
Modulation of Endosome Function, Vesicle Trafficking and Autophagy by Human Herpesviruses

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Endocytosis-Mediated Herpesvirus Entry
Role of Endocytosis in Herpesvirus Entry
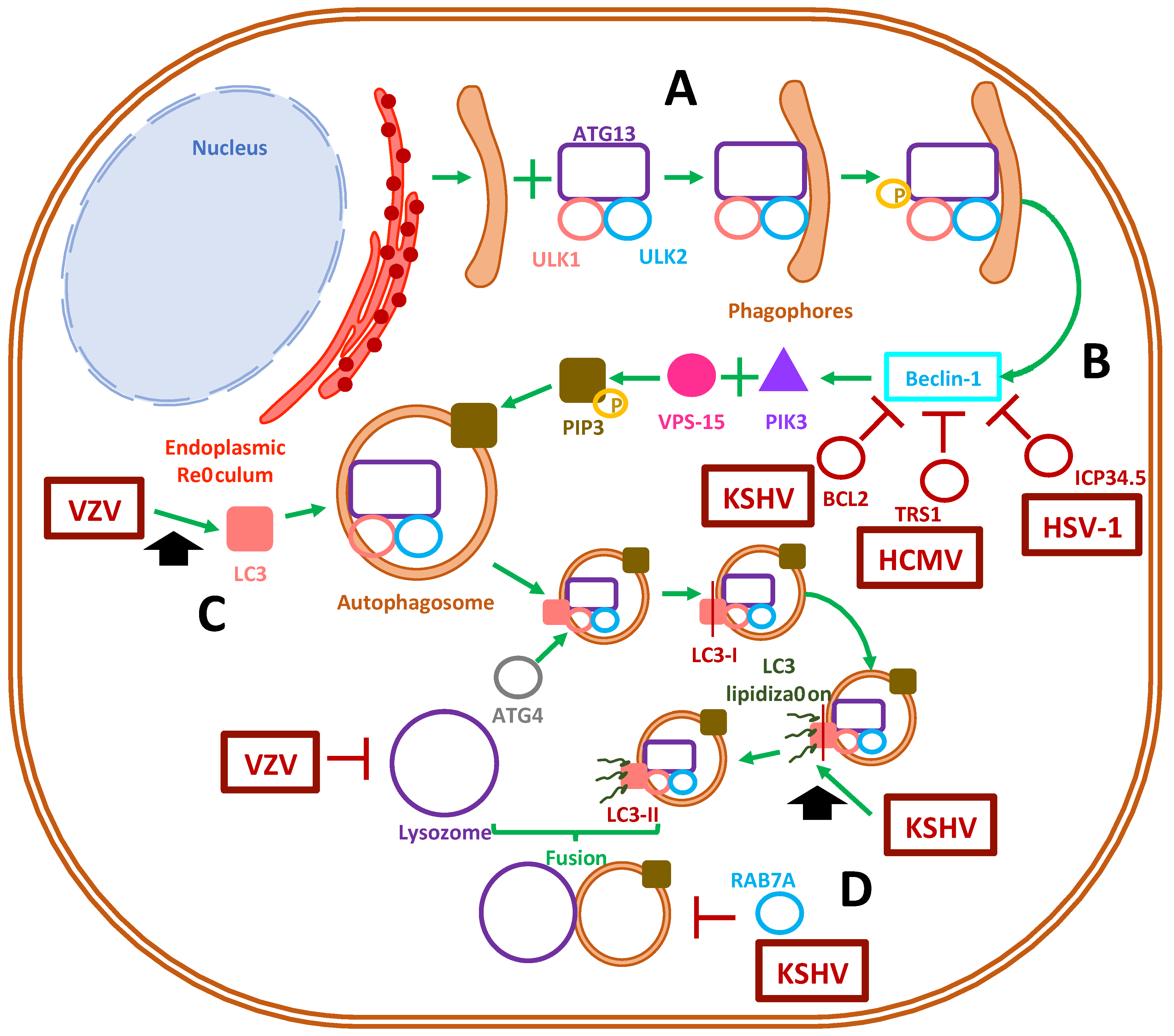
3. Role of Autophagy during Herpesvirus Infections
3.1. Autophagosome Function
3.2. Herpesvirus Modulation of Autophagosome Function
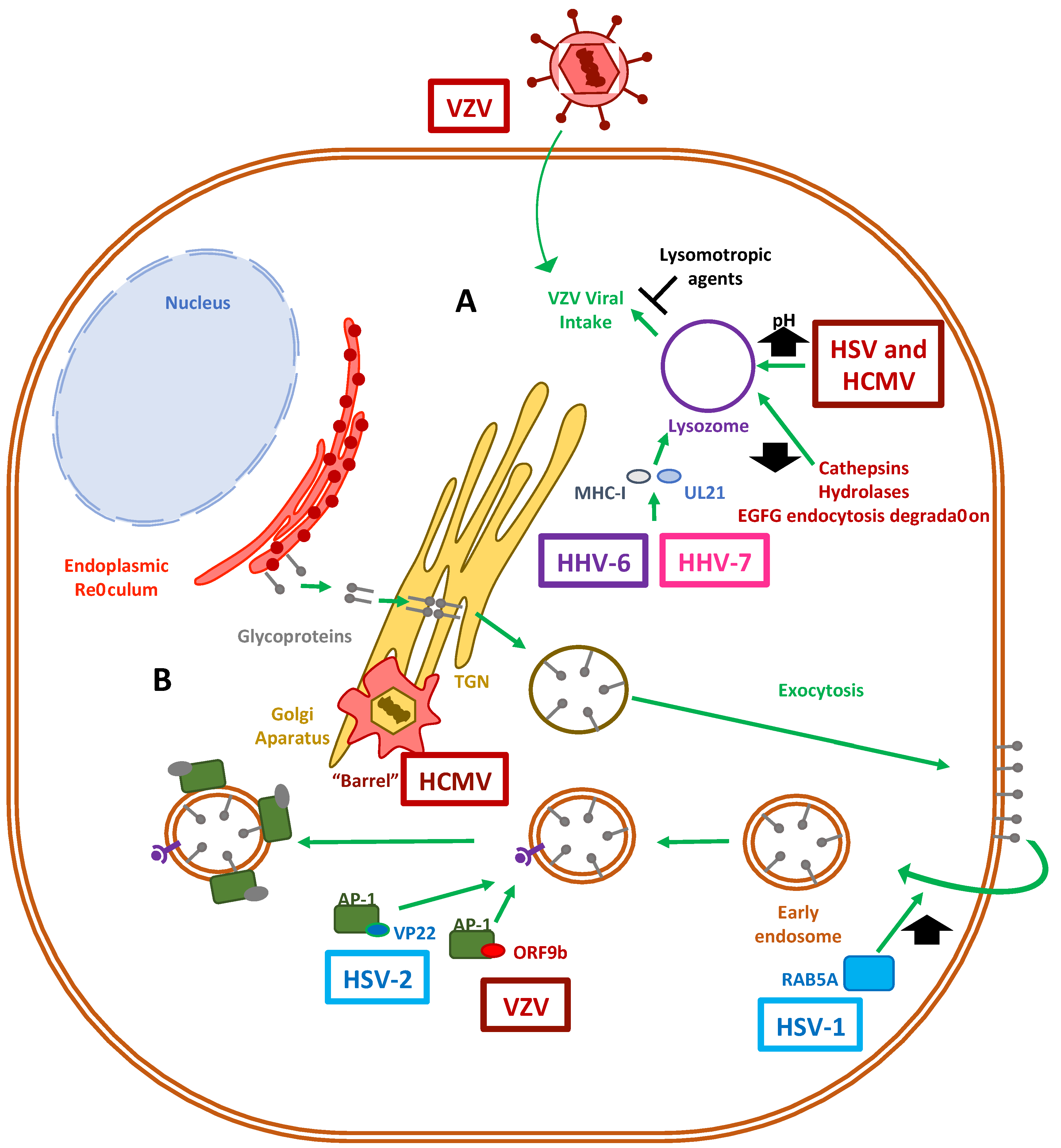
4. Lysosomal and Golgi-Sorting Vesicles during Herpesvirus Exit
4.1. Herpesvirus Modulation of the Lysosomal Pathway
4.2. The Role of the Golgi Apparatus in Herpesvirus Virion Maturation
5. Exocytosis Vesicles Hijacked by Herpesviruses
5.1. Modulation of Exocytosis by Herpesvirus
5.2. Modulation of Endosomal and Exosomal Pathways by Herpesviruses
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rajsbaum, R.; García-Sastre, A. Viral evasion mechanisms of early antiviral responses involving regulation of ubiquitin pathways. Trends Microbiol. 2013, 21, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, Y.; Helenius, A. Virus entry at a glance. J. Cell Sci. 2013, 126, 1289–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-C.; Chathuranga, K.; Lee, J.-S. Intracellular sensing of viral genomes and viral evasion. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tognarelli, E.I.; Palomino, T.F.; Corrales, N.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Herpes Simplex Virus Evasion of Early Host Antiviral Responses. Front. Cell. Infect. Microbiol. 2019, 9, 127. [Google Scholar] [CrossRef]
- Taylor, M.P.; Koyuncu, O.O.; Enquist, L.W. Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Genet. 2011, 9, 427–439. [Google Scholar] [CrossRef] [Green Version]
- Bär, S.; Rommelaere, J.; Nüesch, J.P.F. Vesicular Transport of Progeny Parvovirus Particles through ER and Golgi Regulates Maturation and Cytolysis. PLoS Pathog. 2013, 9, e1003605. [Google Scholar] [CrossRef] [Green Version]
- Votteler, J.; Sundquist, W.I. Virus Budding and the ESCRT Pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Pocock, G.M.; Becker, J.T.; Swanson, C.M.; Ahlquist, P.; Sherer, N.M. HIV-1 and M-PMV RNA Nuclear Export Elements Program Viral Genomes for Distinct Cytoplasmic Trafficking Behaviors. PLoS Pathog. 2016, 12, e1005565. [Google Scholar] [CrossRef] [Green Version]
- Ravindran, M.S.; Bagchi, P.; Cunningham, C.N.; Tsai, B. Opportunistic intruders: How viruses orchestrate ER functions to infect cells. Nat. Rev. Genet. 2016, 14, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Münz, C. The Autophagic Machinery in Viral Exocytosis. Front. Microbiol. 2017, 8, 269. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; Schor, S.; Barouch-Bentov, R.; Einav, S. Viral journeys on the intracellular highways. Cell. Mol. Life Sci. 2018, 75, 3693–3714. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Van Den Worm, S.H.; Zevenhoven-Dobbe, J.C.; Van Der Meer, Y.; Koster, A.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef]
- Amorim, M.J.; Bruce, E.A.; Read, E.K.; Foeglein, Á.; Mahen, R.; Stuart, A.D.; Digard, P. A Rab11- and microtu-bule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol. 2011, 85, 4143–4156. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, J.T.; Swan, D.A.; Corey, L.; Wald, A. Rapid viral expansion and short drug half-life explain the incom-plete effectiveness of current herpes simplex virus 2-directed antiviral agents. Antimicrob. Agents Chemother. 2013, 57, 5820–5829. [Google Scholar] [CrossRef] [Green Version]
- Ju, X.; Yan, Y.; Liu, Q.; Li, N.; Sheng, M.; Zhang, L. Neuraminidase of Influenza A Virus Binds Lysosome-Associated Membrane Proteins Directly and Induces Lysosome Rupture. J. Virol. 2015, 89, 10347–10358. [Google Scholar] [CrossRef] [Green Version]
- Barouch-Bentov, R.; Neveu, G.; Xiao, F.; Beer, M.; Bekerman, E.; Schor, S.; Campbell, J.; Boonyaratanakornkit, J.; Lindenbach, B.; Lu, A.; et al. Hepatitis C Virus Proteins Interact with the Endosomal Sorting Complex Required for Transport (ESCRT) Machinery via Ubiquitination To Facilitate Viral Envelopment. mBio 2016, 7, e01456-16. [Google Scholar] [CrossRef] [Green Version]
- Oliver, S.L.; Zhou, M.; Arvin, A.M. Varicella-zoster virus: Molecular controls of cell fusion-dependent pathogenesis. Biochem. Soc. Trans. 2020, 48, 2415–2435. [Google Scholar] [CrossRef]
- Nowalk, A.; Green, M. Epstein-Barr Virus. Microbiol. Spectr. 2016, 4, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Iizasa, H.; Nanbo, A.; Nishikawa, J.; Jinushi, M.; Yoshiyama, H. Epstein-Barr Virus (EBV)-associated gastric carcinoma. Viruses 2012, 4, 3420–3439. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- Dollard, S.C.; Grosse, S.D.; Ross, D.S. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 2007, 17, 355–363. [Google Scholar] [CrossRef]
- Marsico, C.; Kimberlin, D.W. Congenital Cytomegalovirus infection: Advances and challenges in diagnosis, prevention and treatment. Ital. J. Pediatr. 2017, 43, 1–8. [Google Scholar] [CrossRef]
- Agut, H.; Bonnafous, P.; Gautheret-Dejean, A. Laboratory and Clinical Aspects of Human Herpesvirus 6 Infections. Clin. Microbiol. Rev. 2015, 28, 313–335. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Yamanishi, K. HHV-6A, 6B, and 7: Pathogenesis, Host Response, and Clinical Disease. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Wolz, M.M.; Sciallis, G.F.; Pittelkow, M.R. Human Herpesviruses 6, 7, and 8 from a Dermatologic Perspective. Mayo Clin. Proc. 2012, 87, 1004–1014. [Google Scholar] [CrossRef] [Green Version]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Ou, J.-H.J. Autophagy in viral replication and pathogenesis. Mol. Cells 2010, 29, 71–76. [Google Scholar] [CrossRef]
