
Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure


1
Division of Infectious Diseases, Department of Pediatrics, University of California San Diego, La Jolla, CA 92093, USA
2
Rady Children’s Hospital, San Diego, CA 92123, USA
*
Author to whom correspondence should be addressed.
Cells 2021, 10(7), 1798; https://doi.org/10.3390/cells10071798
Submission received: 21 June 2021
/
Revised: 13 July 2021
/
Accepted: 13 July 2021
/
Published: 16 July 2021
(This article belongs to the Collection Role of Autophagy in Viral Infection)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Effective antiretroviral therapy has led to significant human immunodeficiency virus type 1 (HIV-1) suppression and improvement in immune function. However, the persistence of integrated proviral DNA in latently infected reservoir cells, which drive viral rebound post-interruption of antiretroviral therapy, remains the major roadblock to a cure. Therefore, the targeted elimination or permanent silencing of this latently infected reservoir is a major focus of HIV-1 research. The most studied approach in the development of a cure is the activation of HIV-1 expression to expose latently infected cells for immune clearance while inducing HIV-1 cytotoxicity—the “kick and kill” approach. However, the complex and highly heterogeneous nature of the latent reservoir, combined with the failure of clinical trials to reduce the reservoir size casts doubt on the feasibility of this approach. This concern that total elimination of HIV-1 from the body may not be possible has led to increased emphasis on a “functional cure” where the virus remains but is unable to reactivate which presents the challenge of permanently silencing transcription of HIV-1 for prolonged drug-free remission—a “block and lock” approach. In this review, we discuss the interaction of HIV-1 and autophagy, and the exploitation of autophagy to kill selectively HIV-1 latently infected cells as part of a cure strategy. The cure strategy proposed has the advantage of significantly decreasing the size of the HIV-1 reservoir that can contribute to a functional cure and when optimised has the potential to eradicate completely HIV-1.
Keywords:
HIV-1; autophagy; latency promoting approach; cell death; nanoparticle; SMAC mimetics; apoptosis; autosis1. Introduction
Within days of infection, human immunodeficiency virus type 1 (HIV-1) rapidly disseminates to draining lymph nodes. HIV-1 advances to destroy helper T cells and establish latent HIV-1 reservoirs through the irreversible integration of replication-competent proviral DNA into the genome of resting CD4+ T cells, hematopoietic stem cells, cells from the monocyte-macrophage lineage, and microglia [1,2,3,4,5]. Although antiretroviral therapy (ART) has transformed HIV-1 infection from a lethal and destructive disease into a controllable, but chronic condition, the establishment of HIV-1 latency is unaffected by early initiation of ART [6], and there is no evidence that ART alone is able to eradicate latent HIV-1 [7,8]. Moreover, although ART effectively suppresses HIV-1 replication, persistent residual low-level viremia maintains the latent pool [9,10], and the very low expression of viral proteins enables latently infected cells to evade detection and clearance by the immune system and undergo homeostatic proliferation [11,12,13,14,15]. Additionally, as viral loads rebound to high pre-therapy levels following ART interruption, albeit with significant heterogeneity in the speed of viral rebound (days, weeks, or sometimes years [16,17]), lifelong ART is required for continued viral suppression. Therefore, these latent cellular reservoirs present the major obstacle in preventing the eradication of HIV-1.
As with most viruses, HIV-1 must hijack and exploit host cell pathways for its replication and propagation, including the transcription and translation of viral proteins, and for the assembly of new virions. Biosynthesis within mammalian cells is a balance between anabolism and catabolism. While growth factors and cytokines regulate anabolism, conditions of stress promote catabolism under the regulation of AMPK (5′ AMP-activated protein kinase). Both anabolism- and catabolism-promoting signals converge at the evolutionarily conserved atypical serine/threonine kinase mechanistic target of rapamycin (MTOR) [18,19]. MTOR serves as a master regulator of cellular functions in response to diverse signals, including growth factors, immune signals, DNA damage, oxygen, metabolic cues, lysosomal activities, and infections. MTOR forms two structurally and functionally different complexes: MTOR complex 1 (MTORC1) and MTOR complex 2 (MTORC2). Although MTORC1 and MTORC2 are responsive to different signalling mechanisms and produce differing downstream effects, whereby the activation of either facilitates cell growth and survival [20,21]. MTORC2 functions include actin cytoskeleton organization, control of Na+ transport, and the inhibition of apoptosis [22,23,24], while MTORC1 targets downstream RPS6KB1, EIF4EBP1, and others to promote cellular growth and proliferation via increased protein and lipid synthesis, while repressing catabolic programmes resulting in the inhibition of autophagy. In relation to HIV-1-infection, the hyper-activation of MTORC1 by HIV-1 Tat is necessary for HIV-1 expression [25], and the integrity and function of both MTORC1 and MTORC2 are essential to re-activate HIV-1 from latency [26,27,28].
Autophagy is a catabolic intracellular lysosomal degradation pathway that ensures homeostasis through the elimination of misfolded protein aggregates, sub-cellular organelles, and microbial pathogens thereby promoting innate immunity and stimulating growth and/or survival [29,30,31,32,33,34]. Autophagy can be either non-selective—macroautophagy (hereafter referred to as autophagy) or selective. In selective autophagy, it mediates the degradation of specific cargo such as mitochondria (mitophagy), endoplasmic reticulum (reticulophagy), or invading microorganisms (xenophagy) [35,36]. In the latter, it also extensively interacts with immunometabolism to control infection and inflammation [37]. Both canonical and non-canonical autophagy pathways play pivotal roles in host defence and innate immune responses against HIV-1, and there are numerous reviews covering the mechanism, function, and regulation of both canonical and non-canonical autophagy during HIV-1 infection [38,39,40,41]. Here, we provide a brief summary of the potential role for autophagy as part of an HIV-1 cure strategy.
2. The HIV-1 Latent Reservoir
Although the mechanisms by which integrated HIV-1 proviral DNA becomes quiescent in vivo are not fully understood, hindering the development of effective curative strategies, the current understanding is that it is a multifactorial process that operates at the transcriptional and post-transcriptional levels to repress HIV-1 expression. Regardless of the integration site, the 5′ LTR of the transcriptionally silent provirus is embedded within nucleosomes (nuc) [42]. The short isoform of BRD4 recruits the cellular ATP-dependent chromatin remodelling BRG1/BRM-associated factor (BAF) complex to the proviral genome [43] where it actively positions nuc-1 in a refractory sequence immediately downstream of the HIV-1 transcription start site to generate a repressive chromatin conformation [44]. Although cis-regulatory elements located at the 5′ LTR strongly recruit cellular transcription factors and coactivators to initiate transcription, RNA polymerase II (RNAPII) undergoes promoter-proximal stalling at sites that overlap with nuc-1 after the production of a short nascent RNA transcript that includes the trans-activation response (TAR) element. The recruitment of the multi-subunit negative transcription elongation factors, NELF and DSIF, stabilise the paused RNAPII [45,46,47,48]. To overcome this, HIV-1 Tat binds the TAR element, where it undergoes K28 acetylation by the histone acetyltransferase (HAT) KAT2B, promoting the recruitment of positive transcription elongation factor b (P-TEFb; composed of CDK9 and CCNT1) and the super elongation complex [49,50,51,52,53]. The CDK9 subunit of P-TEFb phosphorylates NELF (which dissociates from TAR) [54], DSIF (converting it to an elongation factor) [55], and the RNAPII C-terminal domain, which unpauses and switches to a highly processive elongation complex [56]. HATs, including EP300 and KAT2A, then converge to acetylate HIV-1 Tat K50/51 [57]. The former facilitates the release of P-TEFb and the recruitment of the chromatin remodelling polybromo-associated BAF (PBAF) complex to the HIV-1 promoter to reposition nuc-1 downstream of the transcription start site [44,58]. The latter mediates the transfer of HIV-1 Tat from the TAR element to RNAPII promoting elongation [59]. Therefore, the availability and abundance of P-TEFb and HIV-1 Tat are crucial for HIV-1 transcription elongation. BCL11B represses P-TEFb recruitment through sequestration of P-TEFb in a complex containing 7SK small nuclear ribonucleoprotein (snRNP) and HEXIM1 thereby preventing efficient HIV-1 transcription elongation [60,61,62]. HIV-1 Vpr counteracts BCL11B-mediated viral gene repression by targeting and promoting the proteasomal degradation of BCL11B [63]. HIV-1 Tat abundance is affected by selective autophagy (discussed below) [64,65], and by proteasomal degradation. While lysine 48-polyubiquitination (K48Ub) of HIV-1 Tat stimulates transactivation [66,67], the K48Ub mark targets proteins for proteolytic degradation through the proteasome [68]. Similarly, the long non-coding RNA NRON also targets HIV-1 Tat to the ubiquitin/proteasome system for degradation [69].
In addition to nucleosome positioning by the BAF complex and decreased availability of HIV-1 Tat and P-TEFb, multiple epigenetic modulators involved in the post-translational acetylation, methylation, phosphorylation, ubiquitination, and crotonylation of histones, and the methylation of DNA work to silence the HIV-1 promoter [70,71]. The most studied of these are methylation and acetylation of lysine residues of histone tails that affect nucleosome stability. For instance, negatively acting NF-κB NFKB1 p50 homodimer, BCL11B, and YY1/LSF recruit HDAC1 to the 5′ LTR resulting in decreased nuc-1 acetylation, thereby increasing nucleosome stability [72,73,74]. BCL11B also recruits the histone methyltransferase SUV39H1, which catalyses the K9 trimethylation of H3 (H3K9me3) [74]. Nuc-1 is further K9 trimethylated by EHMT1, EHMT2, and LSD1 [75,76], which recruits the chromatin reader heterochromatin protein 1 (HP1) isoforms CBX1, CBX3, and CBX5 resulting in chromatin compaction. CBX5 further recruits SUV39H1 propagating the H3K9me3 mark along the HIV-1 promoter to further the heterochromatin environment, resulting in the repression of HIV-1 transcription [74].
Despite the epigenetic transcriptional elongation block, cell-associated abortive TAR element transcripts are detectable even in the absence of replication competent virus in persons receiving fully suppressive ART [9,10,77], suggesting that although viral transcription occurs during latency, downstream mechanisms that block viral RNA elongation, splicing, polyadenylation, and the nuclear export of these viral RNAs are involved in repressing HIV-1 expression [77,78]. Indeed, there is a greater block to HIV-1 transcriptional elongation, completion, and splicing than to transcriptional initiation [77,79]. Therefore, the mechanisms that control the translation, modification, and abundance of proteins involved in these steps are important regulators of HIV-1 expression. In support of this hypothesis, latent HIV-1-infected CD4+ T cells, the major reservoir of latent HIV-1, poorly express MATR3, an essential cofactor of HIV-1 Rev-mediated RNA export, thus decreasing translation [80], and express cellular microRNAs (miR-28, miR-125b, miR-150, miR-223, and miR-382) that potently inhibit HIV-1 production by binding to the 3′ untranslated region of mRNAs, including those of HIV-1 Tat, HIV-1 Rev, CCNT1, and components of the PBAF complex, inhibiting their translation [81,82,83,84].
