1417 (1064–1910) | 1384 (1149–1920) | p1: 0.0230 p2: 0.0500 |
| TGF-β1 | 37,529 (29,794–40,906) | 41,009 (33,963–46,821) | 34083 (30,939–42,905) | p1: 0.4044 p2: >0.999 |
| TNF-α | 15.35 (10.4–32.6) | 24 (13.5–43.3) | 23 (9.3–52.1) | p1: 0.6820 p2: >0.999 |
| TNFSF14 | 36.25 (18.5–52.6) | 47.53 (29.5–68.3) | 42.39 (27.9–81.5) | p1: 0.5673 p2: 0.7790 |
| VEGF | 291.4 (38.20) | 293.3 (38.2) | 263.5 (32.24) | p1: >0.999 p2: >0.999 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flaskamp, L.; Roubal, C.; Uddin, S.; Sotzny, F.; Kedor, C.; Bauer, S.; Scheibenbogen, C.; Seifert, M. Serum of Post-COVID-19 Syndrome Patients with or without ME/CFS Differentially Affects Endothelial Cell Function In Vitro. Cells 2022, 11, 2376. https://doi.org/10.3390/cells11152376
Flaskamp L, Roubal C, Uddin S, Sotzny F, Kedor C, Bauer S, Scheibenbogen C, Seifert M. Serum of Post-COVID-19 Syndrome Patients with or without ME/CFS Differentially Affects Endothelial Cell Function In Vitro. Cells. 2022; 11(15):2376. https://doi.org/10.3390/cells11152376
Chicago/Turabian StyleFlaskamp, Lavinia, Constanze Roubal, Steven Uddin, Franziska Sotzny, Claudia Kedor, Sandra Bauer, Carmen Scheibenbogen, and Martina Seifert. 2022. "Serum of Post-COVID-19 Syndrome Patients with or without ME/CFS Differentially Affects Endothelial Cell Function In Vitro" Cells 11, no. 15: 2376. https://doi.org/10.3390/cells11152376