- Ahmad, I.; Wilson, D.W. HSV-1 Cytoplasmic Envelopment and Egress. Int. J. Mol. Sci. 2020, 21, 5969. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nat. Cell Biol. 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef]
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef] [PubMed]
- Adok, V.A.; Chimini, G. The phagocytosis of apoptotic cells. Semin. Immunol. 2001, 13, 365–372. [Google Scholar]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-Independent Pathways of Endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, a016758. [Google Scholar] [CrossRef] [Green Version]
- Conner, S.D.; Schmid, S.L. Differential requirements for AP-2 in clathrin-mediated endocytosis. J. Cell Biol. 2003, 162, 773–780. [Google Scholar] [CrossRef] [Green Version]
- Sandvig, K.; Pust, S.; Skotland, T.; Van Deurs, B. Clathrin-independent endocytosis: Mechanisms and function. Curr. Opin. Cell Biol. 2011, 23, 413–420. [Google Scholar] [CrossRef]
- Canton, J. Macropinocytosis: New Insights into Its Underappreciated Role in Innate Immune Cell Surveillance. Front. Immunol. 2018, 9, 2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parton, R.G.; Del Pozo, M.A.; Vassilopoulos, S.; Nabi, I.R.; Le Lay, S.; Lundmark, R.; Kenworthy, A.K.; Camus, A.; Blouin, C.M.; Sessa, W.C.; et al. Caveolae: The FAQs. Traffic 2020, 21, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated Transport of Incoming Herpes Simplex Virus 1 Capsids to the Nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef]
- Suazo, P.A.; Ibañez, F.J.; Retamal-Díaz, A.R.; Paz-Fiblas, M.V.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Evasion of early antiviral responses by herpes simplex viruses. Mediat. Inflamm. 2015, 2015, 593757. [Google Scholar] [CrossRef]
- Nicola, A.V.; McEvoy, A.M.; Straus, S.E. Roles for Endocytosis and Low pH in Herpes Simplex Virus Entry into HeLa and Chinese Hamster Ovary Cells. J. Virol. 2003, 77, 5324–5332. [Google Scholar] [CrossRef] [Green Version]
- Nicola, A.V.; Straus, S.E.; Bubić, I.; Wagner, M.; Krmpotić, A.; Saulig, T.; Kim, S.; Yokoyama, W.M.; Jonjić, S.; Koszinowski, U.H. Cellular and Viral Requirements for Rapid Endocytic Entry of Herpes Simplex Virus. J. Virol. 2004, 78, 7536–7544. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar] [CrossRef]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus Entry by Endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, F.M. Diversity of Clathrin Function: New Tricks for an Old Protein. Annu. Rev. Cell Dev. Biol. 2012, 28, 309–336. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, F.M.; Chen, C.-Y.; Knuehl, C.; Towler, M.C.; Wakeham, D.E. Biological Basket Weaving: Formation and Function of Clathrin-Coated Vesicles. Annu. Rev. Cell Dev. Biol. 2001, 17, 517–568. [Google Scholar] [CrossRef] [Green Version]
- Kirchhausen, T.; Owen, D.; Harrison, S.C. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 2014, 6, a016725. [Google Scholar] [CrossRef] [Green Version]
- Akamatsu, M.; Vasan, R.; Serwas, D.; Ferrin, M.A.; Rangamani, P.; Drubin, D.G. Principles of self-organization and load adaptation by the actin cytoskeleton during clathrin-mediated endocytosis. Elife 2020, 9, e49840. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zelhof, A.C. Amphiphysins: Raising the BAR for Synaptic Vesicle Recycling and Membrane Dynamics. Traffic 2002, 3, 452–460. [Google Scholar] [CrossRef]
- Simunovic, M.; Manneville, J.-B.; Renard, H.-F.; Evergren, E.; Raghunathan, K.; Bhatia, D.; Kenworthy, A.K.; Voth, G.A.; Prost, J.; McMahon, H.T.; et al. Friction Mediates Scission of Tubular Membranes Scaffolded by BAR Proteins. Cell 2017, 170, 172–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol. 1994, 75, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.I.; Belval, B.J.; Herold, B.C. Differences in the role of glycoprotein C of HSV-1 and HSV-2 in viral binding may contribute to serotype differences in cell tropism. Virology 1995, 214, 29–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Martinez, W.M.; Spear, P.G. Structural Features of Nectin-2 (HveB) Required for Herpes Simplex Virus Entry. J. Virol. 2001, 75, 11185–11195. [Google Scholar] [CrossRef] [Green Version]
- Stiles, K.M.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Herpes simplex virus gly-coprotein D interferes with binding of herpesvirus entry mediator to its ligands through downregulation and direct com-petition. J. Virol. 2010, 84, 11646–11660. [Google Scholar] [CrossRef] [Green Version]