3. Autophagy-Mediated Restriction of HIV-1
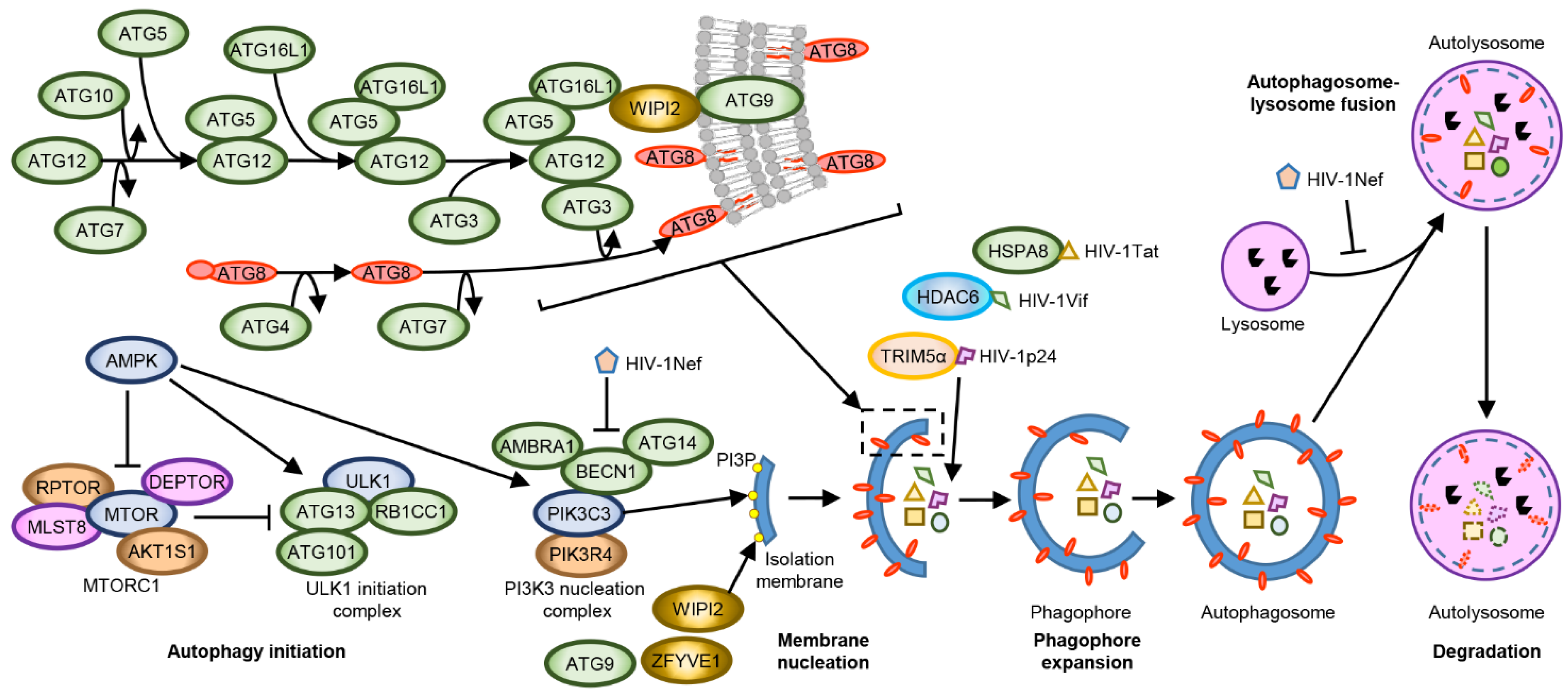
MTORC1 inhibition induces the formation of the ULK1 complex, which drives the formation of the initial autophagosomal precursor membrane structure (the phagophore) by directly activating the PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) complex I (Figure 1). The PIK3C3 complex I is composed of PIK3C3, phosphoinositide-3-kinase regulatory subunit 4 (PIK3R4), Beclin 1 (BECN1), and ATG (autophagy-related protein) 14. This complex translocates to ER sites and produces phosphatidylinositol 3-phosphate (PI3P) which subsequently recruits PI3P-binding proteins, such as WIPI2 and ZFYVE1 [85]. The recruitment of these proteins results in the formation of phagophores, which expand and close through membrane scission forming autophagosomes [86]. The precise mechanisms(s) by which this occurs are not completely known. However, current research suggests that it involves two ubiquitin-like conjugation systems. The first entails the E1-like enzyme ATG7 and the E2-like enzyme ATG3 conjugating the ubiquitin-like ATG8 family proteins (microtubule-associated protein 1 light chain 3 [MAP1LC3 or more commonly LC3] A, LC3B, LC3B2, LC3C, GABARAP [GABA type A receptor associated protein], GABARAP-like [GABARAPL] 1 and GABARAPL2) to the headgroups of membrane lipid phosphatidylethanolamine (PE). The second involves the recruitment of the E3-like ATG12–ATG5-ATG16L1 complex to PI3P-containing phagophores by the WIPI2b isoform. The ATG12–ATG5-ATG16L1 complex then ligates ATG8-PE to nascent autophagosome membranes where they can interact with cargo receptors harbouring LC3-interacting regions (LIRs; a conserved sequence W/F/YxxL/I/V) [85]. ATG16L1, as well as the polyubiquitin-binding autophagy receptors including sequestosome 1 (SQSTM1, p62), optineurin (OPTN), NBR1, and NDP52, which harbour both Ub-binding domains and LIRs also recruit and incorporate ubiquitin-decorated cargos into autophagosomes [87]. After detachment of ATG factors, the isolation membrane closes and syntaxin 17 is recruited to the autophagosomes. These then fuse with lysosomes resulting in the degradation of the engulfed components as well as the ATG8-PE and SQSTM1 associated with the inner membrane [88]. As autophagy is a coordinated pathway of autophagosome formation, sequestration of autophagic cargo, autophagosome-lysosome fusion, followed by the proteolytic degradation of the sequestered cargo, dysregulation or inhibition of any step could lead to the failure of the autophagy cycle that could lead to a disturbance of cellular homeostasis leading to cell death. As an example, inhibition of autophagy by blocking lysosomal degradation can lead to the accumulation of autophagosomes, thus depleting membrane sources.
Host cells can recognise microorganism-specific pathogen-associated molecular pattern (PAMP) molecules through pattern recognition receptors (PRR). These PRR include toll-like receptors (TLR), RIG-I-like receptors (RLR), nucleotide oligomerization domain (NOD)-like receptors (NLR), and C-type lectin receptors (CLR). The most studied PRR in relation to autophagy are the TLR. Humans have 10 TLRs, each recognizing a different PAMP, and are present in many different cell types including CD4+ T cells and macrophages. TLRs 1, 2, 4, 5, 6, and 10 are located at the plasma membrane and recognise bacterial membrane components, whereas TLRs 3, 7, 8, and 9 are predominantly located within endosomes. The phylogenetically and structurally related TLR7/8 recognise GU-rich short single-stranded microbial RNA, such as that found in the U5 region of the HIV-1 genome [89,90]. HIV-1-Env-CD4 interactions mediates the endocytosis of HIV-1 that delivers viral genomic RNA to the endosomal TLR7/8 [91,92]. In addition to mediating the endocytosis of HIV-1, in dendritic cells, HIV-1 Env also activates and exhausts autophagy leading to a rapid shutdown of both autophagy and immunoamphisome production. This results in an increase in both cell-associated HIV-1 and transfer of HIV-1 to CD4+ T cells while also impairing both the innate and adaptive immune responses as autophagy interacts with the MHC-II loading machinery [93,94]. Upon ligand activation, TLR8 associates and interacts with the adapter protein MYD88 that recruits IL-1 receptor-associated kinases, leading to the NF-κB-dependent transcription of numerous pro-inflammatory mediators including IL-6, IL-12, IL-27, TNF, interferon (IFN) γ, and IL-1β in the absence of pyroptosis [95,96,97,98]. Crucially, the NLRP3-inflammasome induced secretion of IL-1β is dependent upon intact autophagy [98,99]. Similarly, exposure of plasmacytoid dendritic cells (pDCs) to infectious or non-infectious HIV-1 or to HIV-1 derived GU-rich HIV-1 RNA sequences induces IFNα production through a TLR7- and autophagy-dependent mechanism [91,100]. In addition to stimulating the release of pro-inflammatory cytokines, TLR7/8 ligation also suppresses HIV-1 replication in acute ex vivo human lymphoid tissue of tonsillar origin and renders peripheral blood mononuclear cells (PBMC) barely permissive to HIV-1 infection [101]. Further studies demonstrated that binding of TLR8 by GU-rich HIV-1 RNA sequences induces autophagy through a cathelicidin antimicrobial peptide and vitamin D-dependent mechanism [92,102], and decreases HIV-1 p24 release from HIV-1 infected macrophages through an autophagy and lysosome dependent mechanism [92,102]. Interestingly, HIV-1 downregulates IRAK4, which is essential for virtually all TLR signalling [103]. People living with HIV-1 (PLWH) have lower levels of vitamin D3 than uninfected individuals [104,105,106,107,108,109,110,111]. Additionally, the concentrations of vitamin D3 decrease during HIV-1 disease progression and correlate with increased markers of inflammation and decreased autophagy and survival rates [110,112,113]. Vitamin D3 is a known autophagy inducer, and studies show that vitamin D3 decreases HIV-1 p24 release from HIV-1 infected macrophages through an autophagy-dependent mechanism [114].
One of the first HIV-1 restriction factors identified was the PRR TRIM5α, which interacts with retroviral capsid proteins in the context of an intact core structure. Although TRIM5α was originally suggested to be a species-specific HIV-1 restriction factor, with rhesus TRIM5α but not human TRIM5α efficiently restricting HIV-1 infection by accelerating premature uncoating of the virus thereby inhibiting nuclear translocation and subsequent integration [115,116,117], recent literature indicates that human TRIM5α also acts as an HIV-1 restriction factor through a different mechanism. While HIV-1 binding to DC-SIGN (CD209) positive dendritic cells leads to disassociation of TRIM5α from CD209, in Langerhans cells, TRIM5α mediates the assembly of an autophagy-activating scaffold to the CLR langerin (CD207), and acts as a receptor for selective autophagic degradation of HIV-1 proteins [118,119,120,121]. Moreover, the TRIM5α-HIV-1 p24 binding promotes the synthesis of unattached K63-linked ubiquitin chains that promotes the recruitment and activation of the MAP3K7-TAB complex in an autophagy-dependent mechanism, leading to the transcription and secretion of cytokines including interferon α/β and IL-6 [118,122,123]. Interestingly, TRIM5α expression is increased in PBMC from long-term non-progressors, and this increase correlates with an increase in HIV-1 gp120 and TRIM5α co-localization in autophagic vacuoles compared with PBMC from typical progressors [124].
Although Sagnier et al. [64] suggested that autophagy selectively degrades HIV-1 Tat through a ubiquitin-independent interaction with SQSTM1, Xu et al. demonstrated that HIV-1 Tat undergoes K63-polyubiquitination, a mark recognised by SQSTM1 and the NBR1 autophagy cargo receptor [125,126,127], complexes with the CMA-specific chaperone HSPA8 and is subjected to K63Ub-selective autophagy mediated by serine hydroxymethyltransferase (SHMT) 1, SHMT2 and the BRCC36/BRISC deubiquitase complex [65].
During HIV-1 infection, the cellular restriction factor APOBEC3G restricts HIV-1 replication through the specific deamination of dCs in newly synthesised (−)-strand viral DNA, resulting in massive G-to-A invalidating hypermutations in the nascent (+)-strand viral DNA genome during reverse transcription. In addition, APOBEC3G also directly blocks reverse transcriptase elongation in a deaminase-independent manner and interferes with the integration of proviral DNA. HIV-1 Vif antagonises the APOBEC3G-mediated host defence system by recruiting APOBEC3G into the E3 ubiquitin ligase complex, thus promoting APOBEC3G degradation through the ubiquitin-proteasome pathway. However, through its C-terminal ubiquitin-binding BUZ (binder of ubiquitin zinc finger) domain, the host factor HDAC6 binds HIV-1 Vif and targets it for autophagic degradation [128]. HDAC6 is a central component of basal autophagy that promotes cortactin-dependent, actin-remodelling machinery to stimulate autophagosome-lysosome fusion and substrate degradation [129], while also facilitating the transport of misfolded proteins to the aggresome [130].
HIV-1 overcomes these innate host autophagic defences through HIV-1 Nef, which blocks autophagy initiation by enhancing the association between BECN1 and its inhibitor BCL2 in a PRKN-dependent mechanism. This blocking is done by inducing the cytoplasmic sequestration of TFEB, an upstream regulator of MTOR, and by affecting autophagosome maturation by preventing the fusion between autophagosomes and lysosomes (Figure 1) [92,131,132].
4. HIV-1 Cure Strategies
Although post-transcriptional regulation of HIV-1 latency is an important multifactorial process, current efforts to eradicate the latent HIV-1 reservoir pool focus on reactivating the transcription of latent HIV-1 proviruses. This approach uses latency-reversing agents (LRAs) followed by a combination of ART, with the expectation that targeted immunotherapy, host immune clearance, and HIV-1-cytolysis will be sufficient to kill latently infected cells expressing viral proteins—the “kick and kill” approach. The LRAs under investigation are various and include histone post-translational modification modulators, NF-κB stimulators, non-histone chromatin modulators, TLR agonists, and MTOR activators [133]. However, with over 160 LRA tested to date, none have shown great promise [133]. The most studied class of LRA are the histone post-translational modification modulators that include the histone deacetylase inhibitors (HDACi) givinostat, panobinostat, romidepsin, and vorinostat. Although these induce HIV-1 expression through the reversal of epigenetic silencing, they do not induce the expression of MATR3 [80], resulting in insufficiently low levels of induced viral expression to cause viral cytopathicity, or their elimination by immune effectors such as cytotoxic T cells. Unfortunately, HDACi impair cytotoxic T cell and natural killer (NK) cell functions [134,135], while also promoting the infection of uninfected CD4+ T cells [136].
The inhibitor of apoptosis protein (IAP) family members BIRC2 and BIRC3, are upregulated in latent HIV-1 infected cells [137,138,139,140] and are important negative regulators of non-canonical NF-κB signalling, and thus 5′ LTR-dependent HIV-1 transcription [141,142]. DIABLO/SMAC mimetics mimic the BIR-binding N-terminal tetrapeptide sequence of DIABLO, a pro-apoptogenic mitochondrial protein released into the cytoplasm in response to apoptotic stimuli to antagonise IAPs. The interaction of DIABLO/SMAC mimetics with BIRC2 and BIRC3 activates their E3 ubiquitin ligase activity promoting their autoubiquitination and proteasomal degradation [143,144,145]. Depletion of BIRC2 using DIABLO/SMAC mimetics can activate non-canonical NF-κB signalling, and can act as a LRA in latent HIV-1 cell line models [140,142,146,147,148,149,150,151]. However, only AZD5582, xevinapant, and ciapavir have shown this ability in primary cells, albeit with mixed efficacy, and none have been tested in PLWH.
