
Extracellular Vesicle Release Promotes Viral Replication during Persistent HCV Infection

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Antibodies, and Chemicals
2.2. Western Blotting
2.3. Confocal Microscopy
2.4. Exosome Isolation and Quantification
2.5. Cryo-TEM for Exosome Characterization
2.6. Transmission Electron Microscopy (TEM)
2.7. MTT Proliferation/Viability Assay
2.8. Enzyme Linked Immunosorbent Assay (ELISA)
2.9. Statistical Analysis
3. Results
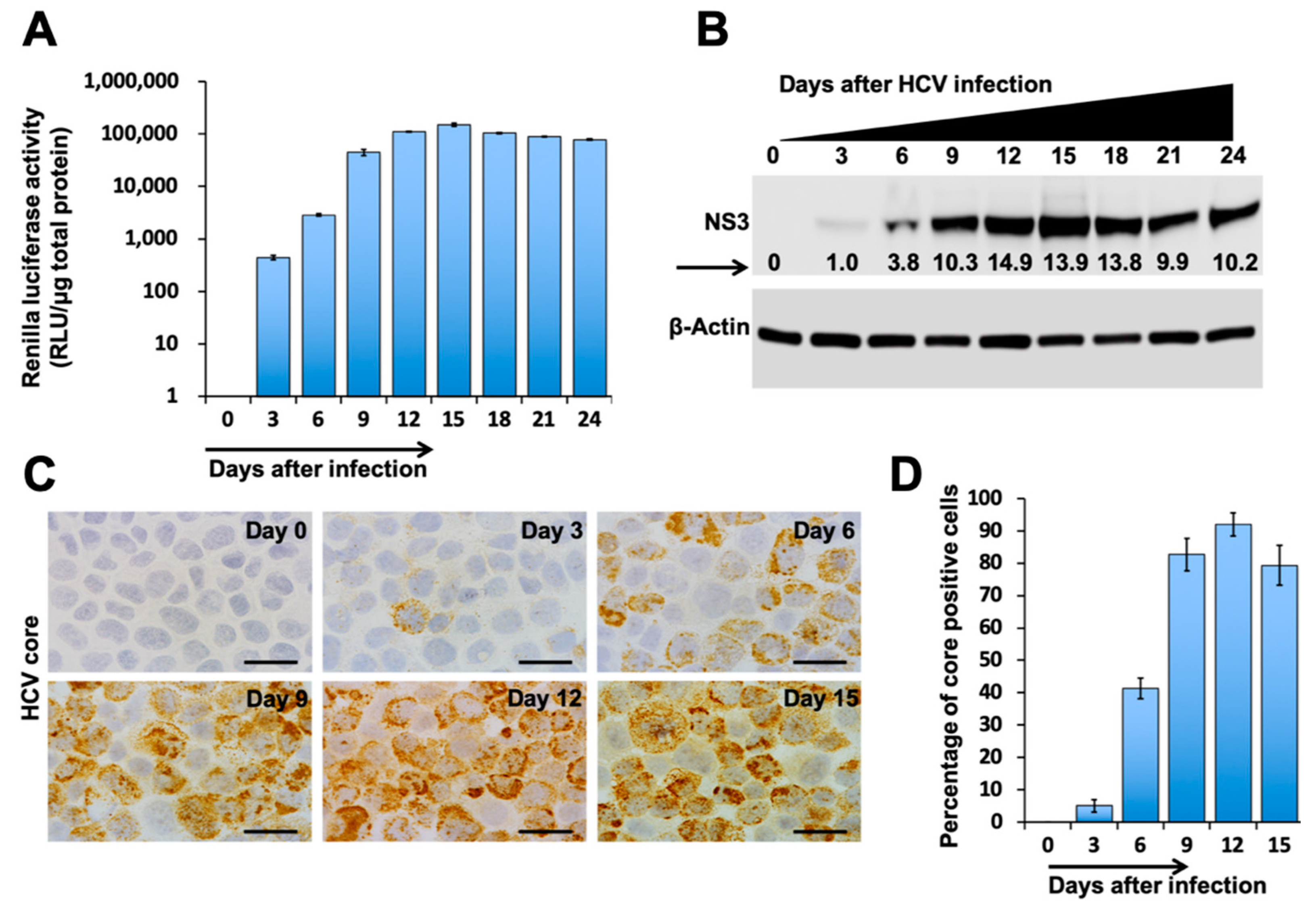
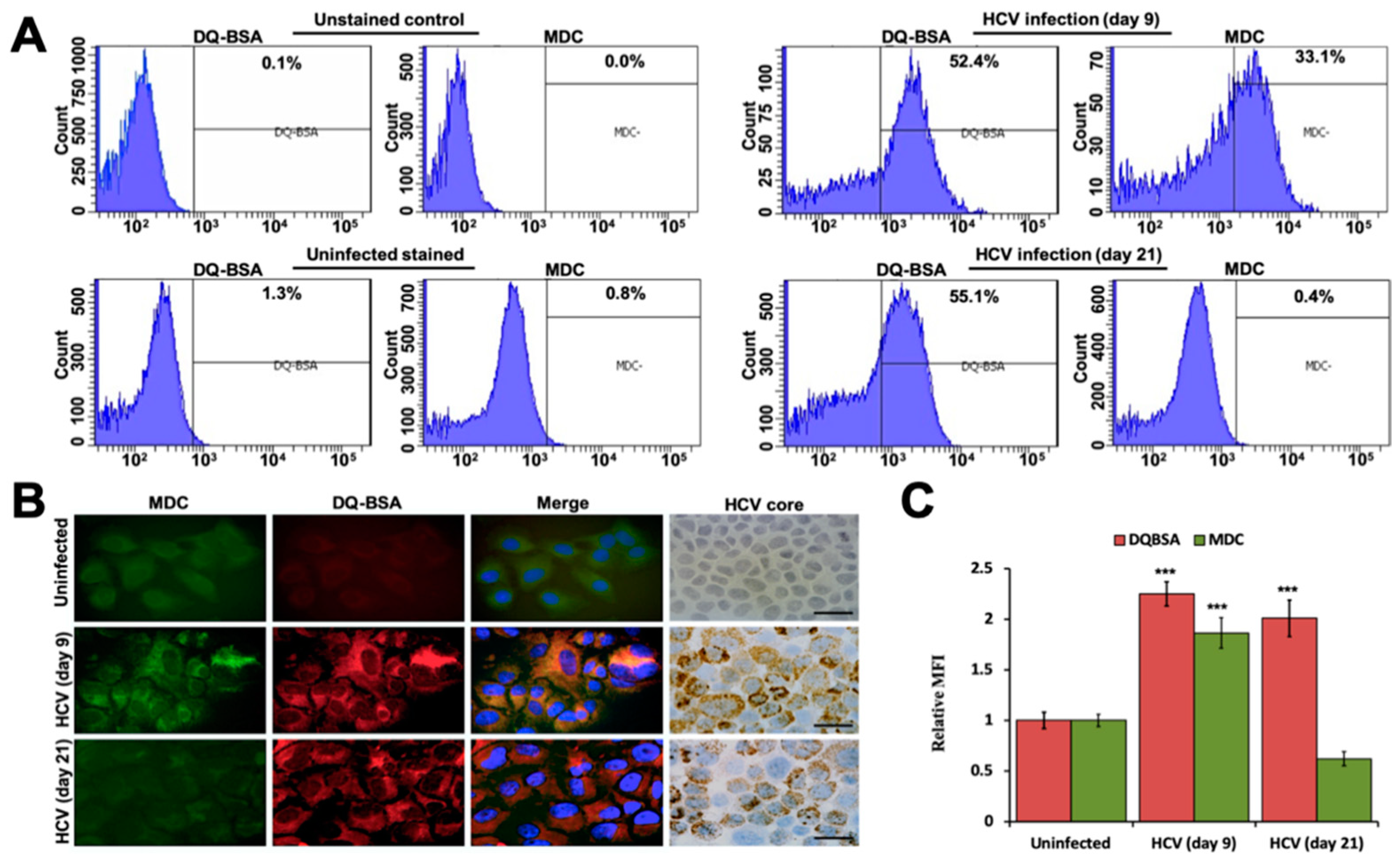
3.1. Autophagic Degradation and Exosome Release Supports Virus-Cell Survival during Persistent HCV Infection
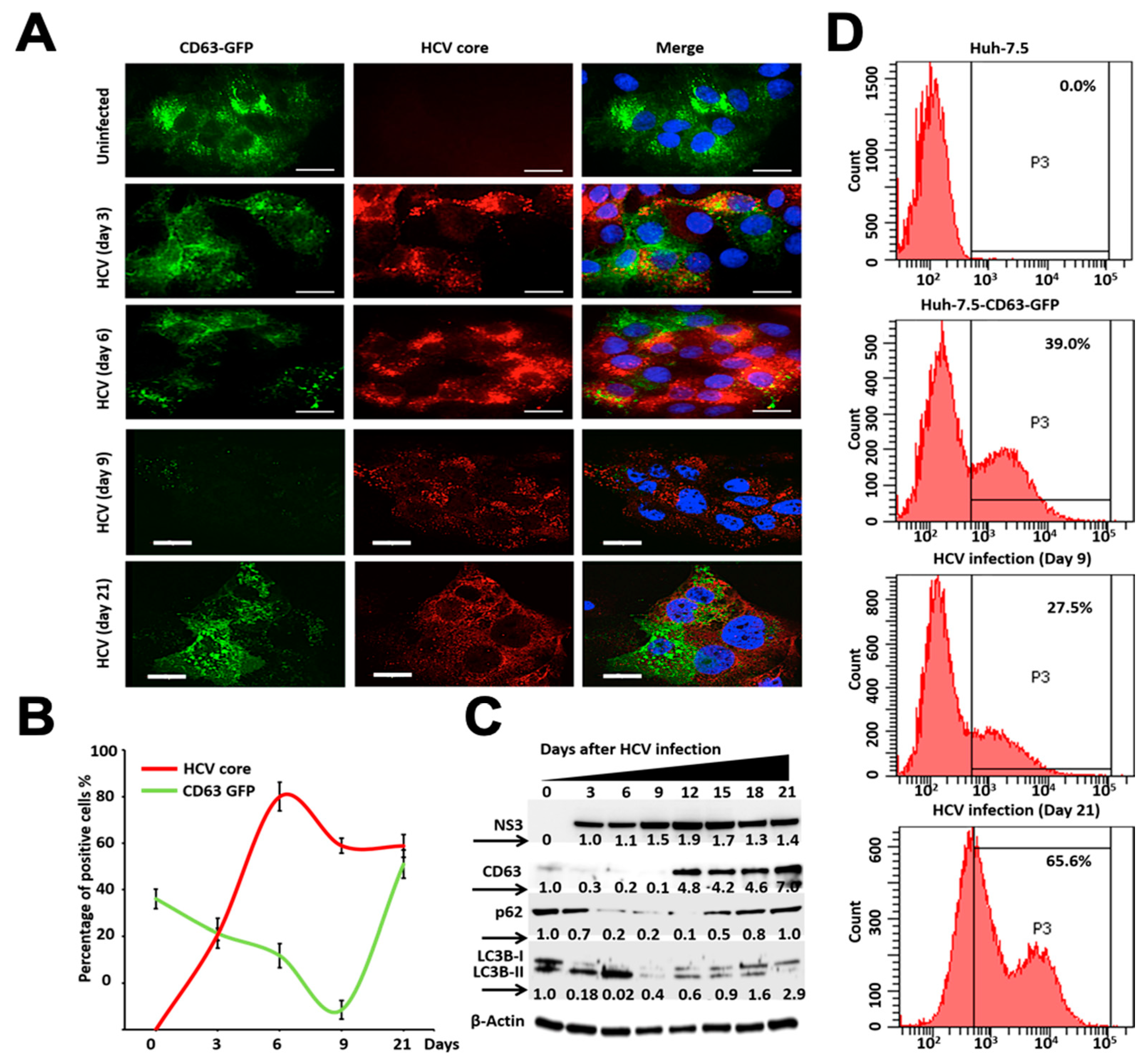
3.2. Persistent HCV Infection Inhibits CD63-Mediated Autophagic Endosome-Lysosomal Degradation
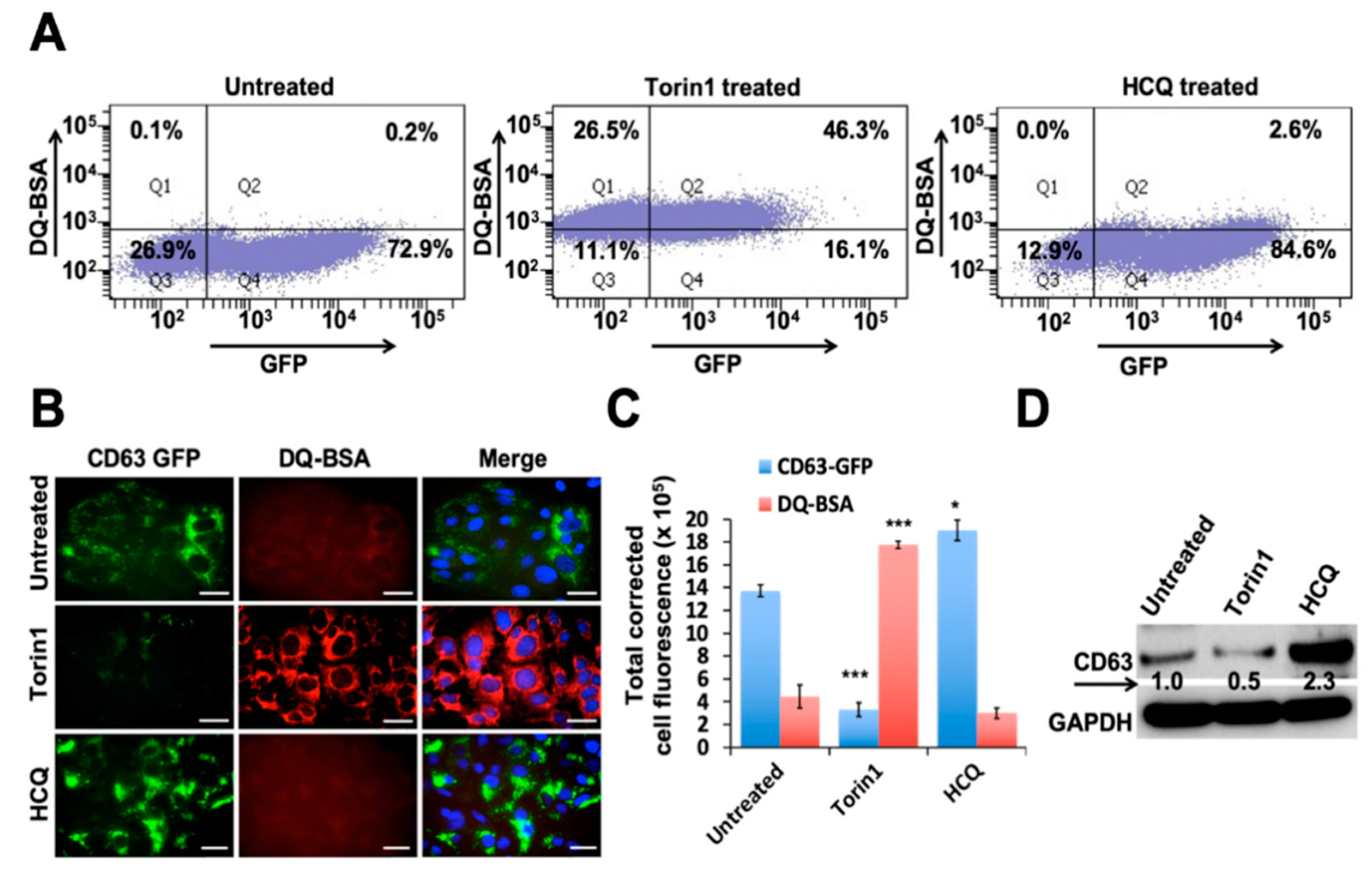
3.3. Autophagy Induction Promotes Degradation of MVBs
3.4. Persistent HCV Replication Decreased Autophagic Vacuoles and Autophagosome-Lysosome Fusion without Affecting Lysosomal Activity
3.5. Ultrastructural Analysis of Huh-7.5 Cells Infected with HCV
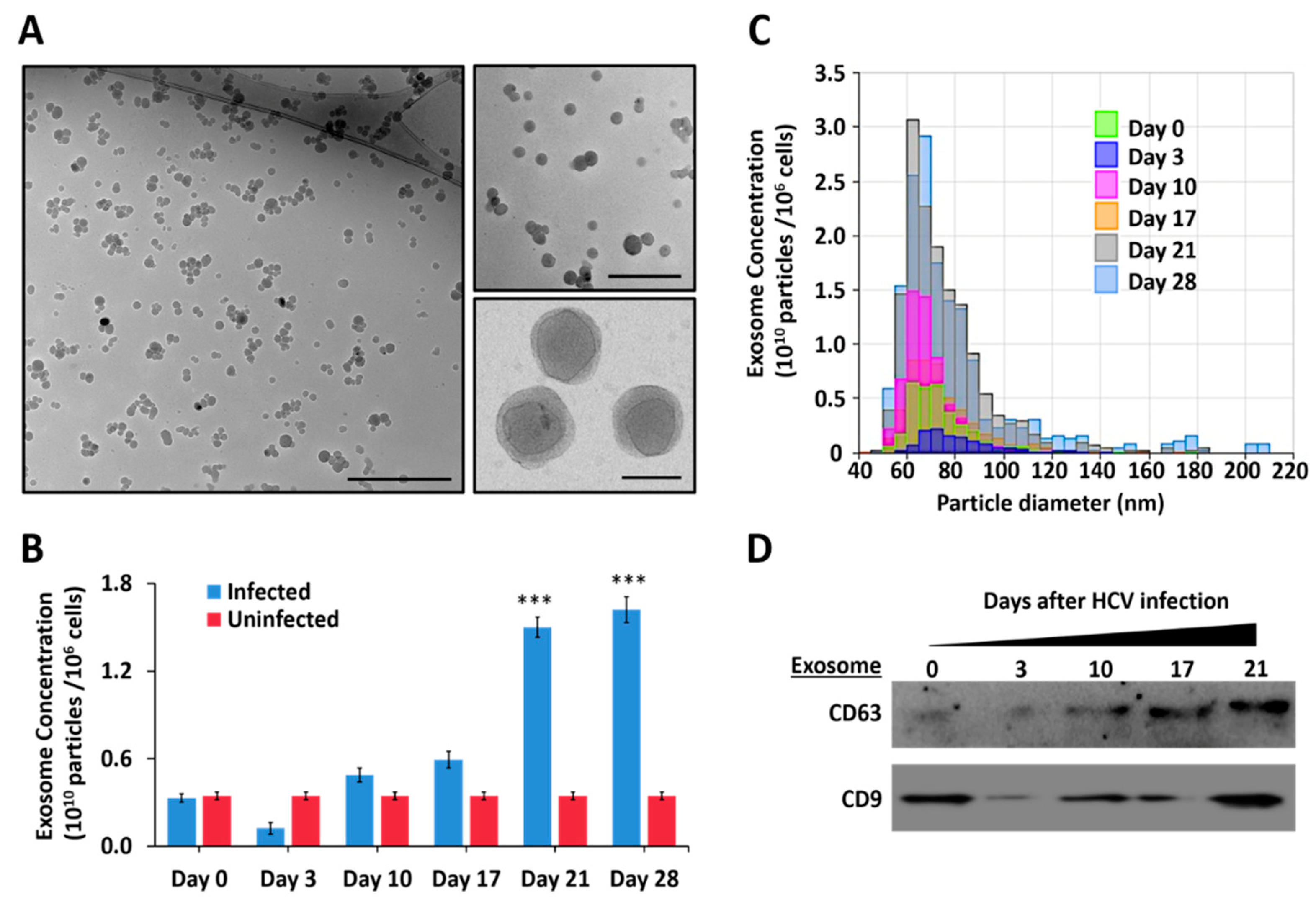
3.6. Persistent HCV Infection Promotes Release of Extracellular Vesicles and Exosomes
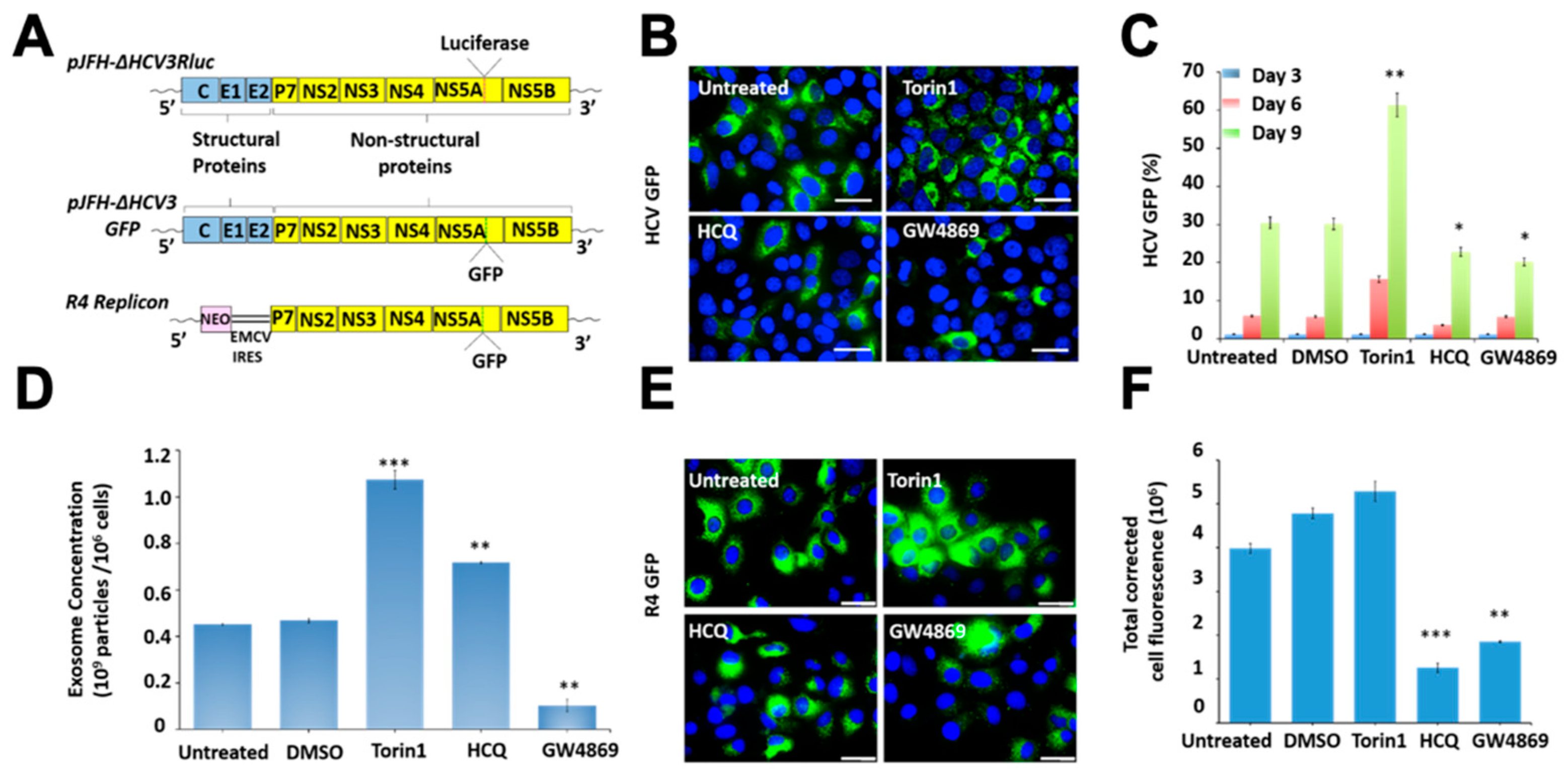
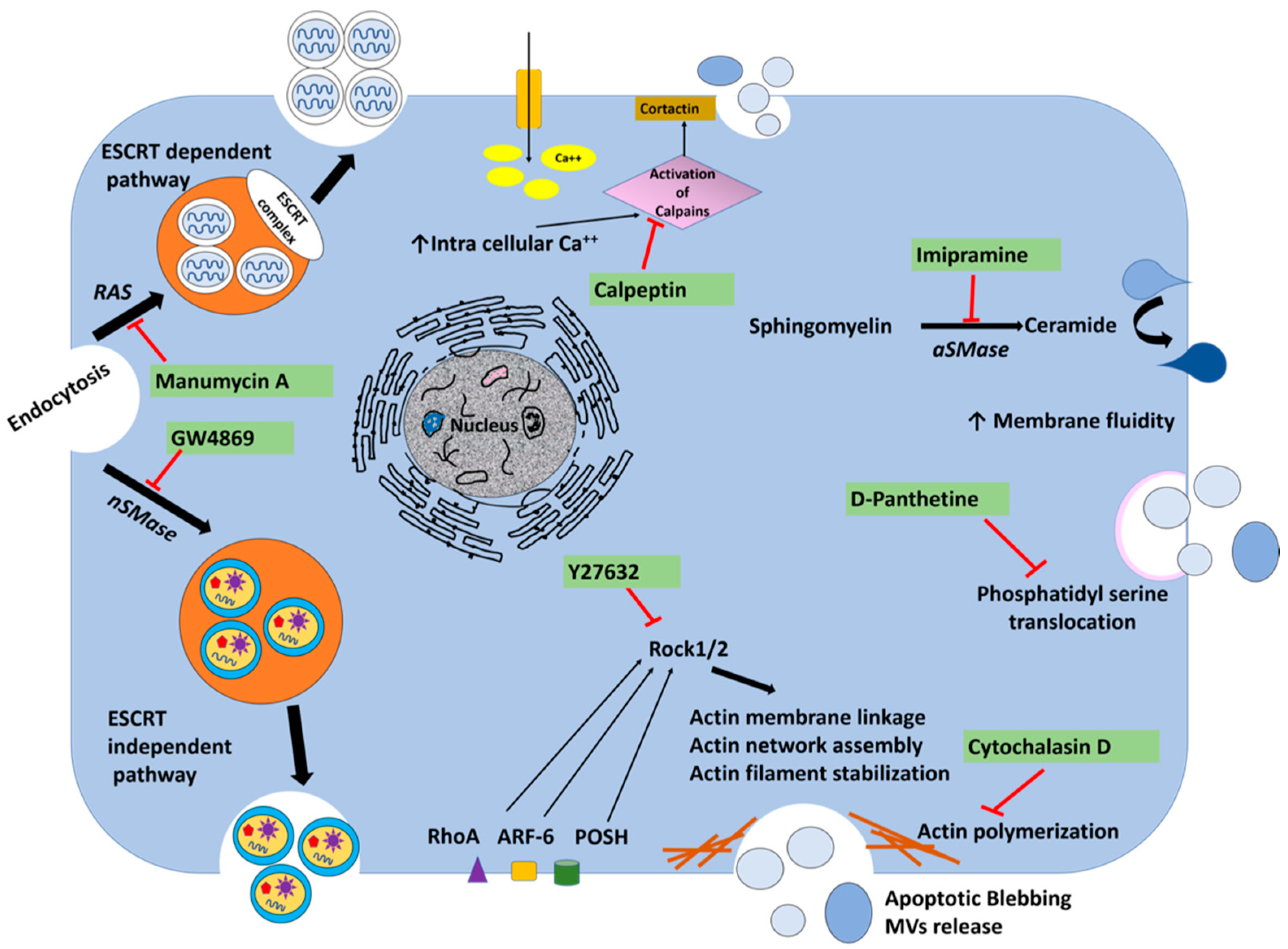
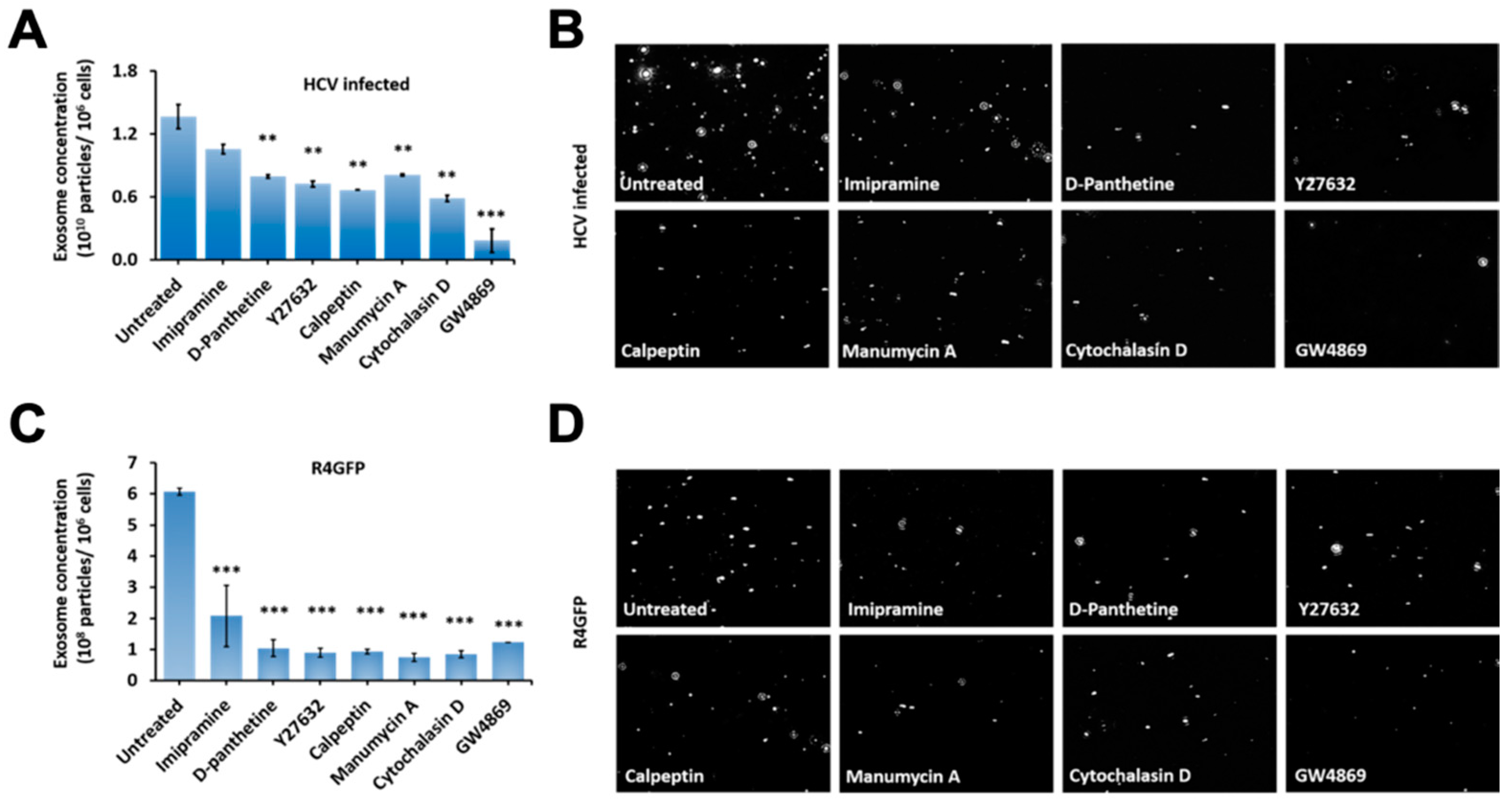
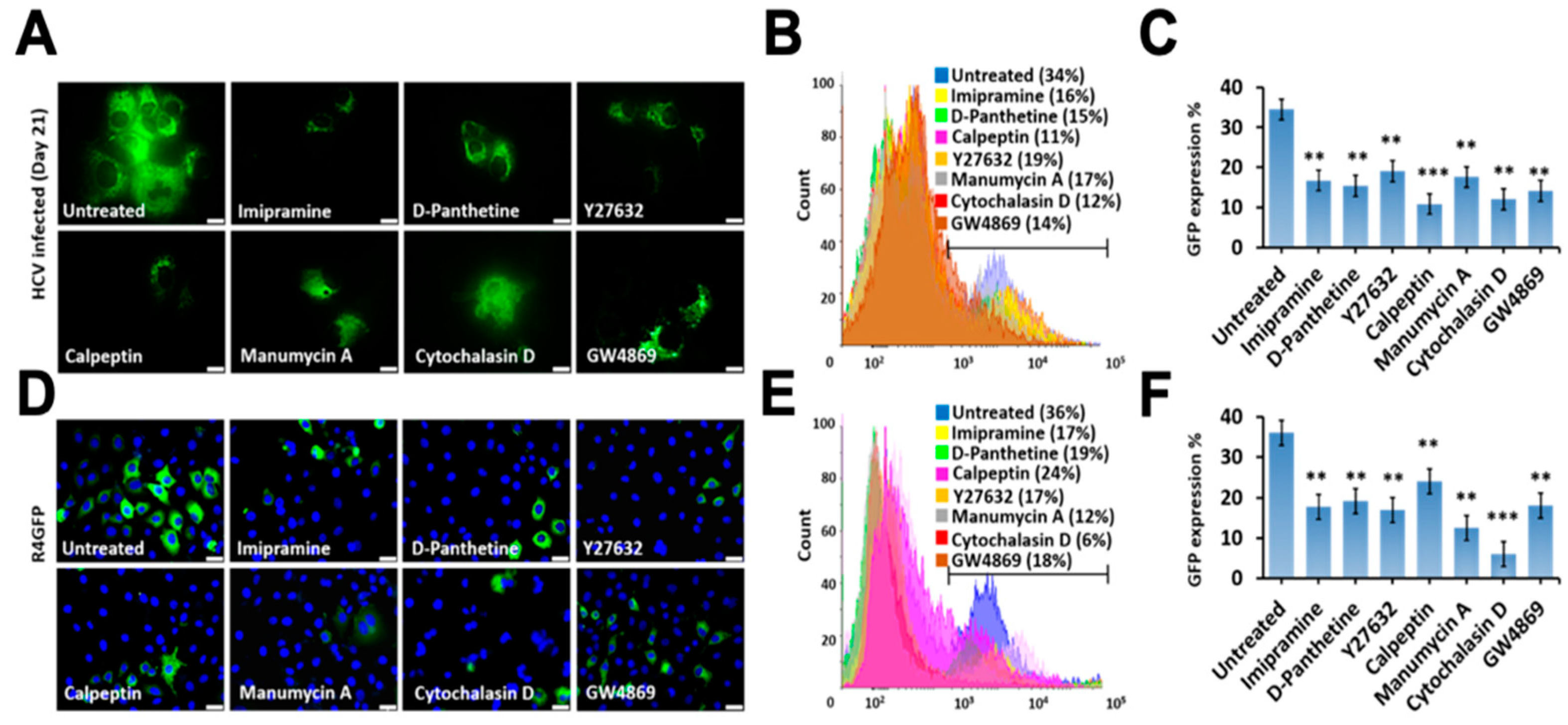
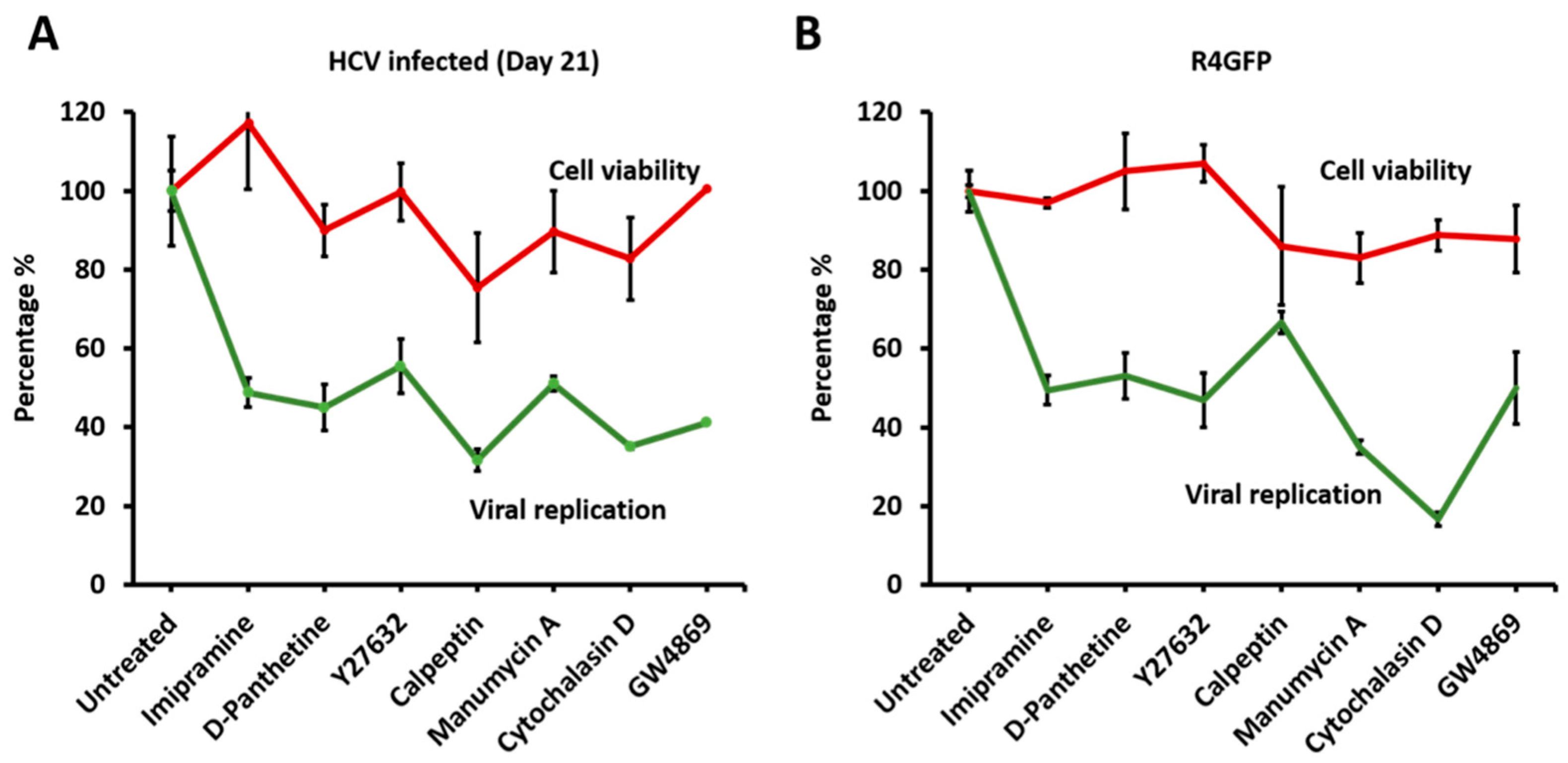
3.7. Release of Extracellular Vesicles Promotes Virus Replication during Persistent HCV Infection