- Karaba, A.H.; Kopp, S.J.; Longnecker, R. Herpesvirus entry mediator and nectin-1 mediate herpes simplex virus 1 infection of the murine cornea. J. Virol. 2011, 85, 10041–10047. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, R.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes Simplex Virus-1 Entry into Cells Mediated by a Novel Member of the TNF/NGF Receptor Family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Backovic, M.; Jardetzky, T.S. Class III Viral Membrane Fusion Proteins. Adv. Exp. Med. Biol. 2011, 714, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.M.; Whitbeck, J.C.; Lou, H.; Heldwein, E.E.; Chowdary, T.K.; Eisenberg, R.J.; Cohen, G.H. Capturing the herpes simplex virus core fusion complex (gB-gH/gL) in an acidic environment. J. Virol. 2011, 85, 6175–6184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.L.; Heldwein, E.E. Mutations in the cytoplasmic tail of herpes simplex virus 1 gH reduce the fuso-genicity of gB in transfected cells. J. Virol. 2013, 87, 10139–10147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dollery, S.J.; Wright, C.C.; Johnson, D.C.; Nicola, A.V. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J. Virol. 2011, 85, 9964–9973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muggeridge, M.I. Glycoprotein B of Herpes Simplex Virus 2 Has More than One Intracellular Conformation and Is Altered by Low pH. J. Virol. 2012, 86, 6444–6456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sari, T.K.; Gianopulos, K.A.; Weed, D.J.; Schneider, S.M.; Pritchard, S.M.; Nicola, A.V. Herpes Simplex Virus Glycoprotein C Regulates Low-pH Entry. mSphere 2020, 5, 10. [Google Scholar]
- Crisci, E.; Ellegård, R.; Nyström, S.; Rondahl, E.; Serrander, L.; Bergström, T.; Sjöwall, C.; Eriksson, K.; Larsson, M. Complement Opsonization Promotes Herpes Simplex Virus 2 Infection of Human Dendritic Cells. J. Virol. 2016, 90, 4939–4950. [Google Scholar] [CrossRef] [Green Version]
- Zerboni, L.; Sen, N.; Oliver, S.L.; Arvin, A.M. Molecular mechanisms of varicella zoster virus pathogenesis. Nat. Rev. Genet. 2014, 12, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Suenaga, T.; Satoh, T.; Somboonthum, P.; Kawaguchi, Y.; Mori, Y.; Arase, H. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. USA 2009, 107, 866–871. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.J.; Zhu, Z.; Gershon, A.A.; Gershon, M.D. Mannose 6-Phosphate Receptor Dependence of Varicella Zoster Virus Infection In Vitro and in the Epidermis during Varicella and Zoster. Cell 2004, 119, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasieka, T.J.; Maresova, L.; Grose, C. A Functional YNKI Motif in the Short Cytoplasmic Tail of Varicella-Zoster Virus Glycoprotein gH Mediates Clathrin-Dependent and Antibody-Independent Endocytosis. J. Virol. 2003, 77, 4191–4204. [Google Scholar] [CrossRef] [Green Version]
- Pasieka, T.J.; Maresova, L.; Shiraki, K.; Grose, C. Regulation of Varicella-Zoster Virus-Induced Cell-to-Cell Fusion by the Endocytosis-Competent Glycoproteins gH and gE. J. Virol. 2004, 78, 2884–2896. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Adland, A.I.; Bell, H.J.; Delecluse, A.B. Features distinguishing Ep-stein-Barr virus infections of epithelial cells and B cells: Viral genome expression, genome maintenance, and genome am-plification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; White, B.E.P.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein–Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.-B.; Zhang, H.; Zhang, J.-P.; Li, Y.; Zhao, B.; Feng, G.-K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.-L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 6240. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.; Hutt-Fletcher, L.M. Epstein-Barr virus enters B cells and epithelial cells by different routes. J. Virol. 1992, 66, 3409–3414. [Google Scholar] [CrossRef] [Green Version]
- Speck, P.; Haan, K.M.; Longnecker, R. Epstein-Barr virus entry into cells. Virology 2000, 277, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, A.N.; Omerović, J.; Popov, B.; Longnecker, R.; Jardetzky, T.S. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 2006, 80, 9444–9454. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, A.N.; Sorem, J.; Longnecker, R.; Jardetzky, T.S. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure 2009, 17, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Compton, T.; Nowlin, D.M.; Cooper, N.R. Initiation of human cytomegalovirus infection requires initial inter-action with cell surface heparan sulfate. Virology 1993, 193, 834–841. [Google Scholar] [CrossRef]
- Carlson, C.; Britt, W.J.; Compton, T. Expression, Purification, and Characterization of a Soluble Form of Human Cytomegalovirus Glycoprotein B. Virology 1997, 239, 198–205. [Google Scholar] [CrossRef]
- Viswanathan, K.; Smith, M.S.; Malouli, D.; Mansouri, M.; Nelson, J.A.; Früh, K. BST2/Tetherin Enhances Entry of Human Cytomegalovirus. PLoS Pathog. 2011, 7, e1002332. [Google Scholar] [CrossRef] [Green Version]
- Soroceanu, L.; Akhavan, A.; Cobbs, C.S. Platelet-derived growth factor-α receptor activation is required for human cytomegalovirus infection. Nat. Cell Biol. 2008, 455, 391–395. [Google Scholar] [CrossRef]
- Feire, A.L.; Koss, H.; Compton, T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. USA 2004, 101, 15470–15475. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.D.Y.; Huang, S.M. Integrin alphavbeta3 is a coreceptor for human cytomegalovi-rus. Nat. Med. 2005, 11, 515–521. [Google Scholar] [CrossRef]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, B.J.; Rainish, B.L.; Chase, M.C.; Borton, J.A.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. Characterization of the Human Cytomegalovirus gH/gL/UL128-131 Complex That Mediates Entry into Epithelial and Endothelial Cells. J. Virol. 2007, 82, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Johnson Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meraner, P.; Lu, P.; Perreira, J.M.; Aker, A.M.; McDougall, W.M.; Zhuge, R.; Chan, G.C.; Gerstein, R.M.; Caposio, P. OR14I1 is a receptor for the human cytomegalovirus pentameric complex and defines viral epithelial cell tropism. Proc. Natl. Acad. Sci. USA 2019, 116, 7043–7052. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Hayashi, M.; Maeki, T.; Yamanishi, K.; Mori, Y. Human Herpesvirus 6 Glycoprotein Complex Formation Is Required for Folding and Trafficking of the gH/gL/gQ1/gQ2 Complex and Its Cellular Receptor Binding. J. Virol. 2011, 85, 11121–11130. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Serada, S.; Kawabata, A.; Ota, M.; Hayashi, E.; Naka, T.; Yamanishi, K.; Mori, Y. CD134 is a cellular receptor specific for human herpesvirus-6B entry. Proc. Natl. Acad. Sci. USA 2013, 110, 9096–9099. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Yang, X.; Akkapaiboon, P.; Okuno, T.; Yamanishi, K. Human Herpesvirus 6 Variant A Glycoprotein H-Glycoprotein L-Glycoprotein Q Complex Associates with Human CD46. J. Virol. 2003, 77, 4992–4999. [Google Scholar] [CrossRef] [Green Version]
- Akkapaiboon, P.; Mori, Y.; Sadaoka, T.; Yonemoto, S.; Yamanishi, K. Intracellular Processing of Human Herpesvirus 6 Glycoproteins Q1 and Q2 into Tetrameric Complexes Expressed on the Viral Envelope. J. Virol. 2004, 78, 7969–7983. [Google Scholar] [CrossRef] [Green Version]
- Maeki, T.; Hayashi, M.; Kawabata, A.; Tang, H.; Yamanishi, K.; Mori, Y. Identification of the Human Herpesvirus 6A gQ1 Domain Essential for Its Functional Conformation. J. Virol. 2013, 87, 7054–7063. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Suenaga, T.; Matsumoto, M.; Seya, T.; Arase, H. Herpesvirus 6 Glycoproteins B (gB), gH, gL, and gQ Are Necessary and Sufficient for Cell-to-Cell Fusion. J. Virol. 2013, 87, 10900–10903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nii, S.; Yoshida, M.; Uno, F.; Kurata, T.; Ikuta, K.; Yamanishi, K. Replication of Human Herpesvirus 6 (HHV-6): Morphological Aspects. Adv. Exp. Med. Biol. 1990, 278, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M.; Zompetta, C.; Angeloni, A.; Ablashi, D.; Salahuddin, S.; Pavan, A.; Torrisi, M.; Frati, L.; Faggioni, A. Infection by Human Herpesvirus 6 (HHV-6) of Human Lymphoid T Cells Occurs Through an Endocytic Pathway. AIDS Res. Hum. Retrovir. 1992, 8, 2031–2037. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Kawabata, A.; Takemoto, M.; Yamanishi, K.; Mori, Y. Human herpesvirus-6 infection induces the reor-ganization of membrane microdomains in target cells, which are required for virus entry. Virology 2008, 378, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Ahlqvist, J.; Donati, D.; Martinelli, E.; Akhyani, N.; Hou, J.; Major, E.O.; Jacobson, S.; Fogdell-Hahn, A. Complete replication cycle and acquisition of tegument in nucleus of human herpesvirus 6A in astrocytes and in T-cells. J. Med. Virol. 2006, 78, 1542–1553. [Google Scholar] [CrossRef]
- Secchiero, P.; Sun, D.; De Vico, A.L.; Crowley, R.W.; Reitz, M.S.; Zauli, G.; Lusso, P.; Gallo, R.C. Role of the extracellular domain of human herpesvirus 7 glycoprotein B in virus binding to cell surface heparan sulfate proteoglycans. J. Virol. 1997, 71, 4571–4580. [Google Scholar] [CrossRef] [Green Version]
- Skrincosky, D.; Hocknell, P.; Whetter, L.; Secchiero, P.; Chandran, B.; Dewhurst, S. Identification and Analysis of a Novel Heparin-Binding Glycoprotein Encoded by Human Herpesvirus 7. J. Virol. 2000, 74, 4530–4540. [Google Scholar] [CrossRef]
- Lusso, P.; Secchiero, P.; Crowley, R.W.; Garzino-Demo, A.; Berneman, Z.N.; Gallo, R.C. CD4 is a critical compo-nent of the receptor for human herpesvirus 7: Interference with human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 1994, 91, 3872–3876. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Wu, L.; Chai, Y.; Qi, J.; Tan, S.; Gao, G.F.; Song, H.; Yan, J. Molecular basis of EphA2 recognition by gHgL from gammaherpesviruses. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.S.; Desrosiers, R.C. Binding of the Kaposi’s sarcoma-associated herpesvirus to the ephrin binding surface of the EphA2 receptor and its inhibition by a small molecule. J. Virol. 2014, 88, 8724–8734. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, X.; Schaller, S.; Jardetzky, T.S.; Longnecker, R. Ephrin Receptor A4 is a New Kaposi’s Sar-coma-Associated Herpesvirus Virus Entry Receptor. mBio 2019, 10, e02892-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, H.; Sharma-Walia, N.; Veettil, M.V.; Sadagopan, S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J. Virol. 2009, 83, 4895–4911. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.M.; ValiyaVeettil, S.; Sadagopan, N.; Chandran, B. c-Cbl-mediated selective virus-receptor translocations into lipid rafts regulate productive Kaposi’s sarcoma-associated herpesvirus infection in endothelial cells. J. Virol. 2011, 85, 12410–12430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veettil, M.V.; Sadagopan, S.; Kerur, N.; Chakraborty, S.; Chandran, B. Chakraborty and B. Chandran Interaction of c-Cbl with myosin IIA regulates Bleb associated macropinocytosis of Kaposi’s sarcoma-associated herpesvirus. PLoS Pathog. 2010, 6, e1001238. [Google Scholar]
- Greene, W.; Gao, S.J. Actin dynamics regulate multiple endosomal steps during Kaposi’s sarcoma-associated herpesvirus entry and trafficking in endothelial cells. PLoS Pathog. 2009, 5, e1000512. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Xiang, Y.; Wang, X.; Wang, Q.; Zhong, M.; Wang, S. Epidermal growth fac-tor receptor-PI3K signaling controls cofilin activity to facilitate herpes simplex virus 1 entry into neuronal cells. mBio 2014, 5, e00958-13. [Google Scholar] [CrossRef] [Green Version]