Agonists against the endosomal TLR3, -7, and -9 have gone through clinical trials for LRA with mixed results [152]. In addition to this potential function, TLR agonists also enhance the generation of HIV-1-specific CD8+ T cells [153], while activating proinflammatory and antimicrobial responses, including the NF-κB-dependent transcription of numerous proinflammatory mediators [154] and the induction of autophagy [155]. As the hyper-activation of MTORC1 by HIV-1 Tat is necessary for HIV-1 production [25], the activation of MTOR by tideglusib, a glycogen synthase kinase 3 inhibitor, was investigated. However, although tideglusib reactivated latent HIV-1 from ex vivo primary CD4+ T cells derived from PLWH on suppressive ART, tideglusib, like other LRAs, failed to reverse latency in vivo [156].
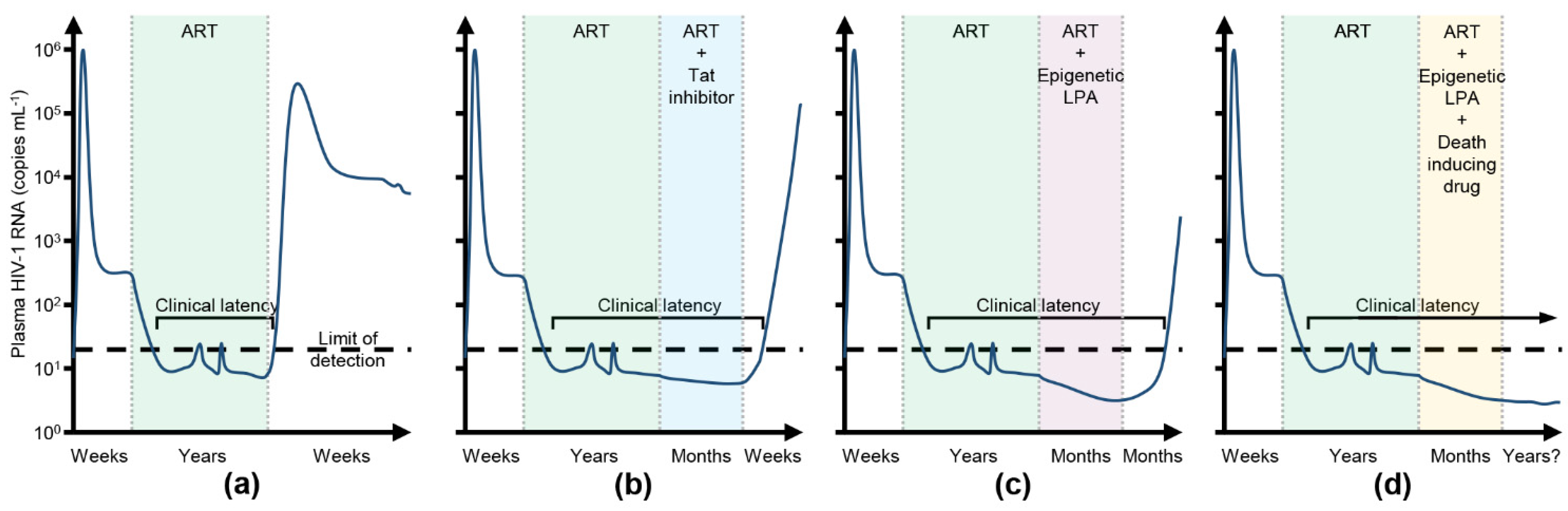
These examples highlight the overall disconnect between the promising in vitro and ex vivo results of LRAs and the poor in vivo findings. Therefore, there is an urgent need for a new approach if a functional HIV-1 cure is to be achieved. In contrast to the “kick and kill” approach, the latency promoting “block and lock” approach is designed to repress HIV-1 expression and release it to levels that can be controlled and cleared by the immune system in the absence of ART—a state of deep latency [157,158]. Deep latency is occasionally observed in some PLWH who are able to control viremia for several years after ART interruption despite possessing weak HIV-1-specific immune responses and low levels of pro-inflammatory markers [16,17,159,160]. Although these post-treatment controllers have relatively small HIV-1 reservoirs, it is theorised that a “block and lock” approach could achieve a similar transcriptionally repressive profile in people initially starting with much larger HIV-1 reservoirs, which result in normal CD4+ T cell counts, the absence of disease progression, and no release of replication competent virus after ART interruption (Figure 2). The “block and lock” approach has a number of advantages over the “kick and kill” approach. As the “block and lock” approach is designed to induce a state of deep latency, the safety concerns related to viral reactivation, drug resistance, and transmission associated with the “kick and kill” approach should be mitigated. Additionally, this approach could be affordable, permanent, and easily implemented in resource-limited settings. Moreover, even if the latency-promoting agents (LPA) in development fail to achieve a long-term functional cure, they could still be of clinical value as a potential new class of ART, as well as being used to uncouple HIV-1 transcription from immune system activation, enhancing clearance by the immune system.
As the control of HIV-1 Tat and P-TEFb translation, modification, turnover, and abundance are important regulators of both HIV-1 replication and latency, HIV-1 Tat, TAR RNA, and P-TEFb are all important targets for LPA development [161]. Unfortunately, the lack of specificity and/or poor pharmacokinetic properties hindered the early development of compounds targeting these proteins [162,163,164,165]. Although these early compounds decreased gross HIV-1 transcription, they had no effect on the integrated pro-virus such that when inhibition was relieved, HIV-1 viremia rapidly rebounded [166,167]. In contrast to these early attempts, an equipotent analogue of the steroidal alkaloid cortistatin A, didehydro-cortistatin A (dCA), has shown promise. dCA binds to the TAR-binding domain of HIV-1 Tat and blocks transcriptional elongation of the HIV-1 promoter [168,169], disrupting the HIV-1 transactivation positive feedback loop, while also instigating the progressive accrual of epigenetic modifications along the HIV-1 5′ LTR. This results in a repressive heterochromatin environment that epigenetically silences the HIV-1 promoter [59,170,171,172,173], even in the presence of an LRA [171]. Other recent promising LPA targeting the HIV-1 Tat/P-TEFb nexus include spironolactone [174], HEXIM1-Tat peptide [175], the BRD4 inhibitor ZL0580 [176,177], and Nullbasic, a 101 amino acid trans-dominant negative HIV-1BH10 Tat in which G/A residues replace the entire basic domain [178,179,180,181].
Although LPA could function to a point where HIV-1 reactivation from latency is unlikely, these drugs do not induce the destruction of the latent reservoir. Unfortunately, the larger the expressed HIV-1 reservoir, the shorter the time to viral rebound after ART interruption [182]. Therefore, combining LPA with drugs that can specifically kill latent HIV-1 infected cells could be advantageous—a “kill, block and lock” (Figure 2d). Although a number of pro-apoptotic compounds, including BCL2 antagonists, DDX3 inhibitors, PLK1 inhibitors, and RLR agonists have been assessed for their potential to specifically induce cell death of HIV-1-infected cells [183,184,185,186,187], they all either require the addition of an LRA to specifically kill HIV-1 infected cells, or induce viral transcription themselves. Thus, they are incompatible with a “block and lock” approach. As the hyper-activation of MTORC1 by HIV-1 Tat is necessary for HIV-1 production [25] and the integrity and function of both MTORC1 and MTORC2 are essential in re-activating HIV-1 from latency [26,27], the inhibition of MTOR is an attractive target for LPA development. The potent and selective ATP-competitive second generation MTOR inhibitors torkinib, torin1, and sapanisertib all repress induced basal (HIV-1 Tat-independent) and (HIV-1 Tat-dependent) HIV-1 gene expression and the reactivation of HIV-1 from in vitro and in vivo models of HIV-1 latency, as well as ex vivo primary CD4+ T cells derived from PLWH on long-term suppressive ART [25,26,188]. Further supporting the use of MTOR inhibitors, the allosteric MTORC1 inhibitors, sirolimus and everolimus inhibit the production of fully infectious HIV-1 particles from a number of different cell types while also limiting intestinal HIV-1 transmission [114,189,190,191]. Additionally, HIV-1-infected adult solid organ transplant recipients receiving these drugs better control HIV-1 replication versus patients receiving alternative immunosuppressants [192,193]. Moreover, sirolimus synergistically enhances the activity of entry inhibitors, such as vicriviroc, aplaviroc and enfuvirtide [194], and inhibits HIV-1 entry through the downregulation of CCR5 [194,195]. Although these MTOR inhibition studies did not assess the role of autophagy, the inhibition of MTOR induces autophagy. Despite being a predominantly cell survival mechanism, under certain conditions autophagy, and the proteins associated with the autophagic process, can promote cell death [196,197]. For instance, in combination with ART and ionizing radiation, sirolimus selectively kills HIV-1-infected myeloid and CD4+ T-cell reservoirs via the inhibition of viral transcription/translation and the induction of autophagy with no deleterious effect on uninfected bystander cells [198].
5. Autophagy as Part of a HIV-1 Cure Approach
Although HIV-1 Nef blocks autophagy initiation and autophagosome maturation, pharmacological inducers of autophagy such as sirolimus can overcome this block leading to the autophagic-degradation of HIV-1 capsid proteins and a decrease in released HIV-1 virions through an ATG5- and autophagy-dependent mechanism [114,199]. Notably, in HIV-1-infected macrophages, the LRA HDACi vorinostat, panobinostat, givinostat and romidepsin and the non-histone chromatin modulating BET inhibitor JQ1 all inhibit MTOR through a yet unidentified mechanism, leading to the dose-dependent autophagic degradation of HIV-1 capsid proteins and a subsequent decrease in HIV-1 release in the absence of cell death [200,201]. Similarly, the dual PI3K/MTOR inhibitor dactolisib (NVP-BEZ235), and the PI3K/MTOR/BRD4 inhibitor SF2523 all inhibit MTOR with similar autophagy-dependent decreases in both intracellular and extracellular HIV-1 p24 abundance in the absence of cell death [201].
Further studies demonstrated that trehalose, a naturally occurring sugar, induces MTOR-dependent and MTOR-independent autophagy in HIV-1 infected macrophages and CD4+ T cells, leading to the degradation of intracellular HIV-1 capsid proteins and an autophagy-dependent reduction in HIV-1 release [202,203]. Trehalose can also decrease HIV-1 entry through the downregulation of CD4+ expression on CD4+ T cells and macrophages and CCR5 on CD4+ T cells, thus, reducing transmission [202]. Interestingly, nelfinavir also inhibits MTOR through the upregulation of the MTOR inhibitor sestrin-2 (SESN2) resulting in autophagy induction [204]. Its role in HIV-1 restriction is yet to be pursued. Finally, the recent discovery of miRAB40, an autophagy-inducing, HIV-1-inhibiting microRNA, may also provide insights into the microRNA-mediated regulation of autophagy-mediated HIV-1 inhibition [205].