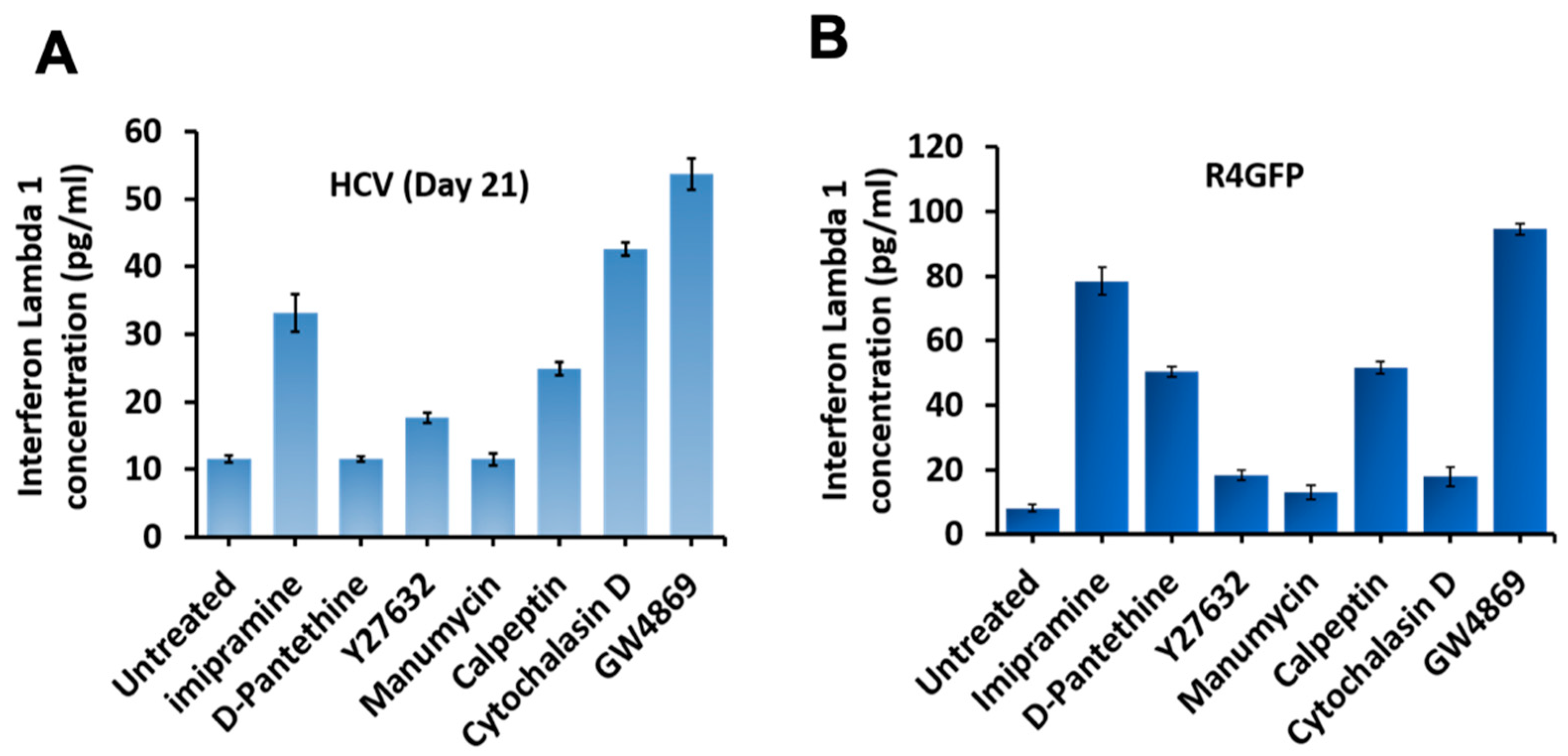
3.8. Inhibition of Extracellular Vesicles Induced Innate Antiviral Response in HCV Culture through Interefron-Lambda (IFNL1) Production
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Serag, H.B.; Kanwal, F.; Davila, J.A.; Kramer, J.; Richardson, P. A new laboratory-based algorithm to predict development of hepatocellular carcinoma in patients with hepatitis C and cirrhosis. Gastroenterology 2014, 146, 1249–1255.e1. [Google Scholar] [CrossRef] [Green Version]
- Gower, E.; Estes, C.; Blach, S.; Razavi-Shearer, K.; Razavi, H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Pawlotsky, J.M.; Feld, J.J.; Zeuzem, S.; Hoofnagle, J.H. From non-A, non-B hepatitis to hepatitis C virus cure. J. Hepatol. 2015, 62, S87–S99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.L. Global control of hepatitis C: Where challenge meets opportunity. Nat. Med. 2013, 19, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, A.; Mondelli, M.U. Hepatitis C: Is eradication possible? Liver Int. 2019, 39, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicks, N.; Gutierrez, K.; Michalak, M.; Bordignon, V.; Agellon, L.B. Endoplasmic reticulum stress, genome damage, and cancer. Front. Oncol. 2015, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C.; et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019–40036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Czaja, M.J. Compensatory mechanisms and the type of injury determine the fate of cells with impaired macroautophagy. Autophagy 