- Shoji-Kawata, S.; Levine, B. Autophagy, antiviral immunity, and viral countermeasures. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 1478–1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, L.; Mostowy, S.; Sancho-Shimizu, V. Autophagy-Virus Interplay: From Cell Biology to Human Disease. Front. Cell Dev. Biol. 2018, 6, 155. [Google Scholar] [CrossRef] [Green Version]
- . Valečka, J.; Almeida, C.R.; Su, B.; Pierre, P.; Gatti, E. Autophagy and MHC-restricted antigen presentation. Mol. Immunol. 2018, 99, 163–170. [Google Scholar] [CrossRef]
- Van Kaer, L.; Parekh, V.V.; Postoak, J.L.; Wu, L. Role of autophagy in MHC class I-restricted antigen presentation. Mol. Immunol. 2019, 113, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-Loading Compartments for Major Histocompatibility Complex Class II Molecules Continuously Receive Input from Autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, S.R.; Simonsen, A. Membrane dynamics in autophagosome biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The Role of Atg Proteins in Autophagosome Formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Yu, J.; Byfield, M.; Pattingre, S.; Levine, B. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy 2005, 1, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation—At the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Agrotis, A.; Pengo, N.; Burden, J.J.; Ketteler, R. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 2019, 15, 976–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Ohsumi, Y. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef] [Green Version]
- Nair, U.; Jotwani, A.; Geng, J.; Gammoh, N.; Richerson, D.; Yen, W.-L.; Griffith, J.; Nag, S.; Wang, K.; Moss, T.; et al. SNARE Proteins Are Required for Macroautophagy. Cell 2011, 146, 290–302. [Google Scholar] [CrossRef] [Green Version]
- Diao, J.; Liu, R.; Rong, Y.; Zhao, M.; Zhang, J.; Lai, Y.; Zhou, Q.; Wilz, L.M.; Li, J.; Vivona, S.; et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nat. Cell Biol. 2015, 520, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Wandelmer, J.; Reggiori, F. Amphisomes: Out of the autophagosome shadow? EMBO J. 2013, 32, 3116–3118. [Google Scholar] [CrossRef] [Green Version]
- Cirone, M. EBV and KSHV Infection Dysregulates Autophagy to Optimize Viral Replication, Prevent Immune Recognition and Promote Tumorigenesis. Viruses 2018, 10, 599. [Google Scholar] [CrossRef] [Green Version]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Desjardins, M. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host. Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Tallóczy, Z.; Jiang, W.; Virgin, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J. Regula-tion of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Tallóczy, Z.; Virgin, H., IV; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar]
- Gobeil, P.A.; Leib, D.A. Herpes simplex virus γ34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 2012, 3, e00267-12. [Google Scholar] [CrossRef] [Green Version]
- Yakoub, A.M.; Shukla, D. Basal Autophagy Is Required for Herpes simplex Virus-2 Infection. Sci. Rep. 2015, 5, 12985. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In Vivo Requirement for Atg5 in Antigen Presentation by Dendritic Cells. Immun. 2010, 32, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.Y.; Sugden, B. The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 2008, 111, 2280–2289. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.N.; Jackson, W.; Laird, D.T.; Culp, T.D.; Grose, C.; Haynes, J.I. Varicella-zoster virus infection induces autophagy in both cultured cells and human skin vesicles. J. Virol. 2009, 83, 5466–5476. [Google Scholar] [CrossRef] [Green Version]
- Graybill, C.; Morgan, M.J.; Levin, M.J.; Lee, K.S. Varicella-zoster virus inhibits autophagosome-lysosome fusion and the degradation stage of mTOR-mediated autophagic flux. Virology 2018, 522, 220–227. [Google Scholar] [CrossRef]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Grose, C. Grose Autophagy and the effects of its inhibition on varicel-la-zoster virus glycoprotein biosynthesis and infectivity. J. Virol. 2014, 88, 890–902. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, E.M.; Girsch, J.; Jackson, W.; Cohen, J.I.; Grose, C. Autophagy Quantification and STAT3 Expression in a Human Skin Organ Culture Model for Innate Immunity to Herpes Zoster. Front. Microbiol. 2018, 9, 2935. [Google Scholar] [CrossRef]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin. Cancer Biol. 2014, 26, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Bose, P.; Patel, K.; Roy, S.G.; Gain, C.; Gowda, H.; Robertson, E.S.; Saha, A. Transcriptional and epigenetic modulation of autophagy promotes EBV oncoprotein EBNA3C induced B-cell survival. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- De Leo, A.; Colavita, F.; Ciccosanti, F.; Fimia, G.M.; Lieberman, P.M.; Mattia, E. Inhibition of autophagy in EBV-positive Burkitt’s lymphoma cells enhances EBV lytic genes expression and replication. Cell Death Dis. 2015, 6, e1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Gilardini Montani, M.S.; Santarelli, R.; Granato, M.; Gonnella, R.; Torrisi, M.R.; Faggioni, A.; Cirone, M. EBV reduces autophagy, intracellular ROS and mitochondria to impair monocyte survival and differentiation. Autophagy 2019, 15, 652–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaumorcel, M.; Lussignol, M.; Mouna, L.; Cavignac, Y.; Fahie, K.; Cotte-Laffitte, J.; Geballe, A.; Brune, W.; Beau, I.; Codogno, P.; et al. The Human Cytomegalovirus Protein TRS1 Inhibits Autophagy via Its Interaction with Beclin 1. J. Virol. 2011, 86, 2571–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taisne, C.; Lussignol, M.; Hernandez, E.; Moris, A.; Mouna, L. Human cytomegalovirus hijacks the autophag-ic machinery and LC3 homologs in order to optimize cytoplasmic envelopment of mature infectious particles. Sci. Rep. 2019, 9, 4560. [Google Scholar] [CrossRef] [Green Version]
