
Why Do Some Vertebrates Have Microchromosomes?

 ,
,
Abstract
:1. Introduction
2. How Can We Clarify Karyotypic Features of Micro- and Macrochromosomes?
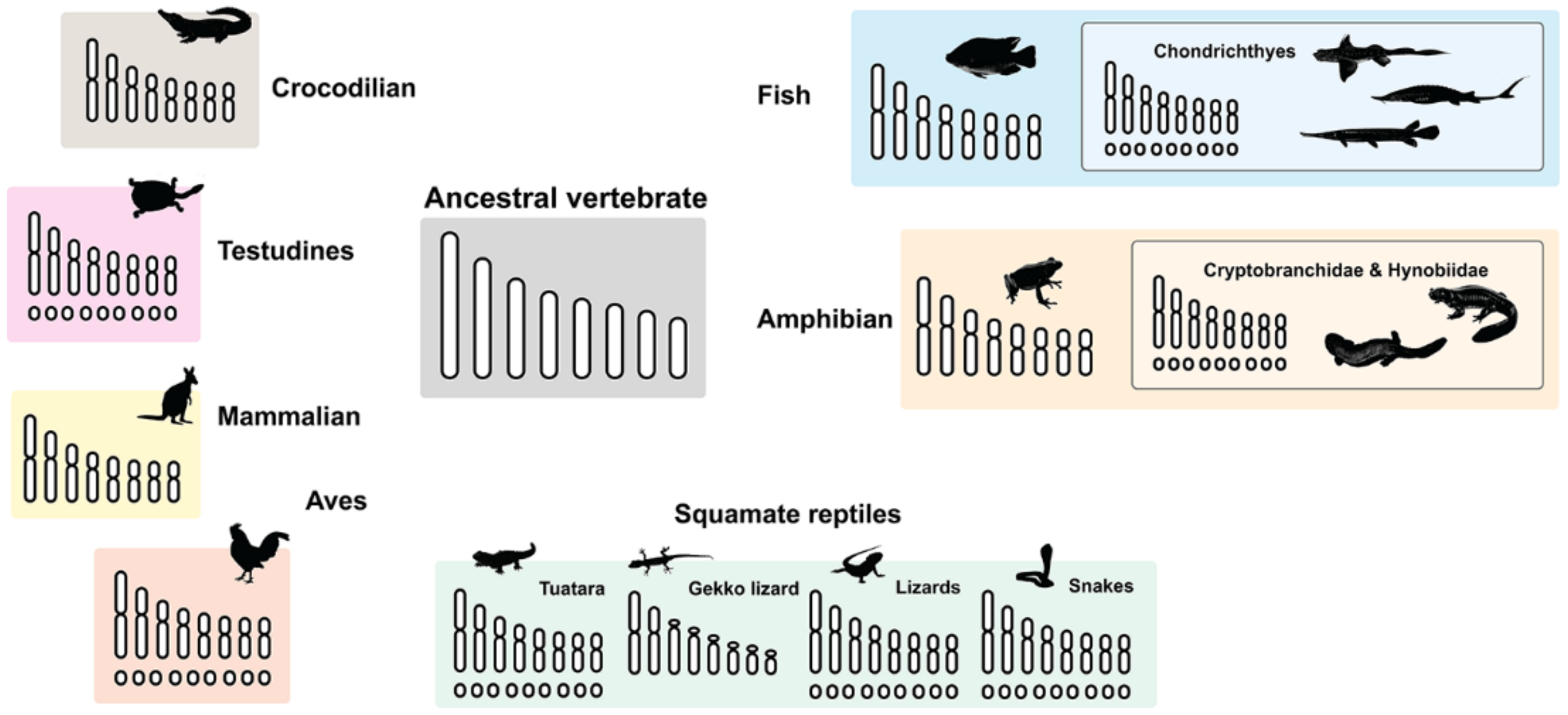
3. Microchromosome Distribution in Vertebrate Lineage
4. Differences in Characteristics of Macro- and Microchromosomes
5. Independent Recombination Frequency between Macro- and Microchromosomes as a Driver to Change Chromosome Structure
6. Nuclear Organization of Macro- and Microchromosomes
7. Distribution of Repeated Sequences between Macro- and Microchromosomes
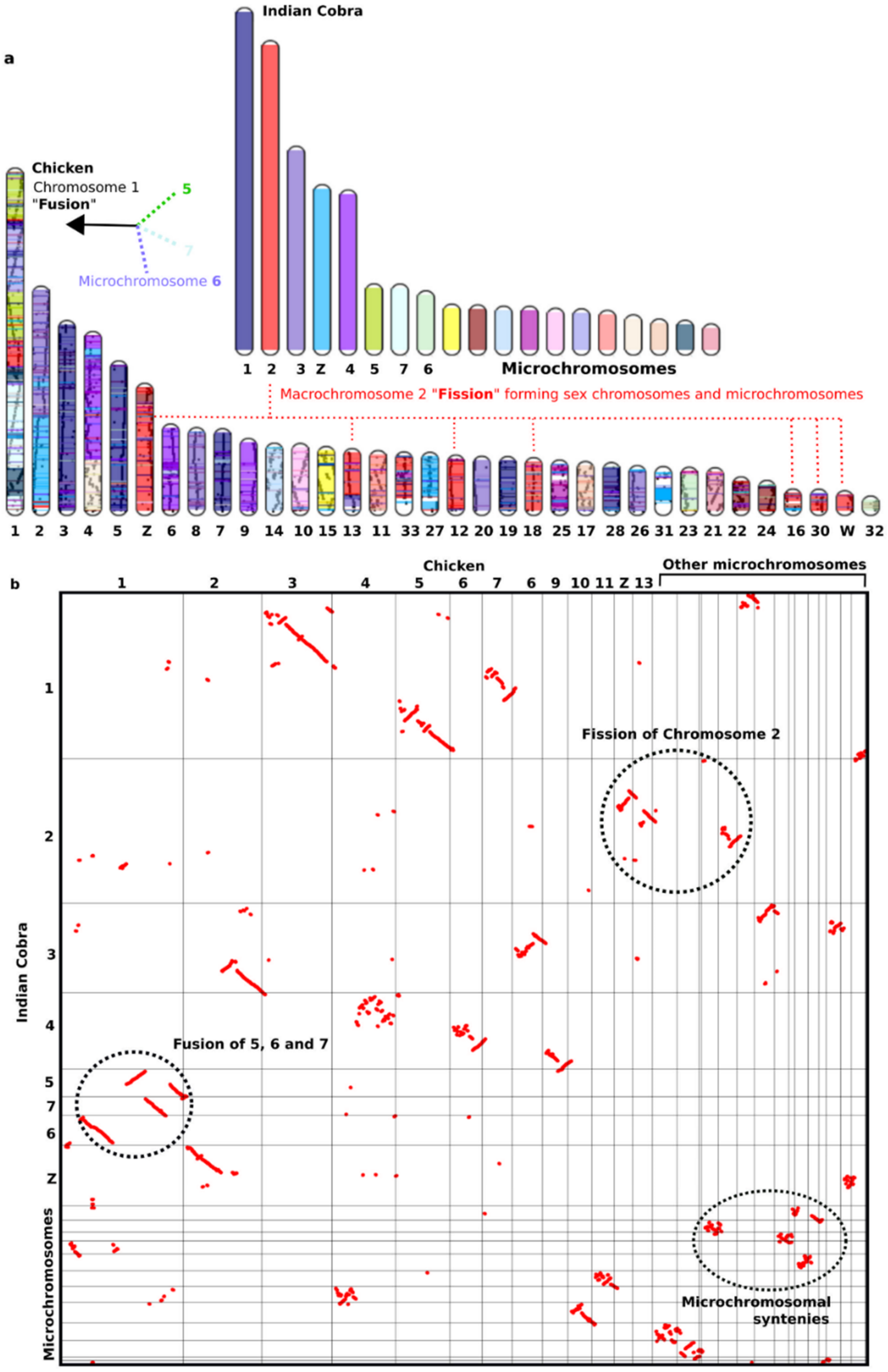
8. Highly Conserved Linkage Homology between Macro- and Microchromosomes and the Fusion-Fission Model of Vertebrate Evolution
9. Natural Selection and Adaptive Value for the Existence of Microchromosomes in Specific Lineages
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Darwin, C. The Origin of Species by Means of Natural Selection, or, the Preservation of Favoured Races in the Struggle for Life; John Murray: London, UK, 1859. [Google Scholar]
- Crandall, K.A.; Hillis, D.M. Rhodopsin evolution in the dark. Nature 1997, 387, 667–668. [Google Scholar] [CrossRef]
- Luk, H.L.; Melaccio, F.; Rinaldi, S.; Gozem, S.; Olivucci, M. Molecular bases for the selection of the chromophore of animal rhodopsins. Proc. Natl. Acad. Sci. USA 2015, 112, 15297–15302. [Google Scholar] [CrossRef] [Green Version]
- Ostrovsky, M.A. Rhodopsin: Evolution and comparative physiology. Paleontol. J. 2017, 51, 562–572. [Google Scholar] [CrossRef]
- Shen, L.; Chen, C.; Zheng, H.; Jin, L. The evolutionary relationship between microbial rhodopsins and metazoan rhodopsins. Sci. World J. 2013, 2013, 435651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burt, D.W. Origin and evolution of avian microchromosomes. Cytogenet. Genome Res. 2002, 96, 97–112. [Google Scholar] [CrossRef]
- Mengden, G.A.; Stock, A.D. Chromosomal evolution in serpentes; a comparison of G and C chromosome banding patterns of some colubrid and boid genera. Chromosoma 1980, 79, 53–64. [Google Scholar] [CrossRef]
- Olmo, E. Trends in the evolution of reptilian chromosomes. Integr. Comp. Biol. 2008, 48, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Srikulnath, K.; Thapana, W.; Muangmai, N. Role of chromosome changes in Crocodylus evolution and diversity. Genom. Inform. 2015, 13, 102. [Google Scholar] [CrossRef] [Green Version]
- Morescalchi, A.; Odierna, G.; Olmo, E. Karyological relationships between the Cryptobranchid salamanders. Experientia 1977, 33, 1579–1581. [Google Scholar] [CrossRef]
- Morescalchi, A.; Odierna, G.; Olmo, E. Karyology of the primitive slamanders, family hynobiidae. Experientia 1979, 35, 1434–1436. [Google Scholar] [CrossRef] [PubMed]
- Stingo, V.; Rocco, L. Chondrichthyan cytogenetics: A comparison with teleosteans. J. Mol. Evol. 1991, 33, 76–82. [Google Scholar] [CrossRef]
- Solovei, I.; Gaginskaya, E.R.; Macgregor, H.C. The arrangement and transcription of telomere DNA sequences at the ends of lampbrush chromosomes of birds. Chromosome Res. 1994, 2, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Boyes, J.W.; van Brink, J.M. Chromosomes of Syrphidae—V. Microchromosomes. Chromosoma 1970, 31, 207–216. [Google Scholar] [CrossRef]
- Baimal, V.; Sene, F.M.; Pereira, M.A.O.R. Heterochromatin and karyotypic differentiation of some neotropical cactus-breeding species of the Drosophila repleta species group. Genetica 1983, 60, 81–92. [Google Scholar] [CrossRef]
- Lukhtanov, V.A.; Dantchenko, A.V. Descriptions of new taxa of the genus Agrodiaetus Hübner, [1822] based on karyotype investigation. Atalanta 2002, 33, 81–107. [Google Scholar]
- Lukhtanov, V.A. Chromosome number evolution in skippers (Lepidoptera, Hesperiidae). Comp. Cytogenet. 2014, 8, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.; Barcellos, S.; Lemos, A.; Garnero, A.; Ribeiro, J.R.I.; Gunski, R. Belostoma estevezae Ribeiro and Alecrim (Heteroptera: Belostomatidae) reveals a new karyotype complement in Belostoma Latreille from mitotic metaphases. Rev. Chil. Entomol. 2019, 45, 31–36. [Google Scholar]
- Groenen, M.A.M.; Cheng, H.H.; Bumstead, N.; Benkel, B.F.; Briles, W.E.; Burke, T.; Burt, D.W.; Crittenden, L.B.; Dodgson, J.; Hillel, J.; et al. A consensus linkage map of the chicken genome. Genome Res. 2000, 10, 137–147. [Google Scholar] [CrossRef]
- Hillier, L.W.; Miller, W.; Birney, E.; Warren, W.; Hardison, R.C.; Ponting, C.P.; Bork, P.; Burt, D.W.; Groenen, M.A.M.; Delany, M.E.; et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef]
- Wallis, J.W.; Aerts, J.; Groenen, M.A.M.; Crooljmans, R.P.M.A.; Layman, D.; Graves, T.A.; Scheer, D.E.; Kremitzki, C.; Fedele, M.J.; Mudd, N.K.; et al. A physical map of the chicken genome. Nature 2004, 432, 761–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auer, H.; Mayr, B.; Lambrou, M.; Schleger, W. An extended chicken karyotype, including the NOR chromosome. Cytogenet. Genome Res. 1987, 45, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Fillon, V. The chicken as a model to study microchromosomes in birds: A review. Genet. Sel. Evol. 1998, 30, 209–219. [Google Scholar] [CrossRef]
- Burt, D.W.; Bruley, C.; Dunn, I.C.; Jones, C.T.; Ramage, A.; Law, A.S.; Morrice, D.R.; Paton, I.R.; Smith, J.; Windsor, D.; et al. The dynamics of chromosome evolution in birds and mammals. Nature 1999, 402, 411–413. [Google Scholar] [CrossRef]
- Axelsson, E.; Webster, M.T.; Smith, N.G.C.; Burt, D.W.; Ellegren, H. Comparison of the chicken and turkey genomes reveals a higher rate of nucleotide divergence on microchromosomes than macrochromosomes. Genome Res. 2005, 15, 120–125. [Google Scholar] [CrossRef] [Green Version]
- Solinhac, R.; Leroux, S.; Galkina, S.; Chazara, O.; Feve, K.; Vignoles, F.; Morisson, M.; Derjusheva, S.; Bed’hom, B.; Vignal, A.; et al. Integrative mapping analysis of chicken microchromosome 16 organization. BMC Genom. 2010, 11, 616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacerdot, C.; Louis, A.; Bon, C.; Berthelot, C.; Roest Crollius, H. Chromosome evolution at the origin of the ancestral vertebrate genome. Genome Biol. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- O’Connor, R.E.; Kiazim, L.; Skinner, B.; Fonseka, G.; Joseph, S.; Jennings, R.; Larkin, D.M.; Griffin, D.K. Patterns of microchromosome organization remain highly conserved throughout avian evolution. Chromosoma 2019, 128, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Nakatani, Y.; Takeda, H.; Kohara, Y.; Morishita, S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 2007, 17, 1254–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoghue, P. Evolution: Divining the nature of the ancestral vertebrate. Curr. Biol. 2017, 27, R277–R279. [Google Scholar] [CrossRef] [Green Version]
- Lynch, V.J.; Wagner, G.P. Multiple chromosomal rearrangements structured the ancestral vertebrate Hox-bearing protochromosomes. PLoS Genet. 2009, 5, e1000349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbs, C.L. A list of the lancelets of the world with diagnoses of five new species of Branchiostoma. Occas. Pap. Mus. Zool. 1922, 105, 1–16. [Google Scholar]
- Putnam, N.H.; Butts, T.; Ferrier, D.E.K.; Furlong, R.F.; Hellsten, U.; Kawashima, T.; Robinson-Rechavi, M.; Shoguchi, E.; Terry, A.; Yu, K.; et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 2008, 453, 1064–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlétaz, F.; Firbas, P.N.; Maeso, I.; Tena, J.J.; Bogdanovic, O.; Perry, M.; Wyatt, C.D.R.; de la Calle-Mustienes, E.; Bertrand, S.; Burguera, D.; et al. Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature 2018, 564, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; de la Torre, D.; Robertson, W.E.; Chin, J.W. Programmed chromosome fission and fusion enable precise large-scale genome rearrangement and assembly. Science 2019, 365, 922–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, T.R. Understanding natural selection: Essential concepts and common misconceptions. Evol. Educ. Outreach 2009, 2, 156–175. [Google Scholar] [CrossRef] [Green Version]
- Gildenhuys, P. Natural selection. In The Stanford Encyclopedia of Philosophy, Winter 2019 ed.; Zalta, E.N., Ed.; Stanford University: Stanford, CA, USA, 2019; Available online: https://plato.stanford.edu/archives/win2019/entries/natural-selection/ (accessed on 22 June 2021).
- Rodionov, A.V. Micro versus macro: A review of structure and functions of avian micro- and macrochromosomes. Russ. J. Genet. 1996, 32, 517–527. [Google Scholar]
- Perry, B.W.; Schield, D.R.; Adams, R.H.; Castoe, T.A. Microchromosomes exhibit distinct features of vertebrate chromosome structure and function with underappreciated ramifications for genome evolution. Mol. Biol. Evol. 2021, 38, 904–910. [Google Scholar] [CrossRef]
- Matthey, R. Caryotypes de mammiferes et d’oiseaux. la question des microchromosomes. quelques reflexions sur l’evolution chromosomique. Arch. Genet. 1975, 48, 12–26. [Google Scholar]
- Tegelström, H.; Ryttman, H. Chromosomes in birds (Aves): Evolutionary implications of macro-and microchromosome numbers and lengths. Hereditas 1981, 94, 225–233. [Google Scholar] [CrossRef]