Although the autophagic degradation of HIV-1 does not have an effect on the transcriptionally latent HIV-1 provirus, autophagy can play a role in controlling latent HIV-1 infection by reducing the reservoir size through the susceptibility of latent HIV-1 infected cells to drug-induced autophagy-dependent cell death. As an example, the autophagy-inducing peptide, Tat–beclin 1, composed of the amino acids 267-284 of BECN1 attached via a diglycine linker to the protein transduction domain of HIV-1 Tat, inhibits HIV-1 replication in primary human macrophages in an autophagy-dependent mechanism [199]. This peptide also restricted in vitro replication of Sindbis virus, chikungunya virus and West Nile Virus, and reduced the mortality of mice infected with West Nile Virus and chikungunya virus through autophagy [199]. Further studies demonstrated that when encapsulated within a lipid-coated hybrid poly(lactic-co-glycolic acid) (PLGA) nanoparticle, the Tat–beclin 1 peptide selectively kills latent HIV-1 infected CD4+ T cells through Na+/K+-ATPase-mediated autophagy-dependent autosis in the absence of increased viral replication [206]. Autosis is an autophagy-dependent, non-necrotic, non-apoptotic cell death characterised by unique biochemical and morphological features. In the first phase (1a), the endoplasmic reticulum becomes dilated and fragmented, and autophagosome, autolysosome, and empty vacuole numbers increase. In the second phase (1b), the perinuclear space (PNS) swells at discrete regions surrounding the inner nuclear membrane and contains cytoplasmic materials. In the last phase (2), the morphology appears necrotic with ER, autophagosome, autolysosome numbers crashing while mitochondria and other cytoplasmic organelles swell. Also observed are a concavity of the nuclear surface and focal ballooning of the PNS, while the plasma membrane becomes porous [207]. Similar to Tat–beclin 1 nanoparticles, a lipid-coated hybrid PLGA nanoparticle encapsulated v-FLIP-α2 peptide (a ten amino acid α2-helix peptide derived from the death effector domain 1 of the K13 protein of human gammaherpesvirus 8 [208]) also induces autosis of both HIV-1-infected resting memory CD4+ T cells and HIV-1-infected macrophages with no deleterious effects on uninfected bystander cells, and in the absence of increased viral replication [206,209]. Nanoparticles are currently being evaluated to deliver small interfering RNAs (siRNAs) to silence gene expression in infected cells, as well as HIV-1 itself [210], and to deliver cargo(s) that directly interfere with and inhibit viral entry [211,212], or to selectively kill HIV-1-infected cells [206]. Interestingly, a biomimetic nanoparticle wrapped in a CD4+ T cell membrane was shown to not only neutralise cell-free HIV-1, but also reduced cell-associated HIV-1 p24 through an autophagy-dependent mechanism [213,214]. Importantly, under certain conditions, nanoparticles can be targeted to occult HIV-1 reservoir sites such as lymphatic tissue or the central nervous system (CNS) through the conjugation of cell-specific ligands to the nanoparticle surface, or with a biomimetic membrane [215,216]. Although there is currently no delivery mechanism approved to deliver RNA or peptides to occult HIV-1 reservoirs in vivo, the regulatory approval of patisiran [217], and the development and emergency use authorization of the COVID-19 mRNA vaccines BNT162b2 and mRNA-1273 [218,219] paves the way for future lipid-based nanoparticle drug research to reach sanctuary sites, including the CNS. In other studies, autophagy inducing PI3K/AKT1 inhibitors, including the clinically available miltefosine, selectively kill HIV-1 infected macrophages [220,221]. Therefore, the selective killing of HIV-1 infected cells, comprising those actively replicating virus and those within the latent reservoirs, has the potential to achieve the complete eradication of HIV-1 infection.
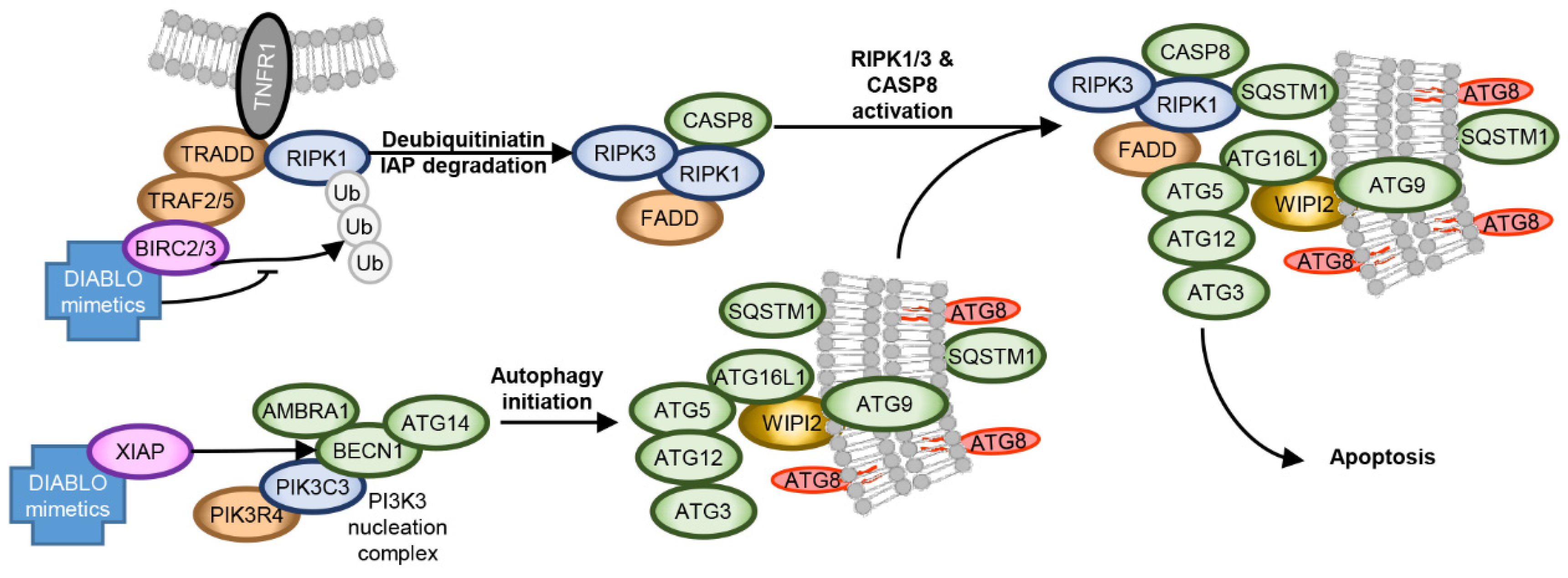
During infection, HIV-1 increases the expression of BIRC2/3/5, XIAP, as well as TREM1 and BCL2 family members, while decreasing the expression of pro-apoptotic proteins [137,138,139,140,147,222,223,224]. In addition to inhibiting 5′ LTR-dependent HIV-1 transcription [141,142], the overexpression of IAPs plays a further important role in the establishment and maintenance of HIV-1 latency by inhibiting HIV-1-mediated cytopathogenesis. They do this through direct antagonism, ubiquitination, and neddylation of apoptosis caspases [225,226], and ubiquitination of BECN1 (reducing autophagy) and RIPK1 [227], suppressing its aberrant activation. RIPK1 is a death domain containing protein that controls differentiation and inflammation transcriptional responses [228]. The scaffolding function of RIPK1 is essential to promote pro-survival NF-κB signalling through the assembly of complex I, while activation of RIPK1 leads to either apoptosis or necroptosis [229]. RIPK1 also plays an important role in the cellular response to low energy levels and mediates AMPK-MTORC1 signalling [230]. Therefore, in addition to promoting the proteasomal degradation of BIRC2/3 and XIAP, the binding of DIABLO/SMAC mimetics to IAP also disinhibits apoptosis caspases and activates RIPK1 (Figure 3) [231]. In HIV-1-infected macrophages and latent HIV-1-infected resting memory CD4+ T cells, DIABLO/SMAC mimetics trigger the proteasomal degradation of IAPs that correlates with the induction of autophagy and cell death [140,147,232]. Importantly, although IAPs similarly induce the degradation of IAPs in uninfected cells, they do not undergo cell death [140,147]. The DIABLO/SMAC mimetic-mediated degradation of IAPs leads to the deubiquitination and activation of RIPK1, which in HIV-1-infected cells leads to the formation of a death-inducing signalling complex (DISC) consisting of pro-apoptotic (FADD, RIPK1, RIPK3, CASP8) and autophagy (ATG5, ATG7, SQSTM1) proteins [140,147]. Notably, inhibition of autophagy initiation, membrane nucleation, phagophore formation and expansion ablated DIABLO/SMAC mimetic-mediated apoptosis, while inhibition of autophagosome closure through membrane scission, autophago-lysosome fusion and degradation had no protective effect. Moreover, the DIABLO/SMAC mimetic-mediated DISC formation and localization to autophagosomes and resultant apoptosis required SQSTM1-mediated RIPK1 tethering to the phagophore membrane. Therefore, autophagosome membranes mediate DIABLO/SMAC mimetic-induced cell death by serving as a scaffold for efficient DISC formation rather than by autophagy degrading intracellular components. Importantly, although the DIABLO/SMAC mimetics AZD5582, xevinapant, and ciapavir are useful as LRA [142,146,148,149,150,151], treatment with birinapant, GDC-0152, and LCL-161 did not increase HIV-1 p24 antigen release indicating that the DIABLO/SMAC mimetics were killing HIV-1-infected cells in the absence of latency reactivation [140,147,232]. Notably, birinapant is also able to induce selective cell death in hepatitis B virus expressing hepatocytes [233,234,235]. Consequently, IAPs are excellent targets for therapeutic exploitation in an HIV-1 “kill, block and lock” cure strategy.
6. Concluding Remarks
The combination of LPA with host-directed or direct-acting autophagy and autosis inducing drugs to eliminate selectively HIV-1 latently infected cells to reduce the viral reservoir can enhance the efficacy of the “block and lock” approach, and when combined with other strategies, could potentially achieve viral eradication. However, reaching isolated body compartments, including lymph nodes and the CNS remains a major challenge to achieving the full potential of these strategies. Currently, numerous drugs have shown considerable potential in vitro and in animal models that could contribute to an HIV-1 cure. Well-designed clinical trials will be required to assess the ability of any of these approaches to penetrate and act throughout the body. As these cure strategies are evaluated, it is likely that the modulation of autophagy will be required to achieve an HIV-1 functional or complete virologic cure.
Author Contributions
Conceptualisation, G.R.C. and S.A.S.; writing—Original Draft, G.R.C. and S.A.S.; writing—review and editing, G.R.C. and S.A.S.; visualisation, G.R.C., funding acquisition, G.R.C. and S.A.S.; resources, S.A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported, in whole or in part, by the National Institute of Mental Health of the NIH (www.nimh.nih.gov; accessed on 14 July 2021) (R01MH128021 to G.R.C.), the National Institute of Neurological Disorders and Stroke of the NIH (www.ninds.nih.gov; accessed on 14 July 2021) (R01NS104015 to S.A.S.), and the International Maternal Pediatric Adolescent AIDS Clinical Trials Network (impaactnetwork.org; accessed on 14 July 2021). Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials (IMPAACT) Network is provided by the National Institute of Allergy and Infectious Diseases (NIAID) of the NIH under award numbers UM1AI068632 (IMPAACT LOC), UM1AI068616 (IMPAACT SDMC), and UM1AI106716 (IMPAACT LC), with co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Institute of Mental Health (NIMH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute HIV-1 infection. N. Engl. J. Med. 2011, 364, 1943–1954. [Google Scholar] [CrossRef] [Green Version]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial cells: The main HIV-1 reservoir in the brain. Front. Cell Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [Green Version]
- Hendricks, C.M.; Cordeiro, T.; Gomes, A.P.; Stevenson, M. The interplay of HIV-1 and macrophages in viral persistence. Front. Microbiol. 2021, 12, 646447. [Google Scholar] [CrossRef]
- Valdebenito, S.; Castellano, P.; Ajasin, D.; Eugenin, E.A. Astrocytes are HIV reservoirs in the brain: A cell type with poor HIV infectivity and replication but efficient cell-to-cell viral transfer. J. Neurochem. 2021, 158, 429–443. [Google Scholar] [CrossRef]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Crooks, A.M.; Bateson, R.; Cope, A.B.; Dahl, N.P.; Griggs, M.K.; Kuruc, J.D.; Gay, C.L.; Eron, J.J.; Margolis, D.M.; Bosch, R.J.; et al. Precise quantitation of the latent HIV-1 reservoir: Implications for eradication strategies. J. Infect. Dis. 2015, 212, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Ramratnam, B.; Mittler, J.E.; Zhang, L.; Boden, D.; Hurley, A.; Fang, F.; Macken, C.A.; Perelson, A.S.; Markowitz, M.; Ho, D.D. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 2000, 6, 82–85. [Google Scholar] [CrossRef]
- Hermankova, M.; Ray, S.C.; Ruff, C.; Powell-Davis, M.; Ingersoll, R.; D’Aquila, R.T.; Quinn, T.C.; Siliciano, J.D.; Siliciano, R.F.; Persaud, D. HIV-1 drug resistance profiles in children and adults with viral load of <50 copies/ml receiving combination therapy. JAMA 2001, 286, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boritz, E.A.; Darko, S.; Swaszek, L.; Wolf, G.; Wells, D.; Wu, X.; Henry, A.R.; Laboune, F.; Hu, J.; Ambrozak, D.; et al. Multiple origins of virus persistence during natural control of HIV infection. Cell 2016, 166, 1004–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Frange, P.; Faye, A.; Avettand-Fenoel, V.; Bellaton, E.; Descamps, D.; Angin, M.; David, A.; Caillat-Zucman, S.; Peytavin, G.; Dollfus, C.; et al. HIV-1 virological remission lasting more than 12 years after interruption of early antiretroviral therapy in a perinatally infected teenager enrolled in the French ANRS EPF-CO10 paediatric cohort: A case report. Lancet HIV 2016, 3, e49–e54. [Google Scholar] [CrossRef] [Green Version]
- Savini, M.; Zhao, Q.; Wang, M.C. Lysosomes: Signaling hubs for metabolic sensing and longevity. Trends Cell Biol. 2019, 29, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Saravia, J.; Raynor, J.L.; Chapman, N.M.; Lim, S.A.; Chi, H. Signaling networks in immunometabolism. Cell Res. 2020, 30, 328–342. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Hall, M.N. Regulation of mTORC2 signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef] [PubMed]