- Chaumorcel, M.; Souquère, S.; Pierron, G.; Codogno, P.; Esclatine, A. Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 2008, 4, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.J.; Yang, Z.; Zhou, Y.; Wood, C. Enhancement of autophagy during lytic replication by the Kaposi’s sar-coma-associated herpesvirus replication and transcription activator. J. Virol. 2010, 84, 7448–7458. [Google Scholar] [CrossRef] [Green Version]
- Pringle, E.S.; Robinson, C.A.; McCormick, C. Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication Inter-feres with mTORC1 Regulation of Autophagy and Viral Protein Synthesis. J. Virol. 2019, 93, 21. [Google Scholar] [CrossRef] [Green Version]
- Romeo, M.A.; Santarelli, R.; Montani, M.S.G.; Gonnella, R.; Benedetti, R.; Faggioni, A.; Cirone, M. Viral Infection and Autophagy Dysregulation: The Case of HHV-6, EBV and KSHV. Cells 2020, 9, 2624. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Li, Q.; Lee, J.-Y.; Lee, S.-H.; Jeong, J.H.; Lee, H.-R.; Chang, H.; Zhou, F.-C.; Gao, S.-J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.C.; Colbert, C.L.; Becker, N.; Wei, Y.; Levine, B. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 2008, 4, 989–997. [Google Scholar] [CrossRef]
- Park, S.; Buck, M.D.; Desai, C.; Zhang, X.; Loginicheva, E.; Martinez, J. Autophagy Genes Enhance Murine Gammaherpesvirus 68 Reactivation from Latency by Preventing Virus-Induced Systemic Inflamma-tion. Cell Host. Microbe 2016, 19, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leib, D.A.; Alexander, D.E.; Cox, D.; Yin, J.; Ferguson, T.A. Interaction of ICP34.5 with Beclin 1 Modulates Herpes Simplex Virus Type 1 Pathogenesis through Control of CD4+ T-Cell Responses. J. Virol. 2009, 83, 12164–12171. [Google Scholar] [CrossRef] [Green Version]
- Romeo, M.A.; Masuelli, L.; Gaeta, A.; Nazzari, C.; Granato, M.; Montani, M.S.G.; Faggioni, A.; Cirone, M. Impact of HHV-6A and HHV-6B lytic infection on autophagy and endoplasmic reticulum stress. J. Gen. Virol. 2019, 100, 89–98. [Google Scholar] [CrossRef]
- Edens, B.M.; Miller, N.; Ma, Y.-C. Impaired Autophagy and Defective Mitochondrial Function: Converging Paths on the Road to Motor Neuron Degeneration. Front. Cell. Neurosci. 2016, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, L.F.; Farías, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.A.; González, P.A. Herpes Simplex Virus Type 1 Infection of the Central Nervous System: Insights Into Proposed Interrelationships With Neurodegenerative Disorders. Front. Cell. Neurosci. 2019, 13, 46. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Morita, E. Differential requirements of mammalian ESCRTs in multivesicular body formation, virus budding and cell division. FEBS J. 2012, 279, 1399–1406. [Google Scholar] [CrossRef]
- Schuh, A.L.; Audhya, A. The ESCRT machinery: From the plasma membrane to endosomes and back again. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 242–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: It’s all in the neck. Nat. Rev. Mol. Cell. Biol. 2010, 11, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, F.J.; Farías, M.A.; Gonzalez-Troncoso, M.P.; Corrales, N.; Duarte, L.F.; Retamal-Díaz, A.; González, P.A. Experimental Dissection of the Lytic Replication Cycles of Herpes Simplex Viruses. Front. Microbiol. 2018, 9, 2406. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Dammer, E.B.; Ren, R.J.; Wang, G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl. Neurodegener 2015, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Wei, B.L.; Denton, P.W.; O’Neill, E.; Luo, T.; Foster, J.L.; Garcia, J.V. Inhibition of lysosome and proteasome func-tion enhances human immunodeficiency virus type 1 infection. J. Virol. 2005, 79, 5705–5712. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.S.; He, R.; Hoffmann, H.-H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef]
- Smith, J.D.; De Harven, E. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. III. Cytochemical localization of lysosomal enzymes in infected cells. J. Virol. 1978, 26, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Kristen, H.; Sastre, I.; Muñoz-Galdeano, T.; Recuero, M.; Aldudo, J.; Bullido, M.J. The lysosome system is severely impaired in a cellular model of neurodegeneration induced by HSV-1 and oxidative stress. Neurobiol. Aging 2018, 68, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnen, R.L.; Mizokami, K.R.; Banfield, B.W.; Cai, G.Y.; Simpson, S.A.; Pizer, L.I.; Levin, M.J. Postentry events are responsible for restriction of productive varicella-zoster virus infection in Chinese hamster ovary cells. J. Virol. 2006, 80, 10325–10334. [Google Scholar] [CrossRef] [Green Version]
- Welsch, S.; Müller, B.; Kräusslich, H.-G. More than one door—Budding of enveloped viruses through cellular membranes. FEBS Lett. 2007, 581, 2089–2097. [Google Scholar] [CrossRef] [Green Version]