- Habermann, F.A.; Cremer, M.; Walter, J.; Kreth, G.; Von Hase, J.; Bauer, K.; Wienberg, J.; Cremer, C.; Cremer, T.; Solovei, I. Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res. 2001, 9, 569–584. [Google Scholar] [CrossRef]
- Maslova, A.V.; Krasikova, A.V. Spatial arrangement of macro-, midi-, and microchromosomes in transcriptionally active nuclei of growing oocytes in birds of the order galliformes. Cell Tissue Biol. 2011, 5, 281–293. [Google Scholar] [CrossRef]
- Young, M.J.; O’Meally, D.; Sarre, S.D.; Georges, A.; Ezaz, T. Molecular cytogenetic map of the central bearded dragon, Pogona vitticeps (Squamata: Agamidae). Chromosome Res. 2013, 21, 361–374. [Google Scholar] [CrossRef]
- Losada, A.; Hirano, T. Shaping the metaphase chromosome: Coordination of cohesion and condensation. BioEssays 2001, 23, 924–935. [Google Scholar] [CrossRef]
- Antonin, W.; Neumann, H. Chromosome condensation and decondensation during mitosis. Curr. Opin. Cell Biol. 2016, 40, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillon, V.; Morisson, M.; Zoorob, R.; Auffray, C.; Douaire, M.; Gellin, J.; Vignal, A. Identification of 16 chicken microchromosomes by molecular markers using two-colour fluorescence in situ hybridization (FISH). Chromosome Res. 1998, 6, 307–313. [Google Scholar] [CrossRef]
- Nishida-Umehara, C.; Ishijima, J.; Ishishita, S.; Yamada, K.; Griffin, D.K.; Yamazaki, T.; Matsuda, Y. Karyotype reorganization with conserved genomic compartmentalization in dot-shaped microchromosomes in the Japanese mountain hawk-eagle (Nisaetus nipalensis orientalis, Accipitridae). Cytogenet. Genome Res. 2013, 141, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Burt, D.W. Parameters of the chicken genome (Gallus gallus). Anim. Genet. 1998, 29, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Pichugin, A.M.; Galkina, S.A.; Potekhin, A.A.; Punina, E.O.; Rautian, M.S.; Rodionov, A.V. Estimation of the minimal size of chicken Gallus gallus domesticus microchromosomes via pulsed-field electrophoresis. Russ. J. Genet. 2001, 37, 535–538. [Google Scholar] [CrossRef]
- Furlong, R.F. Insights into vertebrate evolution from the chicken genome sequence. Genome Biol. 2005, 6, 207. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Muramoto, J.; Stenius, C.; Christian, L.; Kittrell, W.A.; Atkin, N.B. Microchromosomes in holocephalian, chondrostean and holostean fishes. Chromosoma 1969, 26, 35–40. [Google Scholar] [CrossRef]
- Schield, D.R.; Card, D.C.; Hales, N.R.; Perry, B.W.; Pasquesi, G.M.; Blackmon, H.; Adams, R.H.; Corbin, A.B.; Smith, C.F.; Ramesh, B.; et al. The origins and evolution of chromosomes, dosage compensation, and mechanisms underlying venom regulation in snakes. Genome Res. 2019, 29, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Giovannotti, M.; Cerioni, P.N.; Slimani, T.; Splendiani, A.; Paoletti, A.; Fawzi, A.; Olmo, E.; Caputo Barucchi, V. Cytogenetic characterization of a population of Acanthodactylus lineomaculatus Duméril and Bibron, 1839 (Reptilia, Lacertidae), from Southwestern Morocco and insights into sex chromosome evolution. Cytogenet. Genome Res. 2017, 153, 86–95. [Google Scholar] [CrossRef]
- Srikulnath, K.; Matsubara, K.; Uno, Y.; Nishida-Umehara, C.; Olsson, M.; Matsuda, Y. Identification of the linkage group of the Z sex chromosomes of the sand lizard (Lacerta agilis, Lacertidae) and elucidation of karyotype evolution in lacertid lizards. Chromosoma 2014, 123, 563–575. [Google Scholar] [CrossRef]
- Ishijima, J.; Uno, Y.; Nunome, M.; Nishida-Umehara, C.; Kuraku, S.; Matsuda, Y. Molecular cytogenetic characterization of chromosome site-specific repetitive sequences in the Arctic lamprey (Lethenteron camtschaticum, Petromyzontidae). DNA Res. 2017, 24, 93–101. [Google Scholar] [CrossRef]
- Deakin, J.E.; Ezaz, T. Understanding the evolution of reptile chromosomes through applications of combined cytogenetics and genomics approaches. Cytogenet. Genome Res. 2019, 157, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Masabanda, J.S.; Burt, D.W.; O’Brien, P.C.M.; Vignal, A.; Fillon, V.; Walsh, P.S.; Cox, H.; Tempest, H.G.; Smith, J.; Habermann, F.; et al. Molecular cytogenetic definition of the chicken genome: The first complete avian karyotype. Genetics 2004, 166, 1367–1373. [Google Scholar] [CrossRef] [Green Version]
- Griffin, D.K.; Robertson, L.B.W.; Tempest, H.G.; Skinner, B.M. The evolution of the avian genome as revealed by comparative molecular cytogenetics. Cytogenet. Genome Res. 2007, 117, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Umehara, C.; Ishijima, J.; Kosaka, A.; Tanabe, H.; Habermann, F.A.; Griffin, D.K.; Matsuda, Y. Characterization of chromosome structures of Falconinae (Falconidae, Falconiformes, Aves) by chromosome painting and delineation of chromosome rearrangements during their differentiation. Chromosome Res. 2008, 16, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Olmo, E. Rate of chromosome changes and speciation in reptiles. Genetica 2005, 125, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, N.; Adams, D.C. Chromosome number and sex determination coevolve in turtles. Evolution 2011, 65, 1808–1813. [Google Scholar] [CrossRef]
- Montiel, E.E.; Badenhorst, D.; Tamplin, J.; Burke, R.L.; Valenzuela, N. Discovery of the youngest sex chromosomes reveals first case of convergent co-option of ancestral autosomes in turtles. Chromosoma 2017, 126, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.R.D.; Domit, C.; Pucci, M.B.; Gazolla, C.B.; Glugoski, L.; Nogaroto, V.; Vicari, M.R. Heterochromatin and microsatellites detection in karyotypes of four sea turtle species: Interspecific chromosomal differences. Genet. Mol. Biol. 2020, 43, 1–9. [Google Scholar] [CrossRef]
- Gregory, T.R. The bigger the C-value, the larger the cell: Genome size and red blood cell size in vertebrates. Blood Cells Mol. Dis. 2001, 27, 830–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, T.R.; Witt, J.D. Population size and genome size in fishes: A closer look. Genome 2008, 51, 309–313. [Google Scholar] [CrossRef]
- Arai, R. Fish Karyotypes: A Check List; Springer: Tokyo, Japan, 2011. [Google Scholar]
- Morescalchi, A. Adaptation and karyotype in Amphibia. Ital. J. Zool. 1977, 44, 287–294. [Google Scholar] [CrossRef]
- Morescalchi, A. Evolution and karyology of the Amphibians. Ital. J. Zool. 1980, 47, 113–126. [Google Scholar] [CrossRef]
- Voss, S.R.; Kump, D.K.; Putta, S.; Pauly, N.; Reynolds, A.; Henry, R.J.; Basa, S.; Walker, J.A.; Smith, J.J. Origin of amphibian and avian chromosomes by fission, fusion, and retention of ancestral chromosomes. Genome Res. 2011, 21, 1306–1312. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Steinlein, C.; Bogart, J.; Feichtinger, W.; León, P.; Marca, E.; Diaz, L.; Sanz, A.; Chen, S.-H.; Hedges, S. The chromosomes of Terraranan frogs. Cytogenet. Genome Res. 2010, 568, 130–131. [Google Scholar] [CrossRef]
- Perkins, R.D.; Gamboa, J.R.; Jonika, M.M.; Lo, J.; Shum, A.; Adams, R.H.; Blackmon, H. A database of amphibian karyotypes. Chromosome Res. 2019, 27, 313–319. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, Y.-Q.; Zhou, H.; Liu, Y.-F.; Wang, X.-L.; Papenfuss, T.J.; Wake, D.B.; Qu, L.-H. Phylogeny, evolution, and biogeography of Asiatic Salamanders (Hynobiidae). Proc. Natl. Acad. Sci. USA 2006, 103, 7360–7365. [Google Scholar] [CrossRef] [Green Version]
- Organ, C.L.; Shedlock, A.M.; Meade, A.; Pagel, M.; Edwards, S.V. Origin of avian genome size and structure in non-avian dinosaurs. Nature 2007, 446, 180–184. [Google Scholar] [CrossRef]
- Tegelström, H.; Ebenhard, T.; Ryttman, H. Rate of karyotype evolution and speciation in birds. Hereditas 1983, 98, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Kapusta, A.; Suh, A.; Feschotte, C. Dynamics of genome size evolution in birds and mammals. Proc. Natl. Acad. Sci. USA 2017, 114, E1460–E1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson-Smith, M.A.; Trifonov, V. Mammalian karyotype evolution. Nat. Rev. Genet. 2007, 8, 950–962. [Google Scholar] [CrossRef]
- Graphodatsky, A.S.; Trifonov, V.A.; Stanyon, R. The genome diversity and karyotype evolution of mammals. Mol. Cytogenet. 2011, 4, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, T.R.; Nicol, J.A.; Tamm, H.; Kullman, B.; Kullman, K.; Leitch, L.J.; Murray, B.G.; Kapraun, D.F.; Greilhuber, J.; Bennett, M.D. Eukaryotic genome size databases. Nucleic Acids Res. 2007, 35, D332–D338. [Google Scholar] [CrossRef] [Green Version]
- Gregory, T.R. Animal Genome Size Database. 2010. Available online: http://www.genomesize.com/results.php?page=1 (accessed on 22 June 2021).
- Srikulnath, K.; Uno, Y.; Nishida-Umehara, C.; Ota, H.; Matsuda, Y. Karyotype reorganization in the Hokou Gecko (Gekko hokouensis, Gekkonidae): The process of microchromosome disappearance in Gekkota. PLoS ONE 2015, 10, e0134829. [Google Scholar] [CrossRef]
- Pasquesi, G.I.M.; Adams, R.H.; Card, D.C.; Schield, D.R.; Corbin, A.B.; Perry, B.W.; Reyes-Velasco, J.; Ruggiero, R.P.; Vandewege, M.W.; Shortt, J.A. Squamate reptiles challenge paradigms of genomic repeat element evolution set by birds and mammals. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.F.; Singchat, W.; Jehangir, M.; Panthum, T.; Srikulnath, K. Consequence of paradigm shift with repeat landscapes in reptiles: Powerful facilitators of chromosomal rearrangements for diversity and evolution. Genes 2020, 11, 827. [Google Scholar] [CrossRef]