- Morozumi, Y.; Shiozaki, K. Conserved and divergent mechanisms that control TORC1 in yeasts and mammals. Genes 2021, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Lu, M.; Wang, J.; Jones, K.T.; Ives, H.E.; Feldman, M.E.; Yao, L.J.; Shokat, K.M.; Ashrafi, K.; Pearce, D. mTOR complex-2 activates ENaC by phosphorylating SGK1. J. Am. Soc. Nephrol. 2010, 21, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Feehan, R.P.; Shantz, L.M. Negative regulation of the FOXO3a transcription factor by mTORC2 induces a pro-survival response following exposure to ultraviolet-B irradiation. Cell. Signal. 2016, 28, 798–809. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Arora, S.; Ahmed, S.; Banerjea, A.C. Hyperactivation of mammalian target of rapamycin complex 1 by HIV-1 is necessary for virion production and latent viral reactivation. FASEB J. 2017, 31, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR complex controls HIV latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Liao, Q.; Chen, J.; Zhang, L.; He, Q.; Zhu, H.; Zhang, X.; Xu, J. TSC1 and DEPDC5 regulate HIV-1 latency through the mTOR signaling pathway. Emerg. Microbes Infect. 2018, 7, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbay, B.; Shmakova, A.; Vassetzky, Y.; Dokudovskaya, S. Modulation of mTORC1 signaling pathway by HIV-1. Cells 2020, 9, 1090. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [Green Version]
- Benmoussa, K.; Garaude, J.; Acin-Perez, R. How mitochondrial metabolism contributes to macrophage phenotype and functions. J. Mol. Biol. 2018, 430, 3906–3921. [Google Scholar] [CrossRef]
- Zhang, S.; Carriere, J.; Lin, X.; Xie, N.; Feng, P. Interplay between cellular metabolism and cytokine responses during viral infection. Viruses 2018, 10, 521. [Google Scholar] [CrossRef] [Green Version]
- Van den Bossche, J.; Saraber, D.L. Metabolic regulation of macrophages in tissues. Cell. Immunol. 2018, 330, 54–59. [Google Scholar] [CrossRef]
- Russell, D.G.; Huang, L.; VanderVen, B.C. Immunometabolism at the interface between macrophages and pathogens. Nat. Rev. Immunol. 2019, 19, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Gabandé-Rodríguez, E.; Gómez de Las Heras, M.M.; Mittelbrunn, M. Control of inflammation by calorie restriction mimetics: On the crossroad of autophagy and mitochondria. Cells 2019, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Spector, S.A. Inhibition of human immunodeficiency virus type-1 through autophagy. Curr. Opin. Microbiol. 2013, 16, 349–354. [Google Scholar] [CrossRef] [Green Version]
- Leymarie, O.; Lepont, L.; Berlioz-Torrent, C. Canonical and non-canonical autophagy in HIV-1 replication cycle. Viruses 2017, 9, 270. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, Y.; Torresilla, C.; Rassart, E.; Barbeau, B. Implication of different HIV-1 genes in the modulation of autophagy. Viruses 2017, 9, 389. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Estévez-Herrera, J.; Márquez-Arce, D.; Cabrera, C.; Espert, L.; Blanco, J.; Valenzuela-Fernández, A. The interplay of HIV and autophagy in early infection. Front. Microbiol. 2021, 12, 661446. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Paras, P., Jr.; Van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar] [CrossRef]
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.M.; Ott, M. The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes. Mol. Cell 2017, 67, 1001–1012.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Ferdous, A.; Imai, T.; Hirose, S.; Sugimoto, S.; Yano, K.; Hartzog, G.A.; Winston, F.; et al. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev. 1998, 12, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Ping, Y.H.; Rana, T.M. DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef] [Green Version]
- Jadlowsky, J.K.; Wong, J.Y.; Graham, A.C.; Dobrowolski, C.; Devor, R.L.; Adams, M.D.; Fujinaga, K.; Karn, J. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol. Cell. Biol. 2014, 34, 1911–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef] [Green Version]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Smith, E.R.; Takahashi, H.; Lai, K.C.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C.; Shilatifard, A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 2010, 37, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Mori, L.; Valente, S.T. Key players in HIV-1 transcriptional regulation: Targets for a functional cure. Viruses 2020, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell. Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourgeois, C.F.; Kim, Y.K.; Churcher, M.J.; West, M.J.; Karn, J. Spt5 cooperates with human immunodeficiency virus type 1 Tat by preventing premature RNA release at terminator sequences. Mol. Cell. Biol. 2002, 22, 1079–1093. [Google Scholar] [CrossRef] [Green Version]
- Parada, C.A.; Roeder, R.G. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxy-terminal domain. Nature 1996, 384, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Col, E.; Caron, C.; Seigneurin-Berny, D.; Gracia, J.; Favier, A.; Khochbin, S. The histone acetyltransferase, hGCN5, interacts with and acetylates the HIV transactivator, Tat. J. Biol. Chem. 2001, 276, 28179–28184. [Google Scholar] [CrossRef] [Green Version]
- Tréand, C.; du Chéné, I.; Brès, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenet. Chromatin 2019, 12, 23. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Cherrier, T.; Le Douce, V.; Eilebrecht, S.; Riclet, R.; Marban, C.; Dequiedt, F.; Goumon, Y.; Paillart, J.C.; Mericskay, M.; Parlakian, A.; et al. CTIP2 is a negative regulator of P-TEFb. Proc. Natl. Acad. Sci. USA 2013, 110, 12655–12660. [Google Scholar] [CrossRef] [Green Version]
- Eilebrecht, S.; Le Douce, V.; Riclet, R.; Targat, B.; Hallay, H.; Van Driessche, B.; Schwartz, C.; Robette, G.; Van Lint, C.; Rohr, O.; et al. HMGA1 recruits CTIP2-repressed P-TEFb to the HIV-1 and cellular target promoters. Nucleic Acids Res. 2014, 42, 4962–4971. [Google Scholar] [CrossRef]
- Forouzanfar, F.; Ali, S.; Wallet, C.; De Rovere, M.; Ducloy, C.; El Mekdad, H.; El Maassarani, M.; Ait-Ammar, A.; Van Assche, J.; Boutant, E.; et al. HIV-1 Vpr mediates the depletion of the cellular repressor CTIP2 to counteract viral gene silencing. Sci. Rep. 2019, 9, 13154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagnier, S.; Daussy, C.F.; Borel, S.; Robert-Hebmann, V.; Faure, M.; Blanchet, F.P.; Beaumelle, B.; Biard-Piechaczyk, M.; Espert, L. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J. Virol. 2015, 89, 615–625. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Moresco, J.J.; Chang, M.; Mukim, A.; Smith, D.; Diedrich, J.K.; Yates, J.R., 3rd; Jones, K.A. SHMT2 and the BRCC36/BRISC deubiquitinase regulate HIV-1 Tat K63-ubiquitylation and destruction by autophagy. PLoS Pathog. 2018, 14, e1007071. [Google Scholar] [CrossRef]
- Brès, V.; Kiernan, R.E.; Linares, L.K.; Chable-Bessia, C.; Plechakova, O.; Tréand, C.; Emiliani, S.; Péloponèse, J.M.; Jeang, K.T.; Coux, O.; et al. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 2003, 5, 754–761. [Google Scholar] [CrossRef]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making sense of multifunctional proteins: Human immunodeficiency virus type 1 accessory and regulatory proteins and connections to transcription. Annu. Rev. Virol. 2017, 4, 241–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Qin, J.; Li, Y.; Wang, J.; He, Q.; Zhou, J.; Liu, M.; Li, D. Modulation of the stability and activities of HIV-1 Tat by its ubiquitination and carboxyl-terminal region. Cell Biosci. 2014, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016, 7, 11730. [Google Scholar] [CrossRef]
- Lange, U.C.; Verdikt, R.; Ait-Ammar, A.; Van Lint, C. Epigenetic crosstalk in chronic infection with HIV-1. Semin. Immunopathol. 2020, 42, 187–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alamer, E.; Zhong, C.; Hajnik, R.; Soong, L.; Hu, H. Modulation of BRD4 in HIV epigenetic regulation: Implications for finding an HIV cure. Retrovirology 2021, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell. Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; Van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [Green Version]
- Le Douce, V.; Colin, L.; Redel, L.; Cherrier, T.; Herbein, G.; Aunis, D.; Rohr, O.; Van Lint, C.; Schwartz, C. LSD1 cooperates with CTIP2 to promote HIV-1 transcriptional silencing. Nucleic Acids Res. 2012, 40, 1904–1915. [Google Scholar] [CrossRef]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Irwin, D.; Kanazawa, S.; Huang, L.; Romeo, J.; Yen, T.S.; Peterlin, B.M. Transcriptional profiles of latent human immunodeficiency virus in infected individuals: Effects of Tat on the host and reservoir. J. Virol. 2003, 77, 8227–8236. [Google Scholar] [CrossRef] [Green Version]
- Telwatte, S.; Lee, S.; Somsouk, M.; Hatano, H.; Baker, C.; Kaiser, P.; Kim, P.; Chen, T.H.; Milush, J.; Hunt, P.W.; et al. Gut and blood differ in constitutive blocks to HIV transcription, suggesting tissue-specific differences in the mechanisms that govern HIV latency. PLoS Pathog. 2018, 14, e1007357. [Google Scholar] [CrossRef] [Green Version]
- Sarracino, A.; Gharu, L.; Kula, A.; Pasternak, A.O.; Avettand-Fenoel, V.; Rouzioux, C.; Bardina, M.; De Wit, S.; Benkirane, M.; Berkhout, B.; et al. Posttranscriptional regulation of HIV-1 gene expression during replication and reactivation from latency by nuclear matrix protein MATR3. mBio 2018, 9, e02158-18. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Triboulet, R.; Mari, B.; Lin, Y.L.; Chable-Bessia, C.; Bennasser, Y.; Lebrigand, K.; Cardinaud, B.; Maurin, T.; Barbry, P.; Baillat, V.; et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science 2007, 315, 1579–1582. [Google Scholar] [CrossRef] [PubMed]
- Sung, T.L.; Rice, A.P. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5, e1000263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chable-Bessia, C.; Meziane, O.; Latreille, D.; Triboulet, R.; Zamborlini, A.; Wagschal, A.; Jacquet, J.M.; Reynes, J.; Levy, Y.; Saib, A.; et al. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 2009, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, H.C.; Razi, M.; Polson, H.E.; Girardin, S.E.; Wilson, M.I.; Tooze, S.A. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell 2014, 55, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Knorr, R.L.; Lipowsky, R.; Dimova, R. Autophagosome closure requires membrane scission. Autophagy 2015, 11, 2134–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Meier, A.; Alter, G.; Frahm, N.; Sidhu, H.; Li, B.; Bagchi, A.; Teigen, N.; Streeck, H.; Stellbrink, H.J.; Hellman, J.; et al. MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J. Virol. 2007, 81, 8180–8191. [Google Scholar] [CrossRef] [Green Version]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, F.P.; Moris, A.; Nikolic, D.S.; Lehmann, M.; Cardinaud, S.; Stalder, R.; Garcia, E.; Dinkins, C.; Leuba, F.; Wu, L.; et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2010, 32, 654–669. [Google Scholar] [CrossRef] [Green Version]
- Coulon, P.G.; Richetta, C.; Rouers, A.; Blanchet, F.P.; Urrutia, A.; Guerbois, M.; Piguet, V.; Theodorou, I.; Bet, A.; Schwartz, O.; et al. HIV-infected dendritic cells present endogenous MHC class II-restricted antigens to HIV-specific CD4+ T cells. J. Immunol. 2016, 197, 517–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, T.K.; Mickelson, D.J.; Fink, J.; Solberg, J.C.; Inglefield, J.R.; Hook, D.; Gupta, S.K.; Gibson, S.; Alkan, S.S. Toll-like receptor (TLR) 2-9 agonists-induced cytokines and chemokines: I. Comparison with T cell receptor-induced responses. Cell. Immunol. 2006, 243, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Vierbuchen, T.; Bang, C.; Rosigkeit, H.; Schmitz, R.A.; Heine, H. The human-associated archaeon Methanosphaera stadtmanae is recognized through its RNA and induces TLR8-dependent NLRP3 inflammasome activation. Front. Immunol. 2017, 8, 1535. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; To, R.K.; Hanna, J.; Spector, S.A. SARS-CoV-2, SARS-CoV-1, and HIV-1 derived ssRNA sequences activate the NLRP3 inflammasome in human macrophages through a non-classical pathway. iScience 2021, 24, 102295. [Google Scholar] [CrossRef]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Kang, K.H.; Spector, S.A. Production of interferon α by human immunodeficiency virus type 1 in human plasmacytoid dendritic cells is dependent on induction of autophagy. J. Infect. Dis. 2012, 205, 1258–1267. [Google Scholar] [CrossRef]