- Calistri, A.; Sette, P.; Salata, C.; Cancellotti, E.; Forghieri, C.; Comin, A.; Göttlinger, H.; Campadelli-Fiume, G.; Palù, G.; Parolin, C. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require func-tional biogenesis of multivesicular bodies. J. Virol. 2007, 81, 11468–11478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glosson, N.L.; Hudson, A.W. Human herpesvirus-6A and -6B encode viral immunoevasins that downregulate class I MHC molecules. Virology 2007, 365, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Brideau, A.; Enquist, L.; Tirabassi, R. The role of virion membrane protein endocytosis in the herpesvirus life cycle. J. Clin. Virol. 2000, 17, 69–82. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: A tale of two membranes. Curr. Opin. Microbiol. 2006, 9, 423–429. [Google Scholar] [CrossRef]
- Alconada, A.U.; Bauer, B.S.; Hoflack, B. Intracellular traffic of herpes simplex virus glycoprotein gE: Char-acterization of the sorting signals required for its trans-Golgi network localization. J. Virol. 1999, 73, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albecka, A.; Laine, R.F.; Janssen, A.F.; Kaminski, C.F.; Crump, C.M. HSV-1 Glycoproteins Are Delivered to Virus Assembly Sites Through Dynamin-Dependent Endocytosis. Traffic 2015, 17, 21–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollinshead, M.; Johns, H.L.; Sayers, C.L.; Gonzalez-Lopez, C.; Smith, G.L.; Elliott, G. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 2012, 31, 4204–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bearer, E.L.; Wu, C. Herpes Simplex Virus, Alzheimer’s Disease and a Possible Role for Rab GTPases. Front Cell Dev. Biol. 2019, 7, 134. [Google Scholar] [CrossRef] [Green Version]
- Campadelli, G.; Brandimarti, R.; Di Lazzaro, C.; Ward, P.L.; Roizman, B.; Torrisi, M.R. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 1993, 90, 2798–2802. [Google Scholar] [CrossRef] [Green Version]
- Bagdonaite, I.; Norden, R.; Joshi, H.J.; Dabelsteen, S.; Nyström, K.; Vakhrushev, S.Y.; Olofsson, S.; Wandall, H.H. A strategy for O-glycoproteomics of enveloped viruses--the O-glycoproteome of herpes simplex virus type 1. PLoS Pathog. 2015, 11, e1004784. [Google Scholar] [CrossRef] [Green Version]
- Lebrun, M.; Lambert, J.; Riva, L.; Thelen, N.; Rambout, X.; Blondeau, C. Varicella-Zoster Virus ORF9p Binding to Cellular Adaptor Protein Complex 1 Is Im-portant for Viral Infectivity. J. Virol. 2018, 92, 15. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Vasanji, A.; Pellett, P.E. Three-Dimensional Structure of the Human Cytomegalovirus Cytoplasmic Virion Assembly Complex Includes a Reoriented Secretory Apparatus. J. Virol. 2007, 81, 11861–11869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alwine, J.C. The Human Cytomegalovirus Assembly Compartment: A Masterpiece of Viral Manipulation of Cellular Processes That Facilitates Assembly and Egress. PLoS Pathog. 2012, 8, e1002878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Close, W.L.; Glassbrook, J.E.; Gurczynski, S.J.; Pellett, P.E. Infection-Induced Changes Within the Endocytic Recycling Compartment Suggest a Roadmap of Human Cytomegalovirus Egress. Front. Microbiol. 2018, 9, 1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrisi, M.R.; Gentile, M.; Cardinali, G.; Cirone, M.; Zompetta, C.; Lotti, L.V.; Frati, L.; Faggioni, A. Intracellular Transport and Maturation Pathway of Human Herpesvirus 6. Virology 1999, 257, 460–471. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Koike, M.; Moriishi, E.; Kawabata, A.; Tang, H.; Oyaizu, H.; Uchiyama, Y.; Yamanishi, K. Human Herpesvirus-6 Induces MVB Formation, and Virus Egress Occurs by an Exosomal Release Pathway. Traffic 2008, 9, 1728–1742. [Google Scholar] [CrossRef] [Green Version]
- Pawliczek, T.; Crump, C.M. Herpes Simplex Virus Type 1 Production Requires a Functional ESCRT-III Complex but Is Independent of TSG101 and ALIX Expression. J. Virol. 2009, 83, 11254–11264. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.; Wilson, D.W. Seeking Closure: How Do Herpesviruses Recruit the Cellular ESCRT Apparatus? J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Hogue, I.B.; Scherer, J.; Enquist, L.W. Enquist Exocytosis of Alphaherpesvirus Virions, Light Particles, and Glycoproteins Uses Constitutive Secretory Mechanisms. mBio 2016, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-P.; Chen, M.-R. Escape of herpesviruses from the nucleus. Rev. Med. Virol. 2010, 20, 214–230. [Google Scholar] [CrossRef]
- Reynolds, A.E.; Ryckman, B.J.; Baines, J.D.; Zhou, Y.; Liang, L.; Roller, R.J. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 2001, 75, 8803–8817. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Zhou, S.; Gao, S.; Deng, H. Remodeling of host membranes during herpesvirus assembly and egress. Protein Cell 2019, 10, 315–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcotte, S.; Letellier, J.; Lippé, R. Herpes Simplex Virus Type 1 Capsids Transit by the trans-Golgi Network, Where Viral Glycoproteins Accumulate Independently of Capsid Egress. J. Virol. 2005, 79, 8847–8860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerke, S.L.; Roller, R.J. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 2006, 347, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Kawaguchi, Y. Us3 Protein Kinase Encoded by HSV: The Precise Function and Mechanism on Viral Life Cycle. Adv. Exp. Med. Biol. 2018, 1045, 45–62. [Google Scholar] [PubMed]