- Norris, T.B.; Rickards, G.K.; Daugherty, C.H. Chromosomes of tuatara, Sphenodon, a chromosome heteromorphism and an archaic reptilian karyotype. Cytogenet. Genome Res. 2004, 105, 93–99. [Google Scholar] [CrossRef]
- O’Meally, D.; Miller, H.; Patel, H.R.; Marshall Graves, J.A.; Ezaz, T. The first cytogenetic map of the Tuatara, Sphenodon punctatus. Cytogenet. Genome Res. 2009, 127, 213–223. [Google Scholar] [CrossRef]
- Pokorná, M.J.; Altmanová, M.; Kratochvíl, L. Multiple sex chromosomes in the light of female meiotic drive in amniote vertebrates. Chromosome Res. 2014, 22, 35–44. [Google Scholar] [CrossRef]
- Olmo, E.; Signorino, G. Chromorep: A Reptile Chromosomes Database 2005. Available online: http://chromorep.univpm.it (accessed on 26 August 2017).
- Srikulnath, K.; Matsubara, K.; Uno, Y.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S.; Matsuda, Y.; Nishida-Umehara, C. Karyological characterization of the butterfly lizard (Leiolepis reevesii rubritaeniata, Agamidae, Squamata) by molecular cytogenetic approach. Cytogenet. Genome Res. 2009, 125, 213–223. [Google Scholar] [CrossRef]
- Srikulnath, K.; Nishida-Umehara, C.; Matsubara, K.; Uno, Y.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S.; Matsuda, Y. Karyotypic evolution in squamate reptiles: Comparative gene mapping revealed highly conserved linkage homology between the butterfly lizard (Leiolepis reevesii rubritaeniata, Agamidae, Lacertilia) and the Japanese four-striped rat snake (Elaphe quadrivirgata, Colubridae, Serpentes). Chromosome Res. 2009, 17, 975–986. [Google Scholar] [CrossRef]
- Srikulnath, K.; Uno, Y.; Matsubara, K.; Thongpan, A.; Suputtitada, S.; Apisitwanich, S.; Nishida-Umehara, C.; Matsuda, Y. Chromosomal localization of the 18S-28S and 5s rRNA genes and (TTAGGG)n sequences of butterfly lizards (Leiolepis belliana belliana and Leiolepis boehmei, Agamidae, Squamata). Genet. Mol. Biol. 2011, 34, 583–586. [Google Scholar] [CrossRef]
- Srikulnath, K.; Uno, Y.; Nishida-Umehara, C.; Matsuda, Y. Karyotype evolution in monitor lizards: Cross-species chromosome mapping of cDNA reveals highly conserved synteny and gene order in the Toxicofera clade. Chromosome Res. 2013, 21, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Srikulnath, K.; Azad, B.; Singchat, W.; Ezaz, T. Distribution and amplification of interstitial telomeric sequences (ITSs) in Australian dragon lizards support frequent chromosome fusions in Iguania. PLoS ONE 2019, 14, e0212683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uno, Y.; Nishida-Umehara, C.; Tarui, H.; Ishishita, S.; Takagi, C.; Nishimura, O.; Ishijima, J.; Ota, H.; Kosaka, A.; Matsubara, K.; et al. Inference of the protokaryotypes of amniotes and tetrapods and the evolutionary processes of microchromosomes from comparative gene mapping. PLoS ONE 2012, 7, e53027. [Google Scholar] [CrossRef] [PubMed]
- Singchat, W.; O’Connor, R.E.; Tawichasri, P.; Suntronpong, A.; Sillapaprayoon, S.; Suntrarachun, S.; Muangmai, N.; Baicharoen, S.; Peyachoknagul, S.; Chanhome, L.; et al. Chromosome map of the Siamese cobra: Did partial synteny of sex chromosomes in the amniote represent “A hypothetical ancestral super-sex chromosome” or random distribution? BMC Genom. 2018, 19, 939. [Google Scholar] [CrossRef] [PubMed]
- Singchat, W.; Ahmad, S.F.; Laopichienpong, N.; Suntronpong, A.; Panthum, T.; Griffin, D.K.; Srikulnath, K. Snake W sex chromosome: The shadow of ancestral amniote super-sex chromosome. Cells 2020, 9, 2386. [Google Scholar] [CrossRef]
- Singchat, W.; Ahmad, S.F.; Sillapaprayoon, S.; Muangmai, N.; Duengkae, P.; Peyachoknagul, S.; O’Connor, R.E.; Griffin, D.K.; Srikulnath, K. Partial amniote sex chromosomal linkage homologies shared on snake W sex chromosomes support the ancestral super-sex chromosome evolution in amniotes. Front. Genet. 2020, 11, 948. [Google Scholar] [CrossRef] [PubMed]
- Singchat, W.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Indananda, C.; Duengkae, P.; Peyachoknagul, S.; O’Connor, R.E.; Griffin, D.K.; Srikulnath, K. Do sex chromosomes of snakes, monitor lizards, and Iguanian lizards result from multiple fission of an “ancestral amniote super-sex chromosome”? Chromosome Res. 2020, 28, 209–228. [Google Scholar] [CrossRef] [PubMed]
- Pinthong, K.; Tanomtong, A.; Getlekha, N.; Sangpadee, W.; Sangpakdee, K.; Sanoamuang, L. First cytogenetic study of puff-faced water snake, Homalopsis buccata (Squamata, Colubridae) by conventional staining, Ag-NOR banding and GTGbanding techniques. Cytologia 2013, 78, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, K.; Knopp, T.; Sarre, S.D.; Georges, A.; Ezaz, T. Karyotypic analysis and FISH mapping of microsatellite motifs reveal highly differentiated XX/XY sex chromosomes in the pink-tailed worm-lizard (Aprasia parapulchella, Pygopodidae, Squamata). Mol. Cytogenet. 2013, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.M.; Clark, H.F. The somatic chromosomes of five crocodilian s pecies. Cytogenet. Genome Res. 1967, 6, 193–203. [Google Scholar] [CrossRef]
- Kawagoshi, T.; Nishida-Umehara, C.; Ota, H.; Kumazawa, Y.; Endo, H.; Matsuda, Y. Molecular structures of centromeric heterochromatin and karyotypic evolution in the Siamese crocodile (Crocodylus siamensis) (Crocodylidae, Crocodylia). Chromosome Res. 2008, 16, 1119–1132. [Google Scholar] [CrossRef]
- Nirchio, M.; Rossi, A.R.; Foresti, F.; Oliveira, C. Chromosome evolution in fishes: A new challenging proposal from Neotropical species. Neotrop. Ichthyol. 2014, 12, 761–770. [Google Scholar] [CrossRef]
- Salvador, L.B.; Moreira-Filho, O. B chromosomes in Astyanax scabripinnis (Pisces, Characidae). Heredity 1992, 69, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Kavalco, K.F.; De Almeida-Toledo, L.F. Molecular cytogenetics of blind Mexican tetra and comments on the karyotypic characteristics of genus Astyanax (Teleostei, Characidae). Zebrafish 2007, 4, 103–111. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Jehangir, M.; Cardoso, A.L.; Wolf, I.R.; Margarido, V.P.; Cabral-De-Mello, D.C.; O’Neill, R.; Valente, G.T.; Martins, C. B chromosomes of multiple species have intense evolutionary dynamics and accumulated genes related to important biological processes. BMC Genom. 2020, 21, 656. [Google Scholar] [CrossRef]
- Smith, J.J.; Voss, S.R. Gene order data from a model amphibian (Ambystoma): New perspectives on vertebrate genome structure and evolution. BMC Genom. 2006, 7, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Session, A.M.; Uno, Y.; Kwon, T.; Chapman, J.A.; Toyoda, A.; Takahashi, S.; Fukui, A.; Hikosaka, A.; Suzuki, A.; Kondo, M.; et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature 2016, 538, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.J.; Veltsos, P. The diversity and evolution of sex chromosomes in frogs. Genes 2021, 12, 483. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, A.V.; Chelysheva, L.A.; Solovei, I.V.; Myakoshina Yu, A. Chiasma distribution in the lambrush chromosomes of the chicken Gallus gallus domesticus. Hot spots of recombination and their feasible role in proper disjunction of homologous chromosome at the first meiotic division. Genetika 1992, 28, 151–160. [Google Scholar] [PubMed]
- Backström, N.; Forstmeier, W.; Schielzeth, H.; Mellenius, H.; Nam, K.; Bolund, E.; Webster, M.T.; Öst, T.; Schneider, M.; Kempenaers, B.; et al. The recombination landscape of the zebra finch Taeniopygia guttata genome. Genome Res. 2010, 20, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Schield, D.R.; Pasquesi, G.I.M.; Perry, B.W.; Adams, R.H.; Nikolakis, Z.L.; Westfall, A.K.; Orton, R.W.; Meik, J.M.; MacKessy, S.P.; Castoe, T.A.; et al. Snake recombination landscapes are concentrated in functional regions despite PRDM9. Mol. Biol. Evol. 2020, 37, 1272–1294. [Google Scholar] [CrossRef] [PubMed]
- McQueen, H.A.; Fantes, J.; Cross, S.H.; Clark, V.H.; Archibald, A.L.; Bird, A.P. CpG islands of chicken are concentrated on microchromosomes. Nat. Genet. 1996, 12, 321–324. [Google Scholar] [CrossRef]
- Grützner, F.; Zend-Ajusch, E.; Stout, K.; Munsche, S.; Niveleau, A.; Nanda, I.; Schmid, M.; Haaf, T. Chicken microchromosomes are hypermethylated and can be identified by specific painting probes. Cytogenet. Genome Res. 2001, 93, 265–269. [Google Scholar] [CrossRef]
- Andreozzi, L.; Federico, C.; Motta, S.; Saccone, S.; Sazanova, A.L.; Sazanov, A.A.; Smirnov, A.F.; Galkina, S.A.; Lukina, N.A.; Rodionov, A.V.; et al. Compositional mapping of chicken chromosomes and identification of the gene-richest regions. Chromosome Res. 2001, 9, 521–532. [Google Scholar] [CrossRef]