- Schlaepfer, E.; Audige, A.; Joller, H.; Speck, R.F. TLR7/8 triggering exerts opposing effects in acute versus latent HIV infection. J. Immunol. 2006, 176, 2888–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, G.R.; Spector, S.A. Toll-like receptor 8 ligands activate a vitamin D mediated autophagic response that inhibits human immunodeficiency virus type 1. PLoS Pathog. 2012, 8, e1003017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, S.; De Souza, G.A.; Salte, T.; Wiker, H.G.; Asjo, B. HIV induces both a down-regulation of IRAK-4 that impairs TLR signalling and an up-regulation of the antibiotic peptide dermcidin in monocytic cells. Scand. J. Immunol. 2009, 70, 264–276. [Google Scholar] [CrossRef]
- Haug, C.; Müller, F.; Aukrust, P.; Frøland, S.S. Subnormal serum concentration of 1,25-vitamin D in human immunodeficiency virus infection: Correlation with degree of immune deficiency and survival. J. Infect. Dis. 1994, 169, 889–893. [Google Scholar] [CrossRef]
- Haug, C.J.; Aukrust, P.; Haug, E.; Mørkrid, L.; Müller, F.; Frøland, S.S. Severe deficiency of 1,25-dihydroxyvitamin D3 in human immunodeficiency virus infection: Association with immunological hyperactivity and only minor changes in calcium homeostasis. J. Clin. Endocrinol. Metab. 1998, 83, 3832–3838. [Google Scholar] [CrossRef]
- Teichmann, J.; Stephan, E.; Discher, T.; Lange, U.; Federlin, K.; Stracke, H.; Friese, G.; Lohmeyer, J.; Bretzel, R.G. Changes in calciotropic hormones and biochemical markers of bone metabolism in patients with human immunodeficiency virus infection. Metabolism 2000, 49, 1134–1139. [Google Scholar] [CrossRef]
- Teichmann, J.; Stephan, E.; Lange, U.; Discher, T.; Friese, G.; Lohmeyer, J.; Stracke, H.; Bretzel, R.G. Osteopenia in HIV-infected women prior to highly active antiretroviral therapy. J. Infect. 2003, 46, 221–227. [Google Scholar] [CrossRef]
- Müller, N.J.; Fux, C.A.; Ledergerber, B.; Elzi, L.; Schmid, P.; Dang, T.; Magenta, L.; Calmy, A.; Vergopoulos, A.; Bischoff-Ferrari, H.A. High prevalence of severe vitamin D deficiency in combined antiretroviral therapy-naive and successfully treated Swiss HIV patients. AIDS 2010, 24, 1127–1134. [Google Scholar] [CrossRef]
- Pinzone, M.R.; Di Rosa, M.; Malaguarnera, M.; Madeddu, G.; Foca, E.; Ceccarelli, G.; d’Ettorre, G.; Vullo, V.; Fisichella, R.; Cacopardo, B.; et al. Vitamin D deficiency in HIV infection: An underestimated and undertreated epidemic. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 1218–1232. [Google Scholar]
- Viard, J.P.; Souberbielle, J.C.; Kirk, O.; Reekie, J.; Knysz, B.; Losso, M.; Gatell, J.; Pedersen, C.; Bogner, J.R.; Lundgren, J.D.; et al. Vitamin D and clinical disease progression in HIV infection: Results from the EuroSIDA study. AIDS 2011, 25, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, J.E.; Adams, J.S. Vitamin D in HIV-infected patients. Curr. HIV/AIDS Rep. 2011, 8, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, N.; Aguilar-Jiménez, W.; Rugeles, M.T. The potential protective role of vitamin D supplementation on HIV-1 infection. Front. Immunol. 2019, 10, 2291. [Google Scholar] [CrossRef] [PubMed]
- Currò, M.; Visalli, G.; Pellicanò, G.F.; Ferlazzo, N.; Costanzo, M.G.; D’Andrea, F.; Caccamo, D.; Nunnari, G.; Ientile, R. Vitamin D status modulates inflammatory response in HIV+ subjects: Evidence for involvement of autophagy and TG2 expression in PBMC. Int. J. Mol. Sci. 2020, 21, 7558. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Hormonally active vitamin D3 (1α,25-dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J. Biol. Chem. 2011, 286, 18890–18902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [Green Version]
- Black, L.R.; Aiken, C. TRIM5α disrupts the structure of assembled HIV-1 capsid complexes in vitro. J. Virol. 2010, 84, 6564–6569. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.; Pertel, T.; Gray, S.; Robia, S.L.; Bakowska, J.C.; Luban, J.; Campbell, E.M. p62/sequestosome-1 associates with and sustains the expression of retroviral restriction factor TRIM5α. J. Virol. 2010, 84, 5997–6006. [Google Scholar] [CrossRef] [Green Version]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Munch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef]
- Keown, J.R.; Black, M.M.; Ferron, A.; Yap, M.; Barnett, M.J.; Pearce, F.G.; Stoye, J.P.; Goldstone, D.C. A helical LC3-interacting region mediates the interaction between the retroviral restriction factor Trim5α and mammalian autophagy-related ATG8 proteins. J. Biol. Chem. 2018, 293, 18378–18386. [Google Scholar] [CrossRef] [Green Version]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, B.; Chisholm, D.; Kell, A.M.; Mandell, M.A. A non-canonical role for the autophagy machinery in anti-retroviral signaling mediated by TRIM5α. PLoS Pathog. 2020, 16, e1009017. [Google Scholar] [CrossRef] [PubMed]
- Ciccosanti, F.; Corazzari, M.; Casetti, R.; Amendola, A.; Collalto, D.; Refolo, G.; Vergori, A.; Taibi, C.; D’Offizi, G.; Antinori, A.; et al. High levels of TRIM5α are associated with xenophagy in HIV-1-infected long-term nonprogressors. Cells 2021, 10, 1207. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Shaid, S.; Brandts, C.H.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Erpapazoglou, Z.; Walker, O.; Haguenauer-Tsapis, R. Versatile roles of K63-linked ubiquitin chains in trafficking. Cells 2014, 3, 1027–1088. [Google Scholar] [CrossRef] [Green Version]
- Valera, M.S.; de Armas-Rillo, L.; Barroso-González, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernández, S.; Borel, S.; Garcia-Expósito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [Green Version]
- Hyttinen, J.M.; Amadio, M.; Viiri, J.; Pascale, A.; Salminen, A.; Kaarniranta, K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res. Rev. 2014, 18, 16–28. [Google Scholar] [CrossRef]
- Kyei, G.B.; Dinkins, C.; Davis, A.S.; Roberts, E.; Singh, S.B.; Dong, C.; Wu, L.; Kominami, E.; Ueno, T.; Yamamoto, A.; et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 2009, 186, 255–268. [Google Scholar] [CrossRef]
- Castro-Gonzalez, S.; Shi, Y.; Colomer-Lluch, M.; Song, Y.; Mowery, K.; Almodovar, S.; Bansal, A.; Kirchhoff, F.; Sparrer, K.; Liang, C.; et al. HIV-1 Nef counteracts autophagy restriction by enhancing the association between BECN1 and its inhibitor BCL2 in a PRKN-dependent manner. Autophagy 2021, 17, 553–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antivir. Res. 2019, 166, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef] [Green Version]
- Pace, M.; Williams, J.; Kurioka, A.; Gerry, A.B.; Jakobsen, B.; Klenerman, P.; Nwokolo, N.; Fox, J.; Fidler, S.; Frater, J.; et al. Histone deacetylase inhibitors enhance CD4 T cell susceptibility to NK cell killing but reduce NK cell function. PLoS Pathog. 2016, 12, e1005782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucera, M.; Tilton, C.A.; Mao, H.; Dobrowolski, C.; Tabler, C.; Haqqani, A.A.; Karn, J.; Tilton, J.C. The histone deacetylase inhibitor vorinostat (SAHA) increases the susceptibility of uninfected CD4+ T cells to HIV by increasing the kinetics and efficiency of post-entry viral events. J. Virol. 2014, 88, 10803–10812. [Google Scholar] [CrossRef] [Green Version]
- Berro, R.; de la Fuente, C.; Klase, Z.; Kehn, K.; Parvin, L.; Pumfery, A.; Agbottah, E.; Vertes, A.; Nekhai, S.; Kashanchi, F. Identifying the membrane proteome of HIV-1 latently infected cells. J. Biol. Chem. 2007, 282, 8207–8218. [Google Scholar] [CrossRef] [Green Version]
- Swingler, S.; Mann, A.M.; Zhou, J.; Swingler, C.; Stevenson, M. Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 2007, 3, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.L.; Chunduri, H.; Wise, J.; Mindley, R.; Rimland, D. Replication-independent expression of anti-apoptosis marker genes in human peripheral blood mononuclear cells infected with the wild-type HIV-1 and reverse transcriptase variants. Viral Immunol. 2012, 25, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Trout, R.N.; Spector, S.A. SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected resting memory CD4+ T cells. Cell Host Microbe 2018, 24, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 is a negative regulator of HIV-1 transcription and can be targeted by Smac mimetics to promote reversal of viral latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.H.; Du, C. Smac/DIABLO selectively reduces the levels of c-IAP1 and c-IAP2 but not that of XIAP and livin in HeLa cells. J. Biol. Chem. 2004, 279, 16963–16970. [Google Scholar] [CrossRef] [Green Version]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; To, R.K.; Zhang, G.; Spector, S.A. SMAC mimetics induce autophagy-dependent apoptosis of HIV-1-infected macrophages. Cell Death Dis. 2020, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Dashti, A.; Waller, C.; Mavigner, M.; Schoof, N.; Bar, K.J.; Shaw, G.M.; Vanderford, T.H.; Liang, S.; Lifson, J.D.; Dunham, R.M.; et al. SMAC mimetic plus triple-combination bispecific HIVxCD3 retargeting molecules in SHIV.C.CH505-infected, antiretroviral therapy-suppressed rhesus macaques. J. Virol. 2020, 94, e00793-20. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Marsden, M.D.; Teriete, P.; Portillo, A.J.; Heimann, D.; Kim, J.T.; Soliman, M.S.A.; Dimapasoc, M.; Carmona, C.; Celeridad, M.; et al. Pharmacological activation of non-canonical NF-κB signaling activates latent HIV-1 reservoirs in vivo. Cell Rep. Med. 2020, 1, 100037. [Google Scholar] [CrossRef]
- Sarabia, I.; Huang, S.H.; Ward, A.R.; Jones, R.B.; Bosque, A. The intact non-inducible latent HIV-1 reservoir is established in an in vitro primary TCM cell model of latency. J. Virol. 2021, 95, e01297-20. [Google Scholar] [CrossRef]
- Mavigner, M.; Liao, L.E.; Brooks, A.D.; Ke, R.; Mattingly, C.; Schoof, N.; McBrien, J.; Carnathan, D.; Liang, S.; Vanderford, T.H.; et al. CD8 lymphocyte depletion enhances the latency reversal activity of the SMAC mimetic AZD5582 in ART-suppressed SIV-infected rhesus macaques. J. Virol. 2021, 95, e01429-20. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; Bosque, A. Targeting cellular and tissue HIV reservoirs with Toll-like receptor agonists. Front. Immunol. 2019, 10, 2450. [Google Scholar] [CrossRef] [Green Version]
- Lore, K.; Betts, M.R.; Brenchley, J.M.; Kuruppu, J.; Khojasteh, S.; Perfetto, S.; Roederer, M.; Seder, R.A.; Koup, R.A. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J. Immunol. 2003, 171, 4320–4328. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. The TLR and IL-1 signalling network at a glance. J. Cell Sci. 2014, 127, 2383–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, M.A.; Elmaoued, R.A.; Davis, A.S.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramatica, A.; Schwarzer, R.; Brantley, W.; Varco-Merth, B.; Sperber, H.S.; Hull, P.A.; Montano, M.; Migueles, S.A.; Rosenthal, D.; Hogan, L.E.; et al. Evaluating a new class of AKT/mTOR activators for HIV latency reversing activity ex vivo and in vivo. J. Virol. 2021, 95, e02393-20. [Google Scholar] [CrossRef]
- Feorino, P.M.; Butera, S.T.; Folks, T.M.; Schinazi, R.F. Prevention of activation of HIV-1 by antiviral agents in OM-10.1 cells. Antivir. Chem. Chemother. 1993, 4, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Patzold, S.; Schneider, J.; Rudolph, C.; Marme, D.; Schachtele, C. Novel indolocarbazole protein kinase C inhibitors prevent reactivation of HIV-1 in latently infected cells. Antivir. Res. 1993, 22, 273–283. [Google Scholar] [CrossRef]