- Saiz-Ros, N.; Czapiewski, R.; Epifano, I.; Stevenson, A.; Swanson, S.K.; Dixon, C.R.; Zamora, D.B.; McElwee, M.; Vijayakrishnan, S.; Richardson, C.A.; et al. Host Vesicle Fusion Protein VAPB Contributes to the Nuclear Egress Stage of Herpes Simplex Virus Type-1 (HSV-1) Replication. Cells 2019, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Polcicova, K.; Johnson, D.C. Herpes Simplex Virus gE/gI and US9 Proteins Promote Transport of both Capsids and Virion Glycoproteins in Neuronal Axons. J. Virol. 2008, 82, 10613–10624. [Google Scholar] [CrossRef] [Green Version]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991203. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.P.; Melancon, J.M.; Olivier, T.L.; Kousoulas, K.G. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 2004, 78, 13262–13277. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Mott, K.R.; Wawrowsky, K.; Kousoulas, K.G.; Luscher, B.; Ghiasi, H. Binding of Herpes Simplex Virus 1 UL20 to GODZ (DHHC3) Affects Its Palmitoylation and Is Essential for Infectivity and Proper Targeting and Localization of UL20 and Glycoprotein K. J. Virol. 2017, 91, e00945-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.-Y.K.; Crump, C.M. HSV-1 gM and the gK/pUL20 Complex Are Important for the Localization of gD and gH/L to Viral Assembly Sites. Viruses 2015, 7, 915–938. [Google Scholar] [CrossRef] [Green Version]
- Naghavi, M.H.; Walsh, D. Microtubule Regulation and Function during Virus Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naghavi, M.H.; Gundersen, G.G.; Walsh, D. Plus-end tracking proteins, CLASPs, and a viral Akt mimic regulate herpesvirus-induced stable microtubule formation and virus spread. Proc. Natl. Acad. Sci. USA 2013, 110, 18268–18273. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, C.R.; Dingwell, K.S.; Wale, C.; Graham, F.L.; Johnson, D.C. Herpes simplex virus gD and virions accu-mulate in endosomes by mannose 6-phosphate-dependent and -independent mechanisms. J. Virol. 1998, 72, 3330–3339. [Google Scholar] [CrossRef] [Green Version]
- Temme, S.; Eis-Hübinger, A.M.; McLellan, A.D.; Koch, N. The Herpes Simplex Virus-1 Encoded Glycoprotein B Diverts HLA-DR into the Exosome Pathway. J. Immunol. 2010, 184, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Niazy, N.; Temme, S.; Bocuk, D.; Giesen, C.; König, A.; Temme, N. Misdirection of endosomal trafficking mediated by herpes simplex virus-encoded gly-coprotein B. FASEB J. 2017, 31, 1650–1667. [Google Scholar] [CrossRef] [Green Version]
- Grabowska, K.; Wąchalska, M.; Graul, M.; Rychłowski, M.; Bieńkowska-Szewczyk, K.; Lipińska, A.D. Alphaherpesvirus gB Homologs Are Targeted to Extracellular Vesicles, but They Differentially Affect MHC Class II Molecules. Viruses 2020, 12, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, E.M.; Jarosinski, K.W.; Jackson, W.; Carpenter, J.E.; Grose, C. Exocytosis of Varicella-Zoster Virus Virions Involves a Convergence of Endosomal and Autophagy Pathways. J. Virol. 2016, 90, 8673–8685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girsch, J.H.; Jackson, W.; Carpenter, J.E.; Moninger, T.O.; Jarosinski, K.W.; Grose, C. Exocytosis of Progeny Infectious Varicella-Zoster Virus Particles via a Mannose-6-Phosphate Receptor Pathway without Xenophagy following Secondary Envelopment. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Zerboni, L.; Sung, P.; Sommer, M.; Arvin, A. The C-terminus of varicella-zoster virus glycoprotein M contains trafficking motifs that mediate skin virulence in the SCID-human model of VZV pathogenesis. Virology 2018, 523, 110–120. [Google Scholar] [CrossRef]
- Canitano, A.; Venturi, G.; Borghi, M.; Ammendolia, M.G.; Fais, S. Exosomes released in vitro from Epstein–Barr virus (EBV)-infected cells contain EBV-encoded latent phase mRNAs. Cancer Lett. 2013, 337, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Nanbo, A.; Sun, L.; Lin, Z. Extracellular Vesicles in Epstein-Barr Virus’ Life Cycle and Pathogenesis. Microorganisms 2019, 7, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, S.N.; Nkosi, D.; Conlon, M.M.; York, S.B.; Liu, X.; Tremblay, D.C.; Meckes, D.G. CD63 Regulates Epstein-Barr Virus LMP1 Exosomal Packaging, Enhancement of Vesicle Production, and Noncanonical NF-kappaB Signaling. J. Virol. 2017, 91, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepeda, V.; Esteban, M.; Fraile-Ramos, A. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell Microbiol. 2010, 12, 386–404. [Google Scholar] [CrossRef]
- Das, S.; Pellett, P.E. Spatial Relationships between Markers for Secretory and Endosomal Machinery in Human Cytomegalovirus-Infected Cells versus Those in Uninfected Cells. J. Virol. 2011, 85, 5864–5879. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tognarelli, E.I.; Reyes, A.; Corrales, N.; Carreño, L.J.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Modulation of Endosome Function, Vesicle Trafficking and Autophagy by Human Herpesviruses. Cells 2021, 10, 542. https://doi.org/10.3390/cells10030542
Tognarelli EI, Reyes A, Corrales N, Carreño LJ, Bueno SM, Kalergis AM, González PA. Modulation of Endosome Function, Vesicle Trafficking and Autophagy by Human Herpesviruses. Cells. 2021; 10(3):542. https://doi.org/10.3390/cells10030542
Chicago/Turabian StyleTognarelli, Eduardo I., Antonia Reyes, Nicolás Corrales, Leandro J. Carreño, Susan M. Bueno, Alexis M. Kalergis, and Pablo A. González. 2021. "Modulation of Endosome Function, Vesicle Trafficking and Autophagy by Human Herpesviruses" Cells 10, no. 3: 542. https://doi.org/10.3390/cells10030542