- McQueen, H.A.; Siriaco, G.; Bird, A.P. Chicken microchromosomes are hyperacetylated, early replicating, and gene rich. Genome Res. 1998, 8, 621–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.; Bruley, C.K.; Paton, I.R.; Dunn, I.; Jones, C.T.; Windsor, D.; Morrice, D.R.; Law, A.S.; Masabanda, J.; Sazanov, A.; et al. Differences in gene density on chicken macrochromosomes and microchromosomes. Anim. Genet. 2000, 31, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Clifford, V.; Western, P.S.; Wilcox, S.A.; Bell, K.S.; Sinclair, A.H. Cloning and expression of a DAX1 homologue in the chicken embryo. J. Mol. Endocrinol. 2000, 24, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Schmid, M.; Enderle, E.; Schindler, D.; Schempp, W. Chromosome banding and DNA replication patterns in bird karyotypes. Cytogenet. Genome Res. 1989, 52, 139–146. [Google Scholar] [CrossRef]
- Li, X.Q.; Du, D. Variation, evolution, and correlation analysis of C+G content and genome or chromosome size in different kingdoms and phyla. PLoS ONE 2014, 9, e88339. [Google Scholar] [CrossRef] [Green Version]
- Piovesan, A.; Pelleri, M.C.; Antonaros, F.; Strippoli, P.; Caracausi, M.; Vitale, L. On the length, weight and GC content of the human genome. BMC Res. Notes 2019, 12, 106. [Google Scholar] [CrossRef]
- Borůvková, V.; Howell, W.M.; Matoulek, D.; Symonová, R. Quantitative approach to fish cytogenetics in the context of vertebrate genome evolution. Genes 2021, 12, 312. [Google Scholar] [CrossRef]
- Matsubara, K.; Kuraku, S.; Tarui, H.; Nishimura, O.; Nishida-Umehara, C.; Agata, K.; Kumazawa, Y.; Matsuda, Y. Intra-genomic GC heterogeneity in sauropsids: Evolutionary insights from cDNA mapping and GC3 profiling in snake. BMC Genom. 2012, 13, 604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skinner, B.M.; Robertson, L.B.W.; Tempest, H.G.; Langley, E.J.; Ioannou, D.; Fowler, K.E.; Crooijmans, R.P.M.A.; Hall, A.D.; Griffin, D.K.; Völker, M. Comparative genomics in chicken and Pekin duck using FISH mapping and microarray analysis. BMC Genom. 2009, 10, 357. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Nishida-Umehara, C.; Tarui, H.; Kuroiwa, A.; Yamada, K.; Isobe, T.; Ando, J.; Fujiwara, A.; Hirao, Y.; Nishimura, O.; et al. Highly conserved linkage homology between birds and turtles: Bird and turtle chromosomes are precise counterparts of each other. Chromosome Res. 2005, 13, 601–615. [Google Scholar] [CrossRef]
- Bista, B.; Valenzuela, N. Turtle insights into the evolution of the reptilian karyotype and the genomic architecture of sex determination. Genes 2020, 11, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GBIF Secretariat. Pelodiscus sinensis (Wiegmann, 1835). GBIF Backbone Taxonomy. Available online: https://www.gbif.org/species/5220435 (accessed on 22 June 2021).
- Kawagoshi, T.; Uno, Y.; Matsubara, K.; Matsuda, Y.; Nishida-Umehara, C. The ZW micro-sex chromosomes of the Chinese soft-shelled turtle (Pelodiscus sinensis, Trionychidae, Testudines) have the same origin as chicken chromosome 15. Cytogenet. Genome Res. 2009, 125, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.C.; Hillier, L.D.W.; Tomlinson, C.; Minx, P.; Kremitzki, M.; Graves, T.; Markovic, C.; Bouk, N.; Pruitt, K.D.; Thibaud-Nissen, F.; et al. A new chicken genome assembly provides insight into avian genome structure. G3 Genes Genomes Genet. 2017, 7, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Nishida-Umehara, C.; Matsuda, Y. Characterization and chromosomal distribution of novel satellite DNA sequences of the lesser rhea (Pterocnemia pennata) and the greater rhea (Rhea americana). Chromosome Res. 2002, 10, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Nishida-Umehara, C.; Matsuda, Y. Erratum: A new family of satellite DNA sequences as a major component of centromeric heterochromatin in owls (Strigiformes). Chromosoma 2004, 112, 372–373. [Google Scholar] [CrossRef] [Green Version]
- GBIF Secretariat. Trachemys scripta Subsp. elegans (Wied, 1838). GBIF Backbone Taxonomy. Available online: https://www.gbif.org/species/6157026 (accessed on 22 June 2021).
- Laurenti, J.N. Specimen Medicum, Exhibens Synopsin Reptilium Emendatam cum Experimentis Circa Venena et Antidota Reptilium Austracorum, Quod Authoritate et Consensu; Joan. Thomae: Vienna, Austria, 1768; p. 217. [Google Scholar]
- Hughes, S.; Zelus, D.; Mouchiroud, D. Warm-blooded isochore structure in Nile crocodile and turtle. Mol. Biol. Evol. 1999, 16, 1521–1527. [Google Scholar] [CrossRef] [Green Version]
- Chojnowski, J.L.; Franklin, J.; Katsu, Y.; Iguchi, T.; Guillette, L.J.; Kimball, R.T.; Braun, E.L. Patterns of vertebrate isochore evolution revealed by comparison of expressed mammalian, avian, and crocodilian genes. J. Mol. Evol. 2007, 65, 259–266. [Google Scholar] [CrossRef]
- Chojnowski, J.L.; Braun, E.L. Turtle isochore structure is intermediate between amphibians and other amniotes. Integr. Comp. Biol. 2008, 48, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Melodelima, C.; Gautier, C. The GC-heterogeneity of teleost fishes. BMC Genom. 2008, 9, 632. [Google Scholar] [CrossRef] [Green Version]
- Matoulek, D.; Boruvkova, V.; Ocalewicz, K.; Symonova, R. GC and repeats profiling along chromosomes—The future of fish compositional cytogenomics. Genes 2021, 12, 50. [Google Scholar] [CrossRef]
- Holbrook, J.E. North American Herpetology: Or a Description of the Reptiles Inhabiting the United States, 2nd ed.; J. Dobson: Philadelphia, PA, USA, 1842; Volume II, p. 142. [Google Scholar]
- St John, J.A.; Braun, E.L.; Isberg, S.R.; Miles, L.G.; Chong, A.Y.; Gongora, J.; Dalzell, P.; Moran, C.; Bed’hom, B.; Abzhanov, A.; et al. Sequencing three crocodilian genomes to illuminate the evolution of archosaurs and amniotes. Genome Biol. 2012, 13, 415. [Google Scholar] [CrossRef]
- Margres, M.J.; Rautsaw, R.M.; Strickland, J.L.; Mason, A.J.; Schramer, T.D.; Hofmann, E.P.; Stiers, E.; Ellsworth, S.A.; Nystrom, G.S.; Hogan, M.P.; et al. The Tiger Rattlesnake genome reveals a complex genotype underlying a simple venom phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2014634118. [Google Scholar] [CrossRef]
- Boie, H. Merkmale einiger japanischer Lurche. Lsis Oken 1826, 18–19, 203–216. [Google Scholar]
- Cuvier, G.; Voigt, F.S. Das Thierreich, Geordnet Nach Seiner Organisation: Nach der Zweiten Ausgabe Übersetzt und Durch Zusätze Erweitert von F.S. Voigt; Brockhaus: Leipzig, Germany, 1832; Volume 2, p. 539. [Google Scholar]
- Alföldi, J.; Di Palma, F.; Grabherr, M.; Williams, C.; Kong, L.; Mauceli, E.; Russell, P.; Lowe, C.B.; Glor, R.E.; Jaffe, J.D.; et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 2011, 477, 587–591. [Google Scholar] [CrossRef] [Green Version]
- Brandley, M.C.; Wynn, A.; De Queiroz, K. Karyotype and relationships of Anolis desechensis. J. Herpetol. 2006, 40, 136–139. [Google Scholar] [CrossRef]
- Figuet, E.; Ballenghien, M.; Romiguier, J.; Galtier, N. Biased gene conversion and GC-content evolution in the coding sequences of reptiles and vertebrates. Genome Biol. Evol. 2015, 7, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Pyron, R.A.; Burbrink, F.T.; Wiens, J.J. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol. Biol. 2013, 13, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romiguier, J.; Ranwez, V.; Douzery, E.J.P.; Galtier, N. Contrasting GC-content dynamics across 33 mammalian genomes: Relationship with life-history traits and chromosome sizes. Genome Res. 2010, 20, 1001–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, Y.S.; Chai, X.W.; Wang, Z.F.; Nie, Q.H.; Zhang, X.Q. Impact of GC content on gene expression pattern in chicken. Genet. Sel. Evol. 2013, 45, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hunter, N. Meiotic recombination: The essence of heredity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016618. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Smeds, L.; Backström, N.; Husby, A.; Qvarnström, A.; Mugal, C.F.; Olason, P.; Ellegren, H. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol. Ecol. 2014, 23, 4035–4058. [Google Scholar] [CrossRef] [Green Version]
- Borrie, M.S.; Campor, J.S.; Joshi, H.; Gartenberg, M.R. Binding, sliding, and function of cohesin during transcriptional activation. Proc. Natl. Acad. Sci. USA 2017, 114, E1062–E1071. [Google Scholar] [CrossRef] [Green Version]
- Mieczkowski, P.A.; Lemoine, F.J.; Petes, T.D. Recombination between retrotransposons as a source of chromosome rearrangements in the yeast Saccharomyces cerevisiae. DNA Repair 2006, 5, 1010–1020. [Google Scholar] [CrossRef]
- Ito, M.; Kugou, K.; Fawcett, J.A.; Mura, S.; Ikeda, S.; Innan, H.; Ohta, K. Meiotic recombination cold spots in chromosomal cohesion sites. Genes Cells 2014, 19, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Merkenschlager, M. Cohesin: A global player in chromosome biology with local ties to gene regulation. Curr. Opin. Genet. Dev. 2010, 20, 555–561. [Google Scholar] [CrossRef]
- Craig, J.M.; Bickmore, W.A. The distribution of CpG islands in mammalian chromosomes. Nat. Genet. 1994, 7, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Baudat, F.; Massy, B. Regulating double-stranded DNA break repair towards crossover or non-crossover during mammalian meiosis. Chromosome Res. 2007, 15, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Baudat, F.; Imai, Y.; Massy, B. Meiotic recombination in mammals: Localization and regulation. Nat. Rev. Genet. 2013, 14, 794–806. [Google Scholar] [CrossRef] [PubMed]
- GBIF Secretariat. Anser anser (Linnaeus, 1758). GBIF Backbone Taxonomy. Available online: https://www.gbif.org/species/2498036 (accessed on 22 June 2021).