- Namazi, G.; Fajnzylber, J.M.; Aga, E.; Bosch, R.J.; Acosta, E.P.; Sharaf, R.; Hartogensis, W.; Jacobson, J.M.; Connick, E.; Volberding, P.; et al. The control of HIV after antiretroviral medication pause (CHAMP) study: Posttreatment controllers identified from 14 clinical studies. J. Infect. Dis. 2018, 218, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Violari, A.; Cotton, M.F.; Kuhn, L.; Schramm, D.B.; Paximadis, M.; Loubser, S.; Shalekoff, S.; Da Costa Dias, B.; Otwombe, K.; Liberty, A.; et al. A child with perinatal HIV infection and long-term sustained virological control following antiretroviral treatment cessation. Nat. Commun. 2019, 10, 412. [Google Scholar] [CrossRef]
- Kamori, D.; Ueno, T. HIV-1 Tat and viral latency: What we can learn from naturally occurring sequence variations. Front. Microbiol. 2017, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Esquieu, D.; Péloponèse, J.M.; Opi, S.; Grégoire, C.; de Mareuil, J.; Watkins, J.; Campbell, G.; Dunot, J.P.; Sturgis, J.; Witvrouw, M.; et al. Discovery of a Tat HIV-1 inhibitor through computer-aided drug design. Spectroscopy 2003, 17, 639–645. [Google Scholar] [CrossRef]
- Montembault, M.; Vo-Thanh, G.; Deyine, A.; Fargeas, V.; Villiéras, M.; Adjou, A.; Dubreuil, D.; Esquieu, D.; Grégoire, C.; Opi, S.; et al. A possible improvement for structure-based drug design illustrated by the discovery of a Tat HIV-1 inhibitor. Bioorg. Med. Chem. Lett. 2004, 14, 1543–1546. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.N.; Palu, G. Inhibitors of HIV-1 Tat-mediated transactivation. Curr. Med. Chem. 2006, 13, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.D.; Campbell, G.R.; Halimi, H.; Loret, E.P. Homonuclear 1H NMR and circular dichroism study of the HIV-1 Tat Eli variant. Retrovirology 2008, 5, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Li, D.; Lin, M.H.; Li, L.; Harrich, D. Tat-based therapies as an adjuvant for an HIV-1 functional cure. Viruses 2020, 12, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moranguinho, I.; Valente, S.T. Block-And-Lock: New horizons for a cure for HIV-1. Viruses 2020, 12, 1443. [Google Scholar] [CrossRef]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-cortistatin A inhibits HIV-1 by specifically binding to the unstructured basic region of Tat. mBio 2019, 10, e02662-18. [Google Scholar] [CrossRef] [Green Version]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D.V. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio 2015, 6, e00465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, L.U.; Rodríguez-González, J. Modeling the effect of tat inhibitors on HIV latency. J. Theor. Biol. 2019, 473, 20–27. [Google Scholar] [CrossRef]
- Morton, E.L.; Forst, C.V.; Zheng, Y.; DePaula-Silva, A.B.; Ramirez, N.P.; Planelles, V.; D’Orso, I. Transcriptional circuit fragility influences HIV proviral fate. Cell Rep. 2019, 27, 154–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacombe, B.; Morel, M.; Margottin-Goguet, F.; Ramirez, B.C. Specific inhibition of HIV infection by the action of spironolactone in T cells. J. Virol. 2016, 90, 10972–10980. [Google Scholar] [CrossRef] [Green Version]
- Leoz, M.; Kukanja, P.; Luo, Z.; Huang, F.; Cary, D.C.; Peterlin, B.M.; Fujinaga, K. HEXIM1-Tat chimera inhibits HIV-1 replication. PLoS Pathog. 2018, 14, e1007402. [Google Scholar] [CrossRef]
- Niu, Q.; Liu, Z.; Alamer, E.; Fan, X.; Chen, H.; Endsley, J.; Gelman, B.B.; Tian, B.; Kim, J.H.; Michael, N.L.; et al. Structure-guided drug design identifies a BRD4-selective small molecule that suppresses HIV. J. Clin. Investig. 2019, 129, 3361–3373. [Google Scholar] [CrossRef]
- Alamer, E.; Zhong, C.; Liu, Z.; Niu, Q.; Long, F.; Guo, L.; Gelman, B.B.; Soong, L.; Zhou, J.; Hu, H. Epigenetic suppression of HIV in myeloid cells by the BRD4-selective small molecule modulator ZL0580. J. Virol. 2020, 94, e01880-19. [Google Scholar] [CrossRef]
- Meredith, L.W.; Sivakumaran, H.; Major, L.; Suhrbier, A.; Harrich, D. Potent inhibition of HIV-1 replication by a Tat mutant. PLoS ONE 2009, 4, e7769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.H.; Sivakumaran, H.; Apolloni, A.; Wei, T.; Jans, D.A.; Harrich, D. Nullbasic, a potent anti-HIV tat mutant, induces CRM1-dependent disruption of HIV rev trafficking. PLoS ONE 2012, 7, e51466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.H.; Sivakumaran, H.; Jones, A.; Li, D.; Harper, C.; Wei, T.; Jin, H.; Rustanti, L.; Meunier, F.A.; Spann, K.; et al. A HIV-1 Tat mutant protein disrupts HIV-1 Rev function by targeting the DEAD-box RNA helicase DDX1. Retrovirology 2014, 11, 121. [Google Scholar] [CrossRef]
- Lin, M.H.; Apolloni, A.; Cutillas, V.; Sivakumaran, H.; Martin, S.; Li, D.; Wei, T.; Wang, R.; Jin, H.; Spann, K.; et al. A mutant tat protein inhibits HIV-1 reverse transcription by targeting the reverse transcription complex. J. Virol. 2015, 89, 4827–4836. [Google Scholar] [CrossRef] [Green Version]
- Li, J.Z.; Etemad, B.; Ahmed, H.; Aga, E.; Bosch, R.J.; Mellors, J.W.; Kuritzkes, D.R.; Lederman, M.M.; Para, M.; Gandhi, R.T. The size of the expressed HIV reservoir predicts timing of viral rebound after treatment interruption. AIDS 2016, 30, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Kaiser, P.; Lampiris, H.W.; Kim, P.; Yukl, S.A.; Havlir, D.V.; Greene, W.C.; Wong, J.K. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat. Med. 2016, 22, 807–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Vidal, E.; Castellvi, M.; Pujantell, M.; Badia, R.; Jou, A.; Gomez, L.; Puig, T.; Clotet, B.; Ballana, E.; Riveira-Munoz, E.; et al. Evaluation of the innate immune modulator acitretin as a strategy to clear the HIV reservoir. Antimicrob. Agents Chemother. 2017, 61, e01368-17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Hayashi, T.; Jean, M.; Kong, W.; Fiches, G.; Biswas, A.; Liu, S.; Yosief, H.O.; Zhang, X.; Bradner, J.; et al. Inhibition of polo-like kinase 1 (PLK1) facilitates the elimination of HIV-1 viral reservoirs in CD4+ T cells ex vivo. Sci. Adv. 2020, 6, eaba1941. [Google Scholar] [CrossRef]
- Alto, A.; Natesampillai, S.; Chandrasekar, A.P.; Krogman, A.; Misra, A.; Shweta, F.; VanLith, C.; Yao, J.D.; Cummins, N.W.; Badley, A.D. The combination of venetoclax and ixazomib selectively and efficiently kills HIV-infected cell lines but has unacceptable toxicity in primary cell models. J. Virol. 2021, 95, e00138-21. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Lungu, C.; Crespo, R.; Steijaert, T.H.; Gorska, A.; Palstra, R.J.; Prins, H.A.B.; van Ijcken, W.; Mueller, Y.M.; van Kampen, J.J.A.; et al. Selective cell death in HIV-1-infected cells by DDX3 inhibitors leads to depletion of the inducible reservoir. Nat. Commun. 2021, 12, 2475. [Google Scholar] [CrossRef]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, J.; Paquette, J.S.; Fortin, J.F.; Tremblay, M.J. The immunosuppressant rapamycin represses human immunodeficiency virus type 1 replication. Antimicrob. Agents Chemother. 2002, 46, 3447–3455. [Google Scholar] [CrossRef] [Green Version]
- Nardacci, R.; Amendola, A.; Ciccosanti, F.; Corazzari, M.; Esposito, V.; Vlassi, C.; Taibi, C.; Fimia, G.M.; Del Nonno, F.; Ippolito, G.; et al. Autophagy plays an important role in the containment of HIV-1 in nonprogressor-infected patients. Autophagy 2014, 10, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Cloherty, A.P.M.; van Teijlingen, N.H.; Eisden, T.; van Hamme, J.L.; Rader, A.G.; Geijtenbeek, T.B.H.; Schreurs, R.; Ribeiro, C.M.S. Autophagy-enhancing drugs limit mucosal HIV-1 acquisition and suppress viral replication ex vivo. Sci. Rep. 2021, 11, 4767. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, F.; Di Sandro, S.; De Ruvo, N.; Montalti, R.; Ballarin, R.; Guerrini, G.P.; Spaggiari, M.; Guaraldi, G.; Gerunda, G. First report on a series of HIV patients undergoing rapamycin monotherapy after liver transplantation. Transplantation 2010, 89, 733–738. [Google Scholar] [CrossRef]
- Henrich, T.J.; Schreiner, C.; Cameron, C.; Hogan, L.E.; Richardson, B.; Rutishauser, R.L.; Deitchman, A.N.; Chu, S.; Rogers, R.; Thanh, C.; et al. Everolimus, an mTORC1/2 inhibitor, in ART-suppressed individuals who received solid organ transplantation: A prospective study. Am. J. Transplant. 2020, 21, 1765–1779. [Google Scholar] [CrossRef]
- Nicoletti, F.; Lapenta, C.; Donati, S.; Spada, M.; Ranazzi, A.; Cacopardo, B.; Mangano, K.; Belardelli, F.; Perno, C.; Aquaro, S. Inhibition of human immunodeficiency virus (HIV-1) infection in human peripheral blood leucocytes-SCID reconstituted mice by rapamycin. Clin. Exp. Immunol. 2009, 155, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Amoroso, A.; Davis, C.; Le, N.; Reardon, E.; Dominique, J.K.; Klingebiel, E.; Gallo, R.C.; Redfield, R.R. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV β-chemokines: An approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 2003, 100, 10411–10416. [Google Scholar] [CrossRef] [Green Version]
- Goodall, M.L.; Cramer, S.D.; Thorburn, A. Autophagy complexes cell death by necroptosis. Oncotarget 2016, 7, 50818–50819. [Google Scholar] [CrossRef]
- Kriel, J.; Loos, B. The good, the bad and the autophagosome: Exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 2019, 26, 640–652. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.O.; DeMarino, C.; Vo, T.T.; Cowen, M.; Kim, Y.; Pleet, M.L.; Barclay, R.A.; Noren Hooten, N.; Evans, M.K.; Heredia, A.; et al. Low-level ionizing radiation induces selective killing of HIV-1-infected cells with reversal of cytokine induction using mTOR inhibitors. Viruses 2020, 12, 885. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Spector, S.A. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J. Biol. Chem. 2015, 290, 5028–5040. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; Bruckman, R.S.; Herns, S.D.; Joshi, S.; Durden, D.L.; Spector, S.A. Induction of autophagy by PI3K/MTOR and PI3K/MTOR/BRD4 inhibitors suppresses HIV-1 replication. J. Biol. Chem. 2018, 293, 5808–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawat, P.; Hon, S.; Teodorof-Diedrich, C.; Spector, S.A. Trehalose inhibits human immunodeficiency virus type 1 infection in primary human macrophages and CD4+ T lymphocytes through two distinct mechanisms. J. Virol. 2020, 94, e00237-20. [Google Scholar] [CrossRef]
- Sharma, V.; Makhdoomi, M.; Singh, L.; Kumar, P.; Khan, N.; Singh, S.; Verma, H.N.; Luthra, K.; Sarkar, S.; Kumar, D. Trehalose limits opportunistic mycobacterial survival during HIV co-infection by reversing HIV-mediated autophagy block. Autophagy 2021, 17, 476–495. [Google Scholar] [CrossRef] [Green Version]
- Brüning, A.; Rahmeh, M.; Friese, K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol. Oncol. 2013, 7, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, T.; Goswami, S.; Hu, X.; Laverdure, S.; Yang, J.; Qiu, J.; Chen, Q.; Sherman, B.T.; Chang, W. MicroRNA profiles in monocyte-derived macrophages generated by interleukin-27 and human serum: Identification of a novel HIV-inhibiting and autophagy-inducing microRNA. Int. J. Mol. Sci. 2021, 22, 1290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Luk, B.T.; Wei, X.; Campbell, G.R.; Fang, R.H.; Zhang, L.; Spector, S.A. Selective cell death of latently HIV-infected CD4+ T cells mediated by autosis inducing nanopeptides. Cell Death Dis. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Luk, B.T.; Hamidy, M.; Zhang, L.; Spector, S.A. Induction of a Na+/K+-ATPase-dependent form of autophagy triggers preferential cell death of human immunodeficiency virus type-1-infected macrophages. Autophagy 2018, 14, 1359–1375. [Google Scholar] [CrossRef]
- Adesina, S.K.; Akala, E.O. Nanotechnology approaches for the delivery of exogenous siRNA for HIV therapy. Mol. Pharm. 2015, 12, 4175–4187. [Google Scholar] [CrossRef]