2008, 4, 516–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, Y.; Stephens, C.M.; Chava, S.; Heidari, Z.; Panigrahi, R.; Williams, D.D.; Wiltz, K.; Bell, A.; Wilson, W.; Reiss, K.; et al. Chaperone-Mediated Autophagy Promotes Beclin1 Degradation in Persistently Infected Hepatitis C Virus Cell Culture. Am. J. Pathol. 2018, 188, 2339–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, P.I.; Cashikar, A. Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 2012, 28, 337–362. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome-mitochondria interactions are modulated by iron release from transferrin. J. Cell. Biol. 2016, 214, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Bartheld, C.S.; Altick, A.L. Multivesicular bodies in neurons: Distribution, protein content, and trafficking functions. Prog. Neurobiol. 2011, 93, 313–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fader, C.M.; Colombo, M.I. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Cuervo, A.M. Autophagy and human disease: Emerging themes. Curr. Opin. Genet. Dev. 2014, 26, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Wesselborg, S.; Stork, B. Autophagy signal transduction by ATG proteins: From hierarchies to networks. Cell Mol. Life Sci. 2015, 72, 4721–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aydin, Y.; Chedid, M.; Chava, S.; Danielle Williams, D.; Liu, S.; Hagedorn, C.H.; Sumitran-Holgersson, S.; Reiss, K.; Moroz, K.; Lu, H.; et al. Activation of PERK-Nrf2 oncogenic signaling promotes