- Groenen, M.A.M.; Wahlberg, P.; Foglio, M.; Cheng, H.H.; Megens, H.J.; Crooijmans, R.P.M.A.; Besnier, F.; Lathrop, M.; Muir, W.M.; Wong, G.K.S.; et al. A high-density SNP-based linkage map of the chicken genome reveals sequence features correlated with recombination rate. Genome Res. 2009, 19, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torgasheva, A.A.; Borodin, P.M. Immunocytological analysis of meiotic recombination in the gray goose (Anser anser). Cytogenet. Genome Res. 2017, 151, 27–35. [Google Scholar] [CrossRef]
- Motacilla alba Linnaeus, 1758. Available online: https://www.gbif.org/species/9599502 (accessed on 22 June 2021).
- Pigozzi, M.I. Distribution of MLH1 foci on the synaptonemal complexes of chicken oocytes. Cytogenet. Cell Genet. 2001, 95, 129–133. [Google Scholar] [CrossRef]
- Marais, G. Biased gene conversion: Implications for genome and sex evolution. Trends Genet. 2003, 19, 330–338. [Google Scholar] [CrossRef]
- Lercher, M.J.; Urrutia, A.O.; Pavlíček, A.; Hurst, L.D. A unification of mosaic structures in the human genome. Hum. Mol. Genet. 2003, 12, 2411–2415. [Google Scholar] [CrossRef] [Green Version]
- Sémon, M.; Mouchiroud, D.; Duret, L. Relationship between gene expression and GC-content in mammals: Statistical significance and biological relevance. Hum. Mol. Genet. 2005, 14, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Bickmore, W.A. Patterns in the genome. Heredity 2019, 123, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, B.M. Decoding the nucleosome. Cell 1993, 75, 5–8. [Google Scholar] [CrossRef]
- Wade, P.A.; Pruss, D.; Wolffe, A.P. Histone acetylation: Chromatin in action. Trends Biochem. Sci. 1997, 22, 128–132. [Google Scholar] [CrossRef]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, P.; Mitchell, A.; Turner, B.; Perry, P. Antibodies to defined histone epitopes reveal variations in chromatin conformation and underacetylation of centric heterochromatin in human metaphase chromosomes. Chromosoma 1992, 101, 322–332. [Google Scholar] [CrossRef]
- Jeppesen, P.; Turner, B.M. The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell 1993, 74, 281–289. [Google Scholar] [CrossRef]
- Ronzoni, S.; Faretta, M.; Ballarini, M.; Pelicci, P.G.; Minucci, S. New method to detect histone acetylation levels by flow cytometry. Cytom. A 2005, 66, 52–61. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, F.; van Steensel, B. Histone modifications: From genome-wide maps to functional insights. Genome Biol. 2005, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Eyre-Walker, A. Recombination and mammalian genome evolution. Proc. R. Soc. B 1993, 252, 237–243. [Google Scholar] [CrossRef]
- Bielawski, J.P.; Dunn, K.A.; Yang, Z. Rates of nucleotide substitution and mammalian nuclear gene evolution: Approximate and maximum-likelihood methods lead to different conclusions. Genetics 2000, 156, 1299–1308. [Google Scholar] [CrossRef]
- Hurst, L.D.; Williams, E.J.B. Covariation of GC content and the silent site substitution rate in rodents: Implications for methodology and for the evolution of isochores. Gene 2000, 261, 107–114. [Google Scholar] [CrossRef]
- Smith, N.G.C.; Webster, M.T.; Ellegren, H. Deterministic mutation rate variation in the human genome. Genome Res. 2002, 12, 1350–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachidanandam, R.; Weissman, D.; Schmidt, S.C.; Kakol, J.M.; Stein, L.D.; Marth, G.; Sherry, S.; Mullikin, J.C.; Mortimore, B.J.; Willey, D.L.; et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001, 409, 928–934. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Boerwinkle, E. Neighboring-nucleotide effects on single nucleotide polymorphisms: A study of 2.6 million polymorphisms across the human genome. Genome Res. 2002, 12, 1679–1686. [Google Scholar] [CrossRef] [Green Version]
- Lercher, M.J.; Hurst, L.D. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 2002, 18, 337–340. [Google Scholar] [CrossRef]
- Waterston, R.H.; Pachter, L. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, I.; Ebersberger, I.; Ptak, S.E.; Pääbo, S.; Przeworski, M. A neutral explanation for the correlation of diversity with recombination rates in humans. Am. J. Hum. Genet. 2003, 72, 1527–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen-Seaman, M.I.; Furey, T.S.; Payseur, B.A.; Lu, Y.; Roskin, K.M.; Chen, C.F.; Thomas, M.A.; Haussler, D.; Jacob, H.J. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 2004, 14, 528–538. [Google Scholar] [CrossRef] [Green Version]
- McVean, G.A.T.; Myers, S.R.; Hunt, S.; Deloukas, P.; Bentley, D.R.; Donnelly, P. The fine-scale structure of recombination rate variation in the human genome. Science 2004, 304, 581–584. [Google Scholar] [CrossRef] [Green Version]
- Strathern, J.N.; Shafer, B.K.; McGill, C.B. DNA synthesis errors associated with double-strand-break repair. Genetics 1995, 140, 965–972. [Google Scholar] [CrossRef]
- Kong, A.; Gudbjartsson, D.F.; Sainz, J.; Jonsdottir, G.M.; Gudjonsson, S.A.; Richardsson, B.; Sigurdardottir, S.; Barnard, J.; Hallbeck, B.; Masson, G.; et al. A high-resolution recombination map of the human genome. Nat. Genet. 2002, 31, 241–247. [Google Scholar] [CrossRef]
- Meunier, J.; Duret, L. Recombination drives the evolution of GC-content in the human genome. Mol. Biol. Evol. 2004, 21, 984–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, H.; Van Schaik, B.; Van der Mee, M.; Baas, F.; Riggins, G.; Van Sluis, P.; Hermus, M.C.; Van Asperen, R.; Boon, K.; Voûte, P.A.; et al. The human transcriptome map: Clustering of highly expressed genes in chromosomal domains. Science 2001, 291, 1289–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyre-Walker, A.; Hurst, L.D. The evolution of isochores. Nat. Rev. Genet. 2001, 2, 549–555. [Google Scholar] [CrossRef]
- Lercher, M.J.; Urrutia, A.O.; Hurst, L.D. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat. Genet. 2002, 31, 180–183. [Google Scholar] [CrossRef]
- Lercher, M.J.; Chamary, J.; Hurst, L.D. Genomic regionality in rates of evolution is not explained by clustering of genes of comparable expression profile. Genome Res. 2004, 14, 1002–1013. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. International Human Genome Sequencing Consortium, 409 3. J. Am. Med. Inform. Assoc. 2003, 412, 11–12. [Google Scholar]
- Axelsson, E.; Smith, N.G.C.; Sundström, H.; Berlin, S.; Ellegren, H. Male-biased mutation rate and divergence in autosomal, Z-linked and W-linked introns of chicken and turkey. Mol. Biol. Evol. 2004, 21, 1538–1547. [Google Scholar] [CrossRef] [Green Version]
- Chuang, J.H.; Li, H. Functional bias and spatial organization of genes in mutational hot and cold regions in the human genome. PLoS Biol. 2004, 2, e29. [Google Scholar] [CrossRef]
- Webster, M.T.; Smith, N.G.C.; Lercher, M.J.; Ellegren, H. Gene expression, synteny, and local similarity in human noncoding mutation rates. Mol. Biol. Evol. 2004, 21, 1820–1830. [Google Scholar] [CrossRef]
- Robertson, K.D.; Wolffe, A.P. DNA methylation in health and disease. Nat. Rev. Genet. 2000, 1, 11–19. [Google Scholar] [CrossRef]
- Piganeau, G.D.; Mouchiroud, D.L.; Gautier, C. Expected relationship between the silent substitution rate and the GC content: Implications for the evolution of isochores. J. Mol. Evol. 2002, 54, 129–133. [Google Scholar] [CrossRef]
- Nabholz, B.; Glémin, S.; Galtier, N. Strong variations of mitochondrial mutation rate across mammals—The longevity hypothesis. Mol. Biol. Evol. 2008, 25, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Nabholz, B.; Glemin, S.; Galtier, N. The erratic mitochondrial clock: Variations of mutation rate, not population size, affect mtDNA diversity across birds and mammals. BMC Evol. Biol. 2009, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Lourenço, J.M.; Glémin, S.; Chiari, Y.; Galtier, N. The determinants of the molecular substitution process in turtles. J. Evol. Biol. 2013, 26, 38–50. [Google Scholar] [CrossRef]
- Bourgeois, Y.; Boissinot, S. On the population dynamics of junk: A review on the population genomics of transposable elements. Genes 2019, 10, 419. [Google Scholar] [CrossRef] [Green Version]
- Federico, C.; Cantarella, C.D.; Scavo, C.; Saccone, S.; Bed’Hom, B.; Bernardi, G. Avian genomes: Different karyotypes but a similar distribution of the GC-richest chromosome regions at interphase. Chromosome Res. 2005, 13, 785–793. [Google Scholar] [CrossRef]
- Berner, D.; Roesti, M. Genomics of adaptive divergence with chromosome-scale heterogeneity in crossover rate. Mol. Ecol. 2017, 26, 6351–6369. [Google Scholar] [CrossRef]
- Haenel, Q.; Laurentino, T.G.; Roesti, M.; Berner, D. Meta-analysis of chromosome-scale crossover rate variation in eukaryotes and its significance to evolutionary genomics. Mol. Ecol. 2018, 27, 2477–2497. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.; Paolella, G.; Ramos, C.; Lamond, A.I. Spatial organization of large-scale chromatin domains in the nucleus: A magnified view of single chromosome territories. J. Cell Biol. 1997, 139, 1597–1610. [Google Scholar] [CrossRef] [Green Version]
- Sadoni, N.; Langer, S.; Fauth, C.; Bernardi, G.; Cremer, T.; Turner, B.M.; Zink, D. Nuclear organization of mammalian genomes: Polar chromosome territories build up functionally distinct higher order compartments. J. Cell Biol. 1999, 146, 1211–1226. [Google Scholar] [CrossRef] [Green Version]
- GBIF Secretariat. Taeniopygia guttata (Vieillot, 1817). GBIF Backbone Taxonomy. Available online: https://www.gbif.org/species/2493633 (accessed on 22 June 2021).