- Bowman, M.C.; Ballard, T.E.; Ackerson, C.J.; Feldheim, D.L.; Margolis, D.M.; Melander, C. Inhibition of HIV fusion with multivalent gold nanoparticles. J. Am. Chem. Soc. 2008, 130, 6896–6897. [Google Scholar] [CrossRef] [Green Version]
- Glass, J.J.; Yuen, D.; Rae, J.; Johnston, A.P.; Parton, R.G.; Kent, S.J.; De Rose, R. Human immune cell targeting of protein nanoparticles--caveospheres. Nanoscale 2016, 8, 8255–8265. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhang, G.; Ran, D.; Krishnan, N.; Fang, R.H.; Gao, W.; Spector, S.A.; Zhang, L. T-cell-mimicking nanoparticles can neutralize HIV infectivity. Adv. Mater. 2018, 30, e1802233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Campbell, G.R.; Zhang, Q.; Maule, E.; Hanna, J.; Gao, W.; Zhang, L.; Spector, S.A. CD4+ T cell-mimicking nanoparticles broadly neutralize HIV-1 and suppress viral replication through autophagy. mBio 2020, 11, e00903-20. [Google Scholar] [CrossRef]
- Fang, R.H.; Kroll, A.V.; Zhang, L. Nanoparticle-based manipulation of antigen-presenting cells for cancer immunotherapy. Small 2015, 11, 5483–5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teleanu, D.M.; Negut, I.; Grumezescu, V.; Grumezescu, A.M.; Teleanu, R.I. Nanomaterials for drug delivery to the central nervous system. Nanomaterials 2019, 9, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Lucas, A.; Kim, Y.; Rivera-Pabon, O.; Chae, S.; Kim, D.H.; Kim, B. Targeting the PI3K/Akt cell survival pathway to induce cell death of HIV-1 infected macrophages with alkylphospholipid compounds. PLoS ONE 2010, 5, e13121. [Google Scholar] [CrossRef]
- Kim, Y.; Hollenbaugh, J.A.; Kim, D.H.; Kim, B. Novel PI3K/Akt inhibitors screened by the cytoprotective function of human immunodeficiency virus type 1 Tat. PLoS ONE 2011, 6, e21781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Guillemard, E.; Jacquemot, C.; Aillet, F.; Schmitt, N.; Barre-Sinoussi, F.; Israel, N. Human immunodeficiency virus 1 favors the persistence of infection by activating macrophages through TNF. Virology 2004, 329, 371–380. [Google Scholar] [CrossRef] [Green Version]
- Campbell, G.R.; To, R.K.; Spector, S.A. TREM-1 protects HIV-1-infected macrophages from apoptosis through maintenance of mitochondrial function. mBio 2019, 10, e02638-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubrez, L.; Berthelet, J.; Glorian, V. IAP proteins as targets for drug development in oncology. OncoTargets Ther. 2013, 9, 1285–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obexer, P.; Ausserlechner, M.J. X-linked inhibitor of apoptosis protein—A critical death resistance regulator and therapeutic target for personalized cancer therapy. Front. Oncol. 2014, 4, 197. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Zhao, H.; Jin, M.; Zhu, H.; Shan, B.; Geng, J.; Dziedzic, S.A.; Amin, P.; Mifflin, L.; Naito, M.G.; et al. Modulating TRADD to restore cellular homeostasis and inhibit apoptosis. Nature 2020, 587, 133–138. [Google Scholar] [CrossRef]
- Dumétier, B.; Zadoroznyj, A.; Dubrez, L. IAP-mediated protein ubiquitination in regulating cell signaling. Cells 2020, 9, 1118. [Google Scholar] [CrossRef]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Najafov, A.; Luu, H.S.; Mookhtiar, A.K.; Mifflin, L.; Xia, H.G.; Amin, P.P.; Ordureau, A.; Wang, H.; Yuan, J. RIPK1 promotes energy sensing by the mTORC1 pathway. Mol. Cell 2021, 81, 370–385.e7. [Google Scholar] [CrossRef]
- Gao, Z.; Tian, Y.; Wang, J.; Yin, Q.; Wu, H.; Li, Y.M.; Jiang, X. A dimeric Smac/diablo peptide directly relieves caspase-3 inhibition by XIAP. Dynamic and cooperative regulation of XIAP by Smac/Diablo. J. Biol. Chem. 2007, 282, 30718–30727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, S.I.; Matsuda, K.; Tsuchiya, K.; Gatanaga, H.; Oka, S.; Yoshimura, K.; Mitsuya, H.; Maeda, K. Combination of a latency-reversing agent with a Smac mimetic minimizes secondary HIV-1 infection in vitro. Front. Microbiol. 2018, 9, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, G.; Preston, S.; Allison, C.; Cooney, J.; Toe, J.G.; Stutz, M.D.; Ojaimi, S.; Scott, H.W.; Baschuk, N.; Nachbur, U.; et al. Cellular inhibitor of apoptosis proteins prevent clearance of hepatitis B virus. Proc. Natl. Acad. Sci. USA 2015, 112, 5797–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, G.; Allison, C.; Preston, S.; Cooney, J.; Toe, J.G.; Stutz, M.D.; Ojaimi, S.; Baschuk, N.; Nachbur, U.; Torresi, J.; et al. Eliminating hepatitis B by antagonizing cellular inhibitors of apoptosis. Proc. Natl. Acad. Sci. USA 2015, 112, 5803–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, E.; Mackiewicz, L.; Silke, N.; Pellegrini, M.; Silke, J.; Brumatti, G.; Ebert, G. Combinatorial treatment of birinapant and zosuquidar enhances effective control of HBV replication in vivo. Viruses 2020, 12, 901. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The initiation of autophagy is a multistep process. The main regulators of autophagy are MTOR, an inhibitor, and AMP-activated kinase (AMPK) an activator. MTORC1 inhibition induces autophagy through the formation of the ULK1 complex. This complex initiates the formation of the phagophore by directly activating the phosphatidylinositol 3-kinase catalytic subunit type 3 complex I (PIK3C3 complex). The PIK3C3 complex translocates to endoplasmic reticulum sites and produces phosphatidylinositol 3-phosphate (PI3P) which then recruits PI3P-binding proteins such as WIPI2 and ZFYVE1. HIV-1 Nef can inhibit this process by enhancing the association between BECN1 and its inhibitor BCL2. In the ATG12 conjugation system, ATG12 forms a covalent bond with ATG5, which then binds ATG16L1, followed by dimerization (not shown) and interaction with the PI3P-binding complex. The WIPI2b isoform acts immediately upstream of ATG16L1 and recruits ATG12–ATG5-ATG16L1 to PI3P-containing phagophores. The ATG12–ATG5-ATG16L1 complex then promotes conjugation of ATG8 family proteins with phosphatidylethanolamine (PE), which are then incorporated into phagophore membranes, where they interact with cargo receptors harbouring LC3-interacting motifs. ATG16L1, as well as the polyubiquitin-binding autophagy receptors including SQSTM1, OPTN, NBR1, and NDP52 also recruit and incorporate ubiquitin-decorated cargos into autophagosomes including HIV-1 Tat, HIV-1 Vif, and HIV-1 p24. After detachment of ATG factors, the isolation membrane closes through membrane scission, forming the autophagosome. These, then, fuse with lysosomes resulting in the degradation of the engulfed components as well as the ATG8-PE and SQSTM1 associated with the inner membrane. HIV-1 Nef affects autophagosome maturation by preventing the fusion between autophagosomes and lysosomes.
Figure 1.
The initiation of autophagy is a multistep process. The main regulators of autophagy are MTOR, an inhibitor, and AMP-activated kinase (AMPK) an activator. MTORC1 inhibition induces autophagy through the formation of the ULK1 complex. This complex initiates the formation of the phagophore by directly activating the phosphatidylinositol 3-kinase catalytic subunit type 3 complex I (PIK3C3 complex). The PIK3C3 complex translocates to endoplasmic reticulum sites and produces phosphatidylinositol 3-phosphate (PI3P) which then recruits PI3P-binding proteins such as WIPI2 and ZFYVE1. HIV-1 Nef can inhibit this process by enhancing the association between BECN1 and its inhibitor BCL2. In the ATG12 conjugation system, ATG12 forms a covalent bond with ATG5, which then binds ATG16L1, followed by dimerization (not shown) and interaction with the PI3P-binding complex. The WIPI2b isoform acts immediately upstream of ATG16L1 and recruits ATG12–ATG5-ATG16L1 to PI3P-containing phagophores. The ATG12–ATG5-ATG16L1 complex then promotes conjugation of ATG8 family proteins with phosphatidylethanolamine (PE), which are then incorporated into phagophore membranes, where they interact with cargo receptors harbouring LC3-interacting motifs. ATG16L1, as well as the polyubiquitin-binding autophagy receptors including SQSTM1, OPTN, NBR1, and NDP52 also recruit and incorporate ubiquitin-decorated cargos into autophagosomes including HIV-1 Tat, HIV-1 Vif, and HIV-1 p24. After detachment of ATG factors, the isolation membrane closes through membrane scission, forming the autophagosome. These, then, fuse with lysosomes resulting in the degradation of the engulfed components as well as the ATG8-PE and SQSTM1 associated with the inner membrane. HIV-1 Nef affects autophagosome maturation by preventing the fusion between autophagosomes and lysosomes.

Figure 2.
Potential strategy to achieve an HIV-1 cure. (a) Antiretroviral therapy (ART) suppresses HIV-1 plasma viremia, but upon interruption of ART, it quickly rebounds; (b) conventional HIV-1 Tat/P-TEFb inhibitors fail to silence epigenetically the HIV-1 promoter, thus when they are removed, transcription is eventually restored and HIV-1 viremia rebounds; (c) supplementation of ART with a latency-promoting epigenetic silencing drug (LPA) could promote a state of deep latency, but as they do not reduce the viral reservoir, rebound could occur; (d) supplementation of ART with both an LPA and a drug that selectively kills HIV-1 infected cells could decrease the size of the viral reservoir, significantly prolonging the time to viral rebound after ART interruption. Optimization of such a strategy has the potential to completely eliminate the viral reservoirs.
Figure 2.
Potential strategy to achieve an HIV-1 cure. (a) Antiretroviral therapy (ART) suppresses HIV-1 plasma viremia, but upon interruption of ART, it quickly rebounds; (b) conventional HIV-1 Tat/P-TEFb inhibitors fail to silence epigenetically the HIV-1 promoter, thus when they are removed, transcription is eventually restored and HIV-1 viremia rebounds; (c) supplementation of ART with a latency-promoting epigenetic silencing drug (LPA) could promote a state of deep latency, but as they do not reduce the viral reservoir, rebound could occur; (d) supplementation of ART with both an LPA and a drug that selectively kills HIV-1 infected cells could decrease the size of the viral reservoir, significantly prolonging the time to viral rebound after ART interruption. Optimization of such a strategy has the potential to completely eliminate the viral reservoirs.

Figure 3.
DIABO/SMAC mimetic mediated apoptosis. Treatment of latent HIV-infected cells with DIABLO/SMAC mimetics induces the degradation of XIAP and BIRC2/3, inducing autophagy, the deubiquitination of RIPK1, and the disinhibition of caspases. This leads to the formation of a death inducing signalling complex comprising of FADD, RIPK1, RIPK3 and caspase-8 on autophagosomal membranes, resulting in the cleavage and activation of caspase-8, thus enabling activation of the effector caspases (caspase-3 and caspase-7) and the execution of apoptosis.
Figure 3.
DIABO/SMAC mimetic mediated apoptosis. Treatment of latent HIV-infected cells with DIABLO/SMAC mimetics induces the degradation of XIAP and BIRC2/3, inducing autophagy, the deubiquitination of RIPK1, and the disinhibition of caspases. This leads to the formation of a death inducing signalling complex comprising of FADD, RIPK1, RIPK3 and caspase-8 on autophagosomal membranes, resulting in the cleavage and activation of caspase-8, thus enabling activation of the effector caspases (caspase-3 and caspase-7) and the execution of apoptosis.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Campbell, G.R.; Spector, S.A. Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure. Cells 2021, 10, 1798. https://doi.org/10.3390/cells10071798
AMA Style
Campbell GR, Spector SA. Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure. Cells. 2021; 10(7):1798. https://doi.org/10.3390/cells10071798
Chicago/Turabian StyleCampbell, Grant R., and Stephen A. Spector. 2021. "Induction of Autophagy to Achieve a Human Immunodeficiency Virus Type 1 Cure" Cells 10, no. 7: 1798. https://doi.org/10.3390/cells10071798
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.