Mdm2-mediated Rb degradation in persistently infected HCV culture. Sci. Rep. 2017, 7, 9223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, P.K.; Bao, L.; Song, K.; Aboulnasr, F.M.; Baker, D.P.; Shores, N.; Wimley, W.C.; Liu, S.; Hagedorn, C.H.; Fuchs, S.Y.; et al. HCV infection selectively impairs type I but not type III IFN signaling. Am. J. Pathol. 2014, 184, 214–229. [Google Scholar] [CrossRef] [Green Version]
- Bachurski, D.; Schuldner, M.; Nguyen, P.H.; Malz, A.; Reiners, K.S.; Grenzi, P.C.; Babatz, F.; Schauss, A.C.; Hansen, H.P.; Hallek, M.; et al. Extracellular vesicle measurements with nanoparticle tracking analysis—An accuracy and repeatability comparison between NanoSight NS300 and ZetaView. J. Extracell. Vesicles 2019, 8, 1596016. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, C.; Bruce, K.D.; Burgy, O.; Boyd, T.D.; Michel, C.R.; Garcia-Perez, J.E.; Adame, V.; Anton, P.; Bettcher, B.M.; Chial, H.J.; et al. Exosome Isolation by Ultracentrifugation and Precipitation and Techniques for Downstream Analyses. Curr. Protoc. Cell Biol. 2020, 88, e110. [Google Scholar] [CrossRef] [PubMed]
- Chahar, H.S.; Bao, X.; Casola, A. Exosomes and Their Role in the Life Cycle and Pathogenesis of RNA Viruses. Viruses 2015, 7, 3204–3225. [Google Scholar] [CrossRef] [Green Version]
- Van de Loosdrecht, A.A.; Beelen, R.H.; Ossenkoppele, G.J.; Broekhoven, M.G.; Langenhuijsen, M.M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods 1994, 174, 311–320. [Google Scholar] [CrossRef]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [Green Version]