- Calderón, P.L.; Pigozzi, M.I. MLH1-focus mapping in birds shows equal recombination between sexes and diversity of crossover patterns. Chromosome Res. 2006, 14, 605–612. [Google Scholar] [CrossRef]
- Semenov, G.A.; Koblik, E.A.; Red’kin, Y.A.; Badyaev, A.V. Extensive phenotypic diversification coexists with little genetic divergence and a lack of population structure in the White Wagtail subspecies complex (Motacilla alba). J. Evol. Biol. 2018, 31, 1093–1108. [Google Scholar] [CrossRef]
- Rice, J.C.; Allis, C.D. Histone methylation versus histone acetylation: New insights into epigenetic regulation. Curr. Opin. Cell Biol. 2001, 13, 263–273. [Google Scholar] [CrossRef]
- Wade, P.A. Methyl CpG binding proteins: Coupling chromatin architecture to gene regulation. Oncogene 2001, 20, 3166–3173. [Google Scholar] [CrossRef]
- Pyrpasopoulou, A.; Meier, J.; Maison, C.; Simos, G.; Georgatos, S.D. The lamin B receptor (LBR) provides essential chromatin docking sites at the nuclear envelope. EMBO J. 1996, 15, 7108–7119. [Google Scholar] [CrossRef]
- Schermelleh, L.; Solovei, I.; Zink, D.; Cremer, T. Two-color fluorescence labeling of early and mid-to-late replicating chromatin in living cells. Chromosome Res. 2001, 9, 77–80. [Google Scholar] [CrossRef]
- de Leon, F.A.P.; Li, Y.; Weng, Z. Early and late replicative chromosomal banding patterns of Gallus domesticus. J. Hered. 1992, 83, 36–42. [Google Scholar] [CrossRef]
- Berchtold, D.; Fesser, S.; Bachmann, G.; Kaiser, A.; Eilert, J.C.; Frohns, F.; Sadoni, N.; Muck, J.; Kremmer, E.; Eick, D.; et al. Nuclei of chicken neurons in tissues and three-dimensional cell cultures are organized into distinct radial zones. Chromosome Res. 2011, 19, 165–182. [Google Scholar] [CrossRef]
- Sexton, T.; Umlauf, D.; Kurukuti, S.; Fraser, P. The role of transcription factories in large-scale structure and dynamics of interphase chromatin. Semin. Cell Dev. Biol. 2007, 18, 691–697. [Google Scholar] [CrossRef]
- Finlan, L.E.; Sproul, D.; Thomson, I.; Boyle, S.; Kerr, E.; Perry, P.; Ylstra, B.; Chubb, J.R.; Bickmore, W.A. Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet. 2008, 4, e1000039. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Fishman, V.; Battulin, N.; Nuriddinov, M.; Maslova, A.; Zlotina, A.; Strunov, A.; Chervyakova, D.; Korablev, A.; Serov, O.; Krasikova, A. 3D organization of chicken genome demonstrates evolutionary conservation of topologically associated domains and highlights unique architecture of erythrocytes’ chromatin. Nucleic Acids Res. 2019, 47, 648–665. [Google Scholar] [CrossRef] [Green Version]
- Fontdevila, A. Genetic instability and rapid speciation: Are they coupled? Genetica 1992, 86, 247–258. [Google Scholar] [CrossRef]
- Kazazian, J. L1 retrotransposons shape the mammalian genome. Science 2000, 289, 1152–1153. [Google Scholar] [CrossRef]
- Kidwell, M.G.; Holyoake, A.J. Transposon-induced hotspots for genomic instability. Genome Res. 2001, 11, 1321–1322. [Google Scholar] [CrossRef]
- Srikulnath, K.; Matsubara, K.; Uno, Y.; Thongpan, A.; Suputtitada, S.; Nishida-Umehara, C.; Matsuda, Y.; Apisitwanich, S. Genetic relationship of three butterfly lizard species (Leiolepis reevesii rubritaeniata, Leiolepis belliana belliana, Leiolepis boehmei, Agamidae, Squamata) inferred from nuclear gene sequence analyses. Kasetsart J. Nat. Sci. 2010, 44, 424–435. [Google Scholar]
- Ahmad, S.F.; Singchat, W.; Jehangir, M.; Suntronpong, A.; Panthum, T.; Malaivijitnond, S.; Srikulnath, K. Dark matter of primate genomes: Satellite DNA repeats and their evolutionary dynamics. Cells 2020, 9, 2714. [Google Scholar] [CrossRef]
- Fan, H.; Chu, J.Y. A brief review of short tandem repeat mutation. Genom. Proteom. Bioinform. 2007, 5, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Melters, D.P.; Bradnam, K.R.; Young, H.A.; Telis, N.; May, M.R.; Ruby, J.G.; Sebra, R.; Peluso, P.; Eid, J.; Rank, D.; et al. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol. 2013, 14, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.; O’neill, R.J. Centromere repeats: Hidden gems of the genome. Genes 2019, 10, 223. [Google Scholar] [CrossRef] [Green Version]
- Nanda, I.; Schrama, D.; Feichtinger, W.; Haaf, T.; Schartl, M.; Schmid, M. Distribution of telomeric (TTAGGG)n sequences in avian chromosomes. Chromosoma 2002, 111, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Clemente, L.; Mazzoleni, S.; Bellavia, E.P.; Augstenová, B.; Auer, M.; Praschag, P.; Protiva, T.; Velenský, P.; Wagner, P.; Fritz, U.; et al. Interstitial telomeric repeats are rare in turtles. Genes 2020, 11, 657. [Google Scholar] [CrossRef] [PubMed]
- Völker, M.; Backström, N.; Skinner, B.M.; Langley, E.J.; Bunzey, S.K.; Ellegren, H.; Griffin, D.K. Copy number variation, chromosome rearrangement, and their association with recombination during avian evolution. Genome Res. 2010, 20, 503–511. [Google Scholar] [CrossRef] [Green Version]
- Ashley, T.; Ward, D.C. A “hot-spot” of recombination coincides with an interstitial telomeric sequence in the Armenian hamster. Cytogenet. Cell Genet. 1993, 62, 169–171. [Google Scholar] [CrossRef]
- Zakian, V.A.; Pluta, A.F. Telomere formation in yeast. Nature 1989, 338, 468. [Google Scholar] [CrossRef]
- Matsubara, K.; Uno, Y.; Srikulnath, K.; Seki, R.; Nishida-Umehara, C.; Matsuda, Y. Molecular cloning and characterization of satellite DNA sequences from constitutive heterochromatin of the habu snake (Protobothrops flavoviridis, Viperidae) and the Burmese python (Python bivittatus, Pythonidae). Chromosoma 2015, 124, 529–539. [Google Scholar] [CrossRef]
- Matsubara, K.; Tarui, H.; Toriba, M.; Yamada, K.; Nishida-Umehara, C.; Agata, K.; Matsuda, Y. Evidence for different origin of sex chromosomes in snakes, birds, and mammals and step-wise differentiation of snake sex chromosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 18190–18195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezaz, T.; Deakin, J.E. Repetitive sequence and sex chromosome evolution in vertebrates. Adv. Evol. Biol. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Furman, B.L.S.; Metzger, D.C.H.; Darolti, I.; Wright, A.E.; Sandkam, B.A.; Almeida, P.; Shu, J.J.; Mank, J.E.; Fraser, B. Sex chromosome evolution: So many exceptions to the rules. Genome Biol. Evol. 2020, 12, 750–763. [Google Scholar] [CrossRef]
- Garrido-Ramos, M.A. Satellite DNA: An evolving topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef]
- Miga, K.H. Centromeric satellite DNAs: Hidden sequence variation in the human population. Genes 2019, 10, 352. [Google Scholar] [CrossRef] [Green Version]
- Weissensteiner, M.H.; Suh, A. Repetitive DNA: The dark matter of avian genomics. In Avian Genomics in Ecology and Evolution; Kraus, R., Ed.; Springer: Cham, Switzerland, 2019; pp. 93–150. [Google Scholar]
- GBIF Secretariat. Colaptes melanochloros (Gmelin, 1788). GBIF Backbone Taxonomy. Available online: https://www.gbif.org/species/2478272 (accessed on 22 June 2021).
- GBIF Secretariat. Colaptes campestris (Vieillot, 1818). GBIF Backbone Taxonomy. 2021. Available online: https://www.gbif.org/ru/species/2478233 (accessed on 22 June 2021).
- de Oliveira, T.D.; Kretschmer, R.; Bertocchi, N.A.; Degrandi, T.M.; De Oliveira, E.H.C.; De Cioffi, M.B.; Garnero, A.D.V.; Gunski, R.J. Genomic organization of repetitive DNA in woodpeckers (Aves, Piciformes): Implications for karyotype and ZW sex chromosome differentiation. PLoS ONE 2017, 12, e169987. [Google Scholar] [CrossRef]
- Yamada, K.; Nishida-Umehara, C.; Matsuda, Y. Molecular and cytogenetic characterization of site-specific repetitive DNA sequences in the Chinese soft-shelled turtle (Pelodiscus sinensis, Trionychidae). Chromosome Res. 2005, 13, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Wagler, G. Natürliches System der Amphibien, mit Vorangehender Classification der Säugetiere und Vögel. Ein Beitrag zur Vergleichenden Zoologie. 1.0; JG Cotta: München/Stuttgart/Tübingen, Germany, 1830; p. 354. [Google Scholar]
- Gray, J.E. Description of a new species of Staurotypus (S. salvinii) from Guatemala. Ann. Mag. Nat. Hist. 1864, 14, 451–452. [Google Scholar]
- Kawagoshi, T.; Uno, Y.; Nishida-Umehara, C.; Matsuda, Y. The Staurotypus turtles and aves share the same origin of sex chromosomes but evolved different types of heterogametic sex determination. PLoS ONE 2014, 9, e105315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thongchum, R.; Singchat, W.; Laopichienpong, N.; Tawichasri, P.; Kraichak, E.; Prakhongcheep, O.; Sillapaprayoon, S.; Muangmai, N.; Baicharoen, S.; Suntrarachun, S.; et al. Diversity of PBI-DdeI satellite DNA in snakes correlates with rapid independent evolution and different functional roles. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Capriglione, T.; Cardone, A.; Odierna, G.; Olmo, E. Evolution of a centromeric satellite DNA and phylogeny of lacertid lizards. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1991, 100, 641–645. [Google Scholar] [CrossRef]
- Capriglione, T.; Cardone, A.; Odierna, G.; Olmo, E. Further data on the occurrence and evolution of satellite DNA families in the lacertid genome. Chromosome Res. 1994, 2, 327–330. [Google Scholar] [CrossRef]
- Capriglione, T.; De Santo, M.G.; Odierna, G.; Olmo, E. An alphoid-like satellite DNA sequence is present in the genome of a lacertid lizard. J. Mol. Evol. 1998, 46, 240–244. [Google Scholar] [CrossRef]
- Ciobanu, D.; Grechko, V.V.; Darevsky, I.S.; Kramerov, D.A. New satellite DNA in Lacerta s. str. lizards (Sauria: Lacertidae): Evolutionary pathways and phylogenetic impact. J. Exp. Zool. Part B 2004, 302, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Grechko, V.V.; Ciobanu, D.G.; Darevsky, I.S.; Kramerov, D.A. Satellite DNA of lizards of the genus Lacerta s. str. (the Group L. agilis), the family Lacertidae. Dokl. Biochem. Biophys. 2005, 400, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Giovannotti, M.; S’Khifa, A.; Nisi Cerioni, P.; Splendiani, A.; Slimani, T.; Fioravanti, T.; Olmo, E.; Caputo Barucchi, V. Isolation and characterization of two satellite DNAs in Atlantolacerta andreanskyi (Werner, 1929) (Reptilia, Lacertidae). J. Exp. Zool. Part B 2020, 334, 178–191. [Google Scholar] [CrossRef]
- Giovannotti, M.; Cerioni, P.N.; Caputo, V.; Olmo, E. Characterisation of a GC-rich telomeric satellite DNA in Eumeces schneideri Daudin (Reptilia, Scincidae). Cytogenet. Genome Res. 2009, 125, 272–278. [Google Scholar] [CrossRef]
- Giovannotti, M.; Cerioni, P.N.; Splendiani, A.; Ruggeri, P.; Olmo, E.; Barucchi, V.C. Slow evolving satellite DNAs: The case of a centromeric satellite in Chalcides ocellatus (Forskål, 1775) (Reptilia, Scincidae). Amphib. Reptil. 2013, 34, 401–411. [Google Scholar] [CrossRef]
- Matsubara, K.; Sarre, S.D.; Georges, A.; Matsuda, Y.; Graves, J.A.M.; Ezaz, T. Highly differentiated ZW sex microchromosomes in the Australian varanus species evolved through rapid amplification of repetitive sequences. PLoS ONE 2014, 9, e95226. [Google Scholar] [CrossRef] [PubMed]
- Prakhongcheep, O.; Thapana, W.; Suntronpong, A.; Singchat, W.; Pattanatanang, K.; Phatcharakullawarawat, R.; Muangmai, N.; Peyachoknagul, S.; Matsubara, K.; Ezaz, T.; et al. Lack of satellite DNA species-specific homogenization and relationship to chromosomal rearrangements in monitor lizards (Varanidae, Squamata). BMC Evol. Biol. 2017, 17, 193. [Google Scholar] [CrossRef] [Green Version]
- Plohl, M.; Meštrović, N.; Mravinac, B. Satellite DNA evolution. In Repetitive DNA; Garrido-Ramos, M.A., Ed.; Karger: Basel, Switzerland, 2012; Volume 7, pp. 126–152. [Google Scholar] [CrossRef]
- Werner, F. Wissenschaftliche Ergebnisse einer zoologischen Forschungsreise nach Westalgerien und Marokko. In Sitzungsberichte der Akademie der Wissenschaften Mathematisch-Naturwissenschaftliche Klasse; Verlag der Österreichischen Akademie der Wissenschaften: Vienna, Austria, 1929; Volume 138, pp. 1–34. [Google Scholar]
- Deraniyagala, P.E.P. Four new races of the “Kabaragoya” lizard Varanus salvator. Spolia Zeylan. 1944, 24, 59–62. [Google Scholar]
- Chaiprasertsri, N.; Uno, Y.; Peyachoknagul, S.; Prakhongcheep, O.; Baicharoen, S.; Charernsuk, S.; Nishida-Umehara, C.; Matsuda, Y.; Koga, A.; Srikulnath, K. Highly species-specific centromeric repetitive DNA sequences in lizards: Molecular cytogenetic characterization of a novel family of satellite DNA sequences isolated from the water monitor lizard (Varanus salvator macromaculatus, Platynota). J. Hered. 2013, 104, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Hallowell, E. Report upon the Reptilia of the North Pacific Exploring Expedition, under command of Capt. John Rogers, U.S.N. Proc. Acad. Nat. Sci. USA 1861, 12, 480–510. [Google Scholar]
- Kuhl, H. Beiträge zur Zoologie und Vergleichenden Anatomie; Hermannsche Buchhandlung: Frankfurt, Germany, 1820; p. 152. [Google Scholar]
- Schneider, J.G. Historiae Amphibiorum Naturalis et Literariae. Fasciculus Secundus Continens Crocodilos. Scincos. Chamaesauras, Boas. Pseudoboas, Elapes, Angues. Amphisbaenas et Caecilias; Frommanni: Jena, Germany, 1801; p. 374. [Google Scholar]
- Tiersch, T.R.; Wachtel, S.S. On the evolution of genome size of birds. J. Hered. 1991, 82, 363–368. [Google Scholar] [CrossRef]
- Holmquist, G.P. Evolution of chromosome bands: Molecular ecology of noncoding DNA. J. Mol. Evol. 1989, 28, 469–486. [Google Scholar] [CrossRef]
- Vandergon, T.L.; Reitman, M. Evolution of chicken repeat 1 (CR1) elements: Evidence for ancient subfamilies and multiple progenitors. Mol. Biol. Evol. 1994, 11, 886–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhary, B.P.; Raudsepp, T.; Frönicke, L.; Scherthan, H. Emerging patterns of comparative genome organization in some mammalian species as revealed by Zoo-FISH. Genome Res. 1998, 8, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, N.; Sasaki, M. A phylogenetic study of bird karyotypes. Chromosoma 1974, 46, 91–120. [Google Scholar] [CrossRef] [PubMed]
- Belterman, R.H.R.; De Boer, L.E.M. A miscellaneous collection of bird karyotypes. Genetica 1990, 83, 17–29. [Google Scholar] [CrossRef]
- Jones, C.T.; Morrice, D.R.; Paton, I.R.; Burt, D.W. Homologues of genes on human chromosome 15q21→q26 and a chicken microchromosome show conserved synteny and gene order. Mamm. Genome 1997, 8, 436–440. [Google Scholar] [CrossRef]
- Masabanda, J.; Friedl, R.; Sazanov, A.; Lahti, J.M.; Li, H.; Kidd, V.J.; Fries, R. Mapping of five members of the cyclin gene family on chicken chromosomes by FISH. Chromosome Res. 1998, 6, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Sazanov, A.; Masabanda, J.; Ewald, D.; Takeuchi, S.; Tixier-Boichard, M.; Buitkamp, J.; Fries, R. Evolutionarily conserved telomeric location of BBC1 and MC1Ron a microchromosome questions the identity of MC1Rand a pigmentation locus on chromosome 1 in chicken. Chromosome Res. 1998, 6, 651–654. [Google Scholar] [CrossRef] [PubMed]
- De Lucca, E. Karyotype and nucleolus organizing regions in somatic chromosomes of the white-tailed hawk Buteo albicaudatus (Falconiformes: Aves). Cytobios 1985, 42, 7–13. [Google Scholar]
- Modesto, S.P.; Anderson, J.S. The phylogenetic definition of reptilia. Syst. Biol. 2004, 53, 815–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiens, J.J. Systematics and herpetology in the age of genomics. Bioscience 2008, 58, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Painter, T.S. The Y-chromosome in mammals. Science 1921, 53, 503–504. [Google Scholar] [CrossRef] [Green Version]
- Matthey, R.; Rovey, R. The chromosome formula in five species of Chiroptera. Cell. Mol. Life Sci. 1948, 4, 26. [Google Scholar]
- Peccinini, D.; Frota-Pessoa, O.; Ferrari, I. Sex determination of the “pseudo-xo/xx” type in the Brazilian lizard Polychrus sp. (Sauria, Iguanidae). Caryologia 1971, 24, 129–139. [Google Scholar] [CrossRef]
- Matsubara, K.; O’Meally, D.; Sarre, S.D.; Georges, A.; Srikulnath, K.; Ezaz, T. ZW sex chromosomes in Australian dragon lizards (Agamidae) originated from a combination of duplication and translocation in the nucleolar organising region. Genes 2019, 10, 861. [Google Scholar] [CrossRef] [Green Version]
- Schoepff, J.D. Historia Testudinum Iconibus Illustrata; Palm, J.J., Ed.; Harvard University: Cambridge, MA, USA, 1972; Volume xii, pp. 1–32. [Google Scholar]
- Kasai, F.; O’Brien, P.C.M.; Ferguson-Smith, M.A. Reassessment of genome size in turtle and crocodile based on chromosome measurement by flow karyotyping: Close similarity to chicken. Biol. Lett. 2012, 8, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, A.M.; Edwards, S.V. Amniotes (Amniota). In The Timetree of Life; Hedges, S.B., Kumar, S., Eds.; Oxford University Press: New York, NY, USA, 2009; pp. 375–379. [Google Scholar]
- Pokorná, M.J.; Rovatsos, M.; Kratochvíl, L. Sex chromosomes and karyotype of the (nearly) mythical creature, the Gila monster, Heloderma suspectum (Squamata: Helodermatidae). PLoS ONE 2014, 9, e104716. [Google Scholar] [CrossRef]
- Damas, J.; O’Connor, R.; Farré, M.; Lenis, V.P.E.; Martell, H.J.; Mandawala, A.; Fowler, K.; Joseph, S.; Swain, M.T.; Griffin, D.K.; et al. Upgrading short-read animal genome assemblies to chromosome level using comparative genomics and a universal probe set. Genome Res. 2017, 27, 875–884. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, R.E.; Farré, M.; Joseph, S.; Damas, J.; Kiazim, L.; Jennings, R.; Bennett, S.; Slack, E.A.; Allanson, E.; Larkin, D.M.; et al. Chromosome-level assembly reveals extensive rearrangement in saker falcon and budgerigar, but not ostrich, genomes. Genome Biol. 2018, 19, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Huang, L.; Markowitz, T.E.; Blitzblau, H.G.; Chen, D.; Klein, F.; Hochwagen, A. Transcription dynamically patterns the meiotic chromosome-axis interface. eLife 2015, 4, e07424. [Google Scholar] [CrossRef] [PubMed]
- Potter, I.C.; Rothwell, B. The mitotic chromosomes of the lamprey, Petromyzon marinus L. Experientia 1970, 26, 429–430. [Google Scholar] [CrossRef]
- Smith, J.J.; Stuart, A.B.; Sauka-Spengler, T.; Clifton, S.W.; Amemiya, C.T. Development and analysis of a germline BAC resource for the sea lamprey, a vertebrate that undergoes substantial chromatin diminution. Chromosoma 2010, 119, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Covelo-Soto, L.; Morán, P.; Pasantes, J.J.; Pérez-García, C. Cytogenetic evidences of genome rearrangement and differential epigenetic chromatin modification in the sea lamprey (Petromyzon marinus). Genetica 2014, 142, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.J.; Timoshevskaya, N.; Ye, C.; Holt, C.; Keinath, M.C.; Parker, H.J.; Cook, M.E.; Hess, J.E.; Narum, S.R.; Lamanna, F.; et al. The sea lamprey germline genome provides insights into programmed genome rearrangement and vertebrate evolution. Nat. Genet. 2018, 50, 270–277. [Google Scholar] [CrossRef]
- Mehta, T.K.; Raèi, È.; Yamasaki, S.; Lee, A.P.; Lian, M.M.; Tay, B.H.; Tohari, S.; Yanai, S.; Tay, A.; Brenner, S.; et al. Evidence for at least six Hox clusters in the Japanese lamprey (Lethenteron japonicum). Proc. Natl. Acad. Sci. USA 2013, 110, 16044–16049. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, R.E.; Romanov, M.N.; Kiazim, L.G.; Barrett, P.M.; Farré, M.; Damas, J.; Ferguson-Smith, M.; Valenzuela, N.; Larkin, D.M.; Griffin, D.K. Reconstruction of the diapsid ancestral genome permits chromosome evolution tracing in avian and non-avian dinosaurs. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair Hedges, S.; Poling, L.L. A molecular phylogeny of reptiles. Science 1999, 283, 998–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, M.J. Phylogeny of the major tetrapod groups: Morphological data and divergence dates. J. Mol. Evol. 1990, 30, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Romanov, M.N.; Farré, M.; Lithgow, P.E.; Fowler, K.E.; Skinner, B.M.; O’Connor, R.; Fonseka, G.; Backström, N.; Matsuda, Y.; Nishida-Umehara, C.; et al. Reconstruction of gross avian genome structure, organization and evolution suggests that the chicken lineage most closely resembles the dinosaur avian ancestor. BMC Genom. 2014, 15, 1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida-Umehara, C.; Tsuda, Y.; Ishijima, J.; Ando, J.; Fujiwara, A.; Matsuda, Y.; Griffin, D.K. The molecular basis of chromosome orthologies and sex chromosomal differentiation in palaeognathous birds. Chromosome Res. 2007, 15, 721–734. [Google Scholar] [CrossRef]
- Nanda, I.; Karl, E.; Griffin, D.K.; Schartl, M.; Schmid, M. Chromosome repatterning in three representative parrots (Psittaciformes) inferred from comparative chromosome painting. Cytogenet. Genome Res. 2007, 117, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Penny, D. Mass survival of birds across the Cretaceous-tertiary boundary: Molecular evidence. Science 1997, 275, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Burt, D.W. Chromosome rearrangement in evolution. In Encyclopedia of Life Sciences; Nature Publishing Group, MacMillan Publishers: London, UK, 2001. [Google Scholar] [CrossRef]
- Kearney, M.; Hewitt, G. Stasipatric speciation: Resurrecting a system to bury a hypothesis? Mol. Ecol. 2009, 18, 3331–3333. [Google Scholar] [CrossRef]
- Bush, G.L.; Case, S.M.; Wilson, A.C.; Patton, J.L. Rapid speciation and chromosomal evolution in mammals. Proc. Natl. Acad. Sci. USA 1977, 74, 3942–3946. [Google Scholar] [CrossRef] [Green Version]
- Szarski, H. Cell size and the concept of wasteful and frugal evolutionary strategies. J. Theor. Biol. 1983, 21, 201–209. [Google Scholar] [CrossRef]
- Castoe, T.A.; De Koning, A.P.J.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouwens, P.A. On a large Varanus species from the island of Komodo. Bull. Jard. Bot. Buitenzorg 1912, 6, 1–3. [Google Scholar]
- Lind, A.L.; Lai, Y.Y.Y.; Mostovoy, Y.; Holloway, A.K.; Iannucci, A.; Mak, A.C.Y.; Fondi, M.; Orlandini, V.; Eckalbar, W.L.; Milan, M.; et al. Genome of the Komodo dragon reveals adaptations in the cardiovascular and chemosensory systems of monitor lizards. Nat. Ecol. Evol. 2019, 3, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.M.; Gans, C. The chromosomes of the order Crocodilia. Cytogenet. Genome Res. 1970, 9, 81–105. [Google Scholar] [CrossRef]
- De Boer, L.E.M.; Sinoo, R.P. A karyological study of Accipitridae (Aves: Falconiformes), with karyotypic descriptions of 16 species new to cytology. Genetica 1984, 65, 89–107. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell: How Genomes Evolve, 4th ed.; Garland Science: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26836/ (accessed on 22 June 2021).