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Lopez-Otin, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, S.N.; Cheerathodi, M.R.; Nkosi, D.; York, S.B.; Meckes, D.G., Jr. Tetraspanin CD63 Bridges Autophagic and Endosomal Processes to Regulate Exosomal Secretion and Intracellular Signaling of Epstein-Barr Virus LMP1. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Grünvogel, O.; Colasanti, O.; Lee, J.Y.; Klöss, V.; Belouzard, S.; Reustle, A.; Esser-Nobis, K.; Hesebeck-Brinckmann, J.; Mutz, P.; Hoffmann, K.; et al. Secretion of Hepatitis C Virus Replication Intermediates Reduces Activation of Toll-Like Receptor 3 in Hepatocytes. Gastroenterology 2018, 154, 2237–2251.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboulnasr, F.; Hazari, S.; Nayak, S.; Chandra, P.K.; Panigrahi, R.; Ferraris, P.; Chava, S.; Kurt, R.; Song, K.; Dash, A.; et al. IFN-λ Inhibits MiR-122 Transcription through a Stat3-HNF4α Inflammatory Feedback Loop in an IFN-α Resistant HCV Cell Culture System. PLoS ONE 2015, 10, e0141655. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Chen, W.L.; Choi, J.; Wakita, T.; Yen, T.S.; Ou, J.H. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008, 48, 1054–1061. [Google Scholar] [CrossRef] [Green Version]