- Waddington, D.; Springbett, A.J.; Burt, D.W. A chromosome-based model for estimating the number of conserved segments between pairs of species from comparative genetic maps. Genetics 2000, 154, 323–332. [Google Scholar] [CrossRef]
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Variation in recombination frequency and distribution across eukaryotes: Patterns and processes. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160455. [Google Scholar] [CrossRef]
- Coyne, J.A.; Orr, H.A. Speciation; Sinauer Associates: Sunderland, MA, USA, 2004. [Google Scholar]
- Butlin, R.K. Recombination and speciation. Mol. Ecol. 2005, 14, 2621–2635. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Barrientos, D.; Engelstädter, J.; Rieseberg, L.H. Recombination rate evolution and the originof species. Trends Ecol. Evol. 2016, 31, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Payseur, B.A.; Rieseberg, L.H. A genomic perspective on hybridization and speciation. Mol. Ecol. 2016, 25, 2337–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toews, D.P.; Taylor, S.A.; Vallender, R.; Brelsford, A.; Butcher, B.G.; Messer, P.W.; Lovette, I.J. Plumage genes and little else distinguish the genomes of Hybridizing Warblers. Curr. Biol. 2016, 26, 2313–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campagna, L.; Repenning, M.; Silveira, L.F.; Fontana, C.S.; Tubaro, P.L.; Lovette, I.J. Repeated divergent selection on pigmentation genes in a rapid finch radiation. Sci. Adv. 2017, 3, e1602404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poelstra, J.W.; Vijay, N.; Bossu, C.M.; Lantz, H.; Ryll, B.; Müller, I.; Baglione, V.; Unneberg, P.; Wikelski, M.; Grabherr, M.G.; et al. The genomic landscape underlying phenotypic integrity in the face of gene flow in crows. Science 2014, 344, 1410–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stryjewski, K.F.; Sorenson, M.D. Mosaic genome evolution in a recent and rapid avian radiation. Nat. Ecol. Evol. 2017, 1, 1912–1922. [Google Scholar] [CrossRef]
- Yin, W.; Wang, Z.J.; Li, Q.Y.; Lian, J.M.; Zhou, Y.; Lu, B.Z.; Jin, L.J.; Qiu, P.X.; Zhang, P.; Zhu, W.B.; et al. Evolutionary trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nat. Commun. 2016, 7, 13107. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.B.; Ellegren, H. Making sense of genomic islands of differentiation in light of speciation. Nat. Rev. Genet. 2017, 18, 87–100. [Google Scholar] [CrossRef]
- Campbell, C.R.; Poelstra, J.W.; Yoder, A.D. What is speciation genomics? The roles of ecology, gene flow, and genomic architecture in the formation of species. Biol. J. Linn. Soc. 2018, 124, 561–583. [Google Scholar] [CrossRef]
- Muller, H.J. The relation of recombination to mutational advance. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 1964, 1, 2–9. [Google Scholar] [CrossRef]
- Kondrashov, A.S. Deleterious mutations and the evolution of sexual reproduction. Nature 1988, 336, 435–441. [Google Scholar] [CrossRef]
- Haddrill, P.R.; Halligan, D.L.; Tomaras, D.; Charlesworth, B. Reduced efficacy of selection in regions of the Drosophila genome that lack crossing over. Genome Biol. 2007, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hill, W.G.; Robertson, A. The effect of linkage on limits to artificial selection. Genet. Res. 2007, 89, 311–336. [Google Scholar] [CrossRef] [PubMed]
- Dumont, B.L.; Payseur, B.A. Evolution of the genomic rate of recombination in mammals. Evolution 2008, 62, 276–294. [Google Scholar] [CrossRef] [PubMed]
- Martin, S. Global diversity of crocodiles (Crocodilia, Reptilia) in freshwater. In Freshwater Animal Diversity Assessment; Springer: Dordrecht, The Netherlands, 2007; Volume 198, pp. 587–591. [Google Scholar]
- Dawson, D.A.; Åkesson, M.; Burke, T.; Pemberton, J.M.; Slate, J.; Hansson, B. Gene order and recombination rate in homologous chromosome regions of the chicken and a passerine bird. Mol. Biol. Evol. 2007, 24, 1537–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duret, L.; Mouchiroud, D.; Gautier, C. Statistical analysis of vertebrate sequences reveals that long genes are scarce in GC-rich isochores. J. Mol. Evol. 1995, 40, 308–317. [Google Scholar] [CrossRef]
- Likhoshvai, V.A.; Matushkin, Y.G. Nucleotide composition-based prediction of gene expression efficacy. Mol. Biol. 2000, 34, 345–350. [Google Scholar] [CrossRef]
- Elhaik, E.; Pellegrini, M.; Tatarinova, T.V. Gene expression and nucleotide composition are associated with genic methylation level in Oryza sativa. BMC Bioinform. 2014, 15, 23. [Google Scholar] [CrossRef] [Green Version]
- Halder, B.; Malakar, A.K.; Chakraborty, S. Nucleotide composition determines the role of translational efficiency in human genes. Bioinformation 2017, 13, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, M.N.; Uddin, A.; Chakraborty, S. Gene expression, nucleotide composition and codon usage bias of genes associated with human Y chromosome. Genetica 2017, 145, 295–305. [Google Scholar] [CrossRef]
- Zahdeh, F.; Carmel, L. Nucleotide composition affects codon usage toward the 3′-end. PLoS ONE 2019, 14, e0225633. [Google Scholar] [CrossRef]
- Zrimec, J.; Börlin, C.S.; Buric, F.; Muhammad, A.S.; Chen, R.; Siewers, V.; Verendel, V.; Nielsen, J.; Töpel, M.; Zelezniak, A. Deep learning suggests that gene expression is encoded in all parts of a co-evolving interacting gene regulatory structure. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Nordborg, M.; Charlesworth, B.; Charlesworth, D. The effect of recombination on background selection. Genet. Res. 1996, 67, 159–174. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Vertebrate Groups | Diploid Chromosome Range | Macrochromosome Range | Microchromosome Range | Genome Size Range (Gb) | References |
|---|---|---|---|---|---|
| Testudines | 26–68 | 10–36 | 0–56 | 2.7–5.4 | Valenzuela and Adams [63] |
| Crocodilian | 30–42 | - | - | 1.3–3.9 | Srikulnath et al. [9] |
| Fish | 12–168 | 12–60 | 0–144 | 0.3–17.05 | Gregory [66]; Gregory and Witt [67]; Arai [68] |
| Holocephalian, chondrostean and holostean fishes | 58–112 | 24–64 | 34–52 | 2.98–14.8 | Ohno et al. [53] |
| Amphibian | 18–106 | 18–92 | 2–30 | 0.93–137 | Morescalchi [69,70]; Voss et al. [71]; Schmid et al. [72]; Perkins et al. [73] |
| Cryptobranchidae and Hynobiidae | 40–78 | 20–50 | 30–40 | 16.5–56.8 | Morescalchi [10,11]; Zhang et al. [74] |
| Aves | 40–142 | 20–60 | 10–90 | 0.96–2.2 | Organ et al. [75]; Tegelström et al. [42,76]; Kapusta et al. [77] |
| Mammalian | 6–102 | 6–102 | 1.6–6.3 | Ferguson-Smith and Trifonov [78]; Graphodatsky et al. [79]; Kapusta et al. [77] | |
| Squamate Reptiles | 20–68 | 10–42 | 0–56 | 1.1–5.4 | |
| Tuatara | 36 | 28 | 8 | 4.9 | Gregory et al. [80,81]; Srikulnath et al. [82]; Deakin and Ezaz [58]; Pasquesi et al. [83]; Ahmad et al. [84] |
| Gecko lizard | 26–68 | 10–36 | - | ||
| Lizards | 20–62 | 10–38 | 0–28 | 1.03–3.8 | |
| Snakes | 26–50 | 10–38 | 0–36 | 1.3–3.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srikulnath, K.; Ahmad, S.F.; Singchat, W.; Panthum, T. Why Do Some Vertebrates Have Microchromosomes? Cells 2021, 10, 2182. https://doi.org/10.3390/cells10092182
Srikulnath K, Ahmad SF, Singchat W, Panthum T. Why Do Some Vertebrates Have Microchromosomes? Cells. 2021; 10(9):2182. https://doi.org/10.3390/cells10092182
Chicago/Turabian StyleSrikulnath, Kornsorn, Syed Farhan Ahmad, Worapong Singchat, and Thitipong Panthum. 2021. "Why Do Some Vertebrates Have Microchromosomes?" Cells 10, no. 9: 2182. https://doi.org/10.3390/cells10092182