- Taguwa, S.; Kambara, H.; Fujita, N.; Noda, T.; Yoshimori, T.; Koike, K.; Moriishi, K.; Matsuura, Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J. Virol. 2011, 85, 13185–13194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Filzmayer, C.; Ni, Y.; Sültmann, H.; Mutz, P.; Hiet, M.S.; Vondran, F.W.R.; Bartenschlager, R.; Urban, S. Hepatitis D virus replication is sensed by MDA5 and induces IFN-β/λ responses in hepatocytes. J. Hepatol. 2018, 69, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Albertini, S.; Lo Cigno, I.; Calati, F.; De Andrea, M.; Borgogna, C.; Dell’Oste, V.; Landolfo, S.; Gariglio, M. HPV18 Persistence Impairs Basal and DNA Ligand-Mediated IFN-β and IFN-λ. J. Immunol. 2018, 200, 2076–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broggi, A.; Ghosh, S.; Sposito, B.; Spreafico, R.; Balzarini, F.; Lo Cascio, A.; Clementi, N.; De Santis, M.; Mancini, N.; Granucci, F.; et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 2020, 369, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- EL Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Bukong, T.N.; Momen-Heravi, F.; Kodys, K.; Bala, S.; Szabo, G. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog. 2014, 10, e1004424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosset, F.L.; Dreux, M. HCV transmission by hepatic exosomes establishes a productive infection. J. Hepatol. 2014, 60, 674–675. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnaiah, V.; Thumann, C.; Fofana, I.; Habersetzer, F.; Pan, Q.; de Ruiter, P.E.; Willemsen, R.; Demmers, J.A.; Stalin Raj, V.; Jenster, G.; et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc. Natl. Acad. Sci. USA 2013, 110, 13109–13113. [Google Scholar] [CrossRef] [Green Version]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Decembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari, F.V. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. USA 2010, 107, 7431–7436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwood, N.M.; Golden-Mason, L.; Cheng, L.; Rosen, H.R.; Mengshol, J.A. HCV-infected cells and differentiation increase monocyte immunoregulatory galectin-9 production. J. Leukoc. Biol. 2016, 99, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, X.J.; Ma, C.J.; Wang, J.M.; Wu, X.Y.; Niki, T.; Hirashima, M.; Moorman, J.P.; Yao, Z.Q. HCV-infected hepatocytes drive CD4+ CD25+ Foxp3+ regulatory T-cell development through the Tim-3/Gal-9 pathway. Eur. J. Immunol. 2013, 43, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Charrier, A.; Zhou, Y.; Chen, R.; Yu, B.; Agarwal, K.; Tsukamoto, H.; Lee, L.J.; Paulaitis, M.E.; Brigstock, D.R. Epigenetic regulation of connective tissue growth factor by MicroRNA-214 delivery in exosomes from mouse or human hepatic stellate cells. Hepatology 2014, 59, 1118–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chen, R.; Kemper, S.; Charrier, A.; Brigstock, D.R. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of Twist1. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G491–G499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devhare, P.B.; Sasaki, R.; Shrivastava, S.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Exosome-Mediated Intercellular Communication between Hepatitis C Virus-Infected Hepatocytes and Hepatic Stellate Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gressner, O.A.; Lahme, B.; Demirci, I.; Gressner, A.M.; Weiskirchen, R. Differential effects of TGF-beta on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J. Hepatol. 2007, 47, 699–710. [Google Scholar] [CrossRef]
- Wang, R.; Ding, Q.; Yaqoob, U.; de Assuncao, T.M.; Verma, V.K.; Hirsova, P.; Cao, S.; Mukhopadhyay, D.; Huebert, R.C.; Shah, V.H. Exosome Adherence and Internalization by Hepatic Stellate Cells Triggers Sphingosine 1-Phosphate-dependent Migration. J. Biol. Chem. 2015, 290, 30684–30696. [Google Scholar] [CrossRef] [Green Version]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aydin, Y.; Koksal, A.R.; Reddy, V.; Lin, D.; Osman, H.; Heidari, Z.; Rhadhi, S.M.; Wimley, W.C.; Parsi, M.A.; Dash, S. Extracellular Vesicle Release Promotes Viral Replication during Persistent HCV Infection. Cells 2021, 10, 984. https://doi.org/10.3390/cells10050984
Aydin Y, Koksal AR, Reddy V, Lin D, Osman H, Heidari Z, Rhadhi SM, Wimley WC, Parsi MA, Dash S. Extracellular Vesicle Release Promotes Viral Replication during Persistent HCV Infection. Cells. 2021; 10(5):984. https://doi.org/10.3390/cells10050984
Chicago/Turabian StyleAydin, Yucel, Ali Riza Koksal, Venu Reddy, Dong Lin, Hanadi Osman, Zahra Heidari, Sadeq Mutlab Rhadhi, William C Wimley, Mansour A Parsi, and Srikanta Dash. 2021. "Extracellular Vesicle Release Promotes Viral Replication during Persistent HCV Infection" Cells 10, no. 5: 984. https://doi.org/10.3390